

Abstract
Although K-Ras, Cdc42, and PAK4 signaling are commonly deregulated in cancer, only a few studies have sought to comprehensively examine the spectrum of phosphorylation-mediated signaling downstream of each of these key signaling nodes. In this study, we completed a label-free quantitative analysis of oncogenic K-Ras, activated Cdc42, and PAK4-mediated phosphorylation signaling, and report relative quantitation of 2152 phosphorylated peptides on 1062 proteins. We define the overlap in phosphopeptides regulated by K-Ras, Cdc42, and PAK4, and find that perturbation of these signaling components affects phosphoproteins associated with microtubule depolymerization, cytoskeletal organization, and the cell cycle. These findings provide a resource for future studies to characterize novel targets of oncogenic K-Ras signaling and validate biomarkers of PAK4 inhibition.
The Ras oncoproteins are small monomeric GTPases that transduce mitogenic signals from cell surface receptor tyrosine kinases (RTKs) to intracellular serine/threonine kinases. Approximately thirty percent of human tumors harbor a somatic gain-of-function mutation in one of three RAS genes, resulting in the constitutive activation of Ras signaling and the aberrant hyperactivation of growth-promoting effector pathways (1). Designing therapeutic agents that directly target Ras has been challenging (2, 3), and thus clinical development efforts have focused on targeting effector pathways downstream of Ras. The Raf-MEK-ERK and PI3K-Akt effector pathways have been extensively studied and several small molecule inhibitors targeting these pathways are currently under clinical evaluation (4, 5). However, biochemical studies and mouse models indicate that several additional effector pathways are essential for Ras-driven transformation and tumorigenesis (6–11). Hence, a comprehensive characterization of these effector pathways may reveal additional druggable targets.
The Rho GTPase Cdc42 lies downstream of Ras (12–14) and regulates many cellular processes that are commonly perturbed in cancer, including migration, polarization, and proliferation (15) (Fig. 1A). Importantly, Cdc42 is overexpressed in several types of human cancer (16–20) and is required for Ras-driven cellular transformation (13, 21, 22). Recent studies show that genetic ablation of Cdc42 impairs Ras-driven tumorigenesis (13), indicating the potential of Cdc42 and its effectors as drug targets in Ras mutant tumors.
Fig. 1.
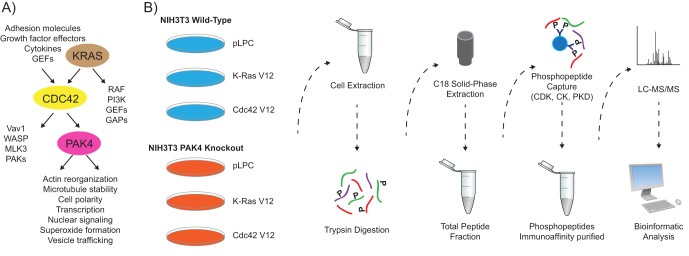
Experimental workflow. A, K-Ras is a small GTPase that regulates the activity of a variety of downstream proteins including the Rho GTPase Cdc42. The PAK4 serine/threonine kinase is a direct effector of Cdc42 and regulates actin reorganization, microtubule stability, and cell polarity. B, To measure large-scale phosphorylation changes induced by constitutive K-Ras or Cdc42 signaling or PAK4 ablation, the quantitative label-free PTMscan® approach was employed (Cell Signaling Technology). Briefly, for each condition extracted proteins were digested with trypsin and separated from non-peptide material by solid-phase extraction with Sep-Pak C18 cartridges. Three phosphorylation motif antibodies were used serially to isolate phosphorylated peptides in independent immunoaffinity purifications (CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], PKD substrate motif l-X-R-X-X-p[S T]). The samples were run in duplicate and tandem mass spectra were collected with an LTQ-Orbitrap hybrid mass spectrometer. pLPC is an empty vector control.
In particular, the p21-activated kinases (PAKs) are Cdc42 effectors that have generated significant interest (23, 24), as they are central components of key oncogenic signaling pathways and regulate cytoskeletal organization, cell migration, and nuclear signaling (25). The PAK family is comprised of six members and is subdivided into two groups (Groups I and II) based on sequence and structural homology. Group I PAKs (PAK1–3) are relatively well characterized, however, much less is known regarding the function and regulation of Group II PAKs (PAK4–6). The kinase domains of Group I and II PAKs share only about 50% identity, suggesting the two groups may recognize distinct substrates and govern unique cellular processes (26).
The Group II PAK family member PAK4 is of particular interest as it is overexpressed or genetically amplified in several lung, colon, prostate, pancreas, and breast tumor cell lines and samples (26–30). Furthermore, functional studies have implicated PAK4 in cell transformation, cell invasion, and migration (27, 31). Xenograft studies in athymic mice show an important role for PAK4 in mediating Cdc42- or K-Ras-driven tumor formation, highlighting a critical role for Pak4 downstream of these GTPases (32). Given its roles in transformation, tumorigenesis, and oncogenic signaling, there is significant interest in targeting PAK4 therapeutically (23). PAK4 binds and phosphorylates several proteins involved in cytoskeletal organization and apoptosis, including Lim domain kinase 1 (LIMK1) (33), guanine nucleotide exchange factor-H1 (GEF-H1) (34), Raf-1 (35), and Bad (36). However, the Group I PAK family member PAK1 also phosphorylates several of these PAK4 targets (37). Thus, there remains a need to identify robust and selective pharmacodynamic biomarkers for PAK4 inhibition.
Despite the importance of PAK4 and its upstream regulators in cancer development, few studies have sought to comprehensively characterize the spectrum of K-Ras, Cdc42, or PAK4 mediated phosphorylation signaling (37–39). Recent developments in mass spectrometry allow the in-depth identification and quantitation of thousands of phosphorylation sites (40–43). The majority of large-scale efforts have aimed to identify the basal phosphoproteomes of different species (44, 45) or tissues (46) to characterize global steady-state phosphorylation. However, this methodology can also be applied to quantify perturbed phosphorylation regulation in cancer signaling pathways (40, 47–49), and has the potential to reveal novel biomarkers of oncogenic signaling.
In this study, we completed a label-free quantitative analysis of K-Ras, Cdc42, and PAK4 phosphorylation signaling using the PTMScan® method, which has proven as robust and reproducible quantitation technology (50, 51). We quantified phosphorylation levels in wild-type and PAK4 knockout NIH3T3 cells expressing oncogenic K-Ras, activated Cdc42, or an empty vector control to elucidate the molecular pathways and functions modulated by these key signaling proteins. We report relative quantitation of 2152 phosphorylated peptides on 1062 proteins among the different conditions, and find that many of the regulated phosphoproteins are associated with microtubule depolymerization, cytoskeletal organization, and the cell cycle. To our knowledge, our study is the first to examine the overlap among signaling networks regulated by K-Ras, Cdc42, and PAK4, and provides a resource for future studies to further interrogate the perturbation of this signaling pathway.
MATERIALS AND METHODS
Culturing of NIH3T3 Cells
Wild-type and Pak4−/− NIH3T3 cells expressing either K-Ras V12, Cdc42 V12, or an empty pLPC vector control have been described previously (32). Cell lines were maintained at 37 °C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 4 mm l-glutamine.
Immunoblotting
To prepare lysates for immunoblot analysis, cells were washed once with cold PBS and lysed in 1x Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA) supplemented with 1 mm PMSF, as described by the manufacturer. After addition of lysis buffer, samples were rocked at 4 °C for 10 - 30 min, scraped, collected, and cleared by centrifugation at 13,000 rpm for 10 min at 4 °C. Protein levels were quantified by the BCA1 Protein Assay Kit (Pierce Biotechnology, Waltham, MA), normalized to equal concentrations and boiled in LDS sample buffer (Invitrogen, Carlsbad, CA) supplemented with DTT for 10 min at 70 °C.
Equal amounts of protein extracts were resolved using SDS-polyacrylamide gel electrophoresis (NuPAGE, Invitrogen) and transferred to a nitrocellulose membrane. After blocking for at least 1 h at room temperature with blocking buffer (Rockland Immunochemicals, Gilbertsville, Pa), membranes were incubated with primary antibodies overnight at 4 °C. Antibodies for phospho-AKT, phospho-eEF2, eEF2, phospho-ERK, ERK, phospho-p90RSK, p90RSK, phospho-S6, S6, phospho-TSC2, and TSC2, were obtained from Cell Signaling. The β-actin antibody was obtained from Sigma, the AKT antibody was obtained from Millipore, and the PAK4 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Specific antigen-antibody interaction was detected with a secondary antibody labeled with either IRDye800 (Rockland Immunochemicals) or Alexa Fluor 680 (Molecular Probes, Eugene, OR) and was visualized by the LI-COR Odyssey Imaging System.
PreScreen Immunoblot Profiling
A kinome-wide view of cellular phosphorylation was obtained by probing NIH3T3 protein lysates with 17 phospho-motif antibodies (KinomeView®, Cell Signaling Technology, www.cellsignal.com/services/kinomeview.html). The following phospho-motif antibodies were employed in immunoblot profiling: AKT substrate motif R-X-X-p[S T], AKT substrate motif R-X-R-X-X-p[S T], ATM/R substrate motif p[S T]-Q, ATM/R substrate motif p[S T]-Q-G, CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], MAPK substrate motif P-X-pS-P, MAPK substrate motif P-X-pT-P, PDK1 docking motif [F Y]-p[S T]-[F Y], PKA substrate motif [K R]-[K R]-X-p[S T], PKC substrate motif [K R]-X-pS-X-[K R], PKD substrate motif l-X-R-X-X-p[S T], PLK binding motif S-pT-P, phospho-motif pT-P-E, phospho-motif pT-P, phospho-motif pT-X-R, phosphotyrosine motif pY (“X” represents any amino acid, and “p” reflects the phosphorylated site). Based on the results of immunoblot profiling, three antibodies (CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], PKD substrate motif l-X-R-X-X-p[S T]) were selected for use in phosphoproteomic studies.
Phosphoproteomic Screen Sample Preparation
To prepare lysates for the phosphoproteomic screen, cells were washed once with cold PBS and lysed in Urea Lysis Buffer (20 mm HEPES pH 8.0, 9.0 m urea, 1 mm sodium orthovanadate, 2.5 mm sodium pyrophosphate, 1 mm β–glycerol-phosphate). For each experimental condition, ten 150 mm dishes of cells grown to 70%–80% confluency were lysed in a total of 10 ml Urea Lysis Buffer. Lysates were snap frozen on dry ice/ethanol until further processing.
Protein Extraction and Digestion
Sonication of cell lysates was applied at 15W output power twice for 20 s, and once for 15 s. The resulting sonicated lysates were centrifuged at 20,000 × g for 15 min to remove insoluble material. The yielded protein extracts were reduced and carboxamidomethylated.
After normalizing total protein for each cell line, proteins were digested overnight using trypsin. Resulting peptides were separated from non-peptide material by solid-phase extraction with Sep-Pak C18 cartridges. Lyophilized peptides were re-dissolved, and phosphopeptides were enriched by serial immunoaffinity purifications, using slurries of the appropriate immobilized motif antibody in the following order: CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], PKD substrate motif l-X-R-X-X-p[S T]. After elution from antibody-resin into a total volume of 100 μl in 0.15% TFA, peptides were concentrated with C18 spin tips immediately prior to LC-MS analysis.
Mass Spectrometric Analysis
The samples were run in duplicate to generate analytical replicates and increase the number of MS/MS identifications from each sample. Peptides were loaded directly onto a 10 cm × 75 μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. The column was developed with a 90-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nL/min. Tandem mass spectra were collected with an LTQ-Orbitrap VELOS hybrid mass spectrometer running XCalibur 2.0.7 SP1 using a top 20 method, a dynamic exclusion repeat count of 1 and a repeat duration of 30 s. Real time recalibration of mass error was performed using lock mass (52) with a singly charged polysiloxane ion (m/z = 371.101237). MS spectra were collected in the Orbitrap component of the mass spectrometer, and MS/MS spectra were collected in the LTQ. A mass accuracy of ±50 ppm was used for precursor ions and 1 Da for product ions.
Data Analysis
Data processing was performed as described previously (52). MS/MS spectra were processed using SEQUEST 3G and the SORCERER 2 platform from Sage-N Research (v4.0, Milpitas CA) (53). Searches were performed against the NCBI Mus musculus FASTA database updated on September 6th, 2010 (release 43) containing 36,483 sequences. Reverse decoy databases were included for all searches to estimate false positive rates, and peptide assignments were obtained using a 5% false positive discovery rate in the Peptide Prophet module of SORCERER 2. Up to 4 missed cleavages were allowed and peptides with one non-tryptic terminus (not cleaved after K or R) were allowed. Cysteine carboxamidomethylation was specified as a static modification, and methionine oxidation and STY phosphorylation was allowed. Results were further narrowed using mass accuracy (−/+ 5ppm) filters and the presence of the intended phosphorylation motif for the antibodies used. The false positive discovery rate on motif-containing peptides was less than 1%. All data may be downloaded from ProteomeCommons.org Tranche using the following hash: fHn2alo+HazR6KYuU965W5pUgqn7/6PmbZzBDLur4KXfPkBdS3ZVUiL5UgMd2GCHv7Sabhassy+GdUkRKoZPBeMlJY4AAAAAAAATKw==.
Label-Free Quantitation
For each MS/MS spectrum the corresponding parent ion intensities were obtained from the extracted ion chromatogram of the corresponding LCMS data file of each sample condition. Changes in phosphorylated peptide intensities were measured by taking the ratio of averaged raw intensities between two specified conditions. Resulting raw ratios were converted to log2 ratios and normalized by applying an offset correction to force the median log2 ratio of all quantified peptides to zero. To quantify peptide levels throughout all samples, we searched for phosphorylated peptide ions in the ion chromatogram files on the basis of their chromatographic retention times and their mass-to-charge (m/z) ratios for all phosphorylated peptides identified by MS/MS in at least one sample. The retention time window used was variable and based on the systematic retention time deviation pattern seen from the extracted ion chromatograms, and the m/z range used was also variable and dependent on the mass error narrowing performed in a previous step. For each peptide ion, the corresponding retention time, observed m/z ratio, and intensity measurement was retrieved. Peak intensity measurements for peptide ions with a fold change above 2.5 were manually reviewed by examining their corresponding extraction ion chromatogram, to make sure that the automated process selected the correct chromatographic peak from which to derive the corresponding intensity measurement. Manually validated intensities are reported in supplemental Table S1 in bold.
Bioinformatics Analysis
Statistical calculations and bioinformatic analyses were performed using the R software environment (http://www.R-project.org). To map Uniprot accessions to Entrez gene identifiers and retrieve further annotation data, we used Ensembl BiomaRt. Phosphorylated peptides with minimum threefold intensity changes induced by PAK4 knockout in pLPC control samples were defined as ‘reversed’ by K-Ras or Cdc42 activation, if there was no intensity change (less than threefold) or opposite changes between K-Ras/Cdc42 activated PAK4 knockout and pLPC treated wild-type samples. In addition the intensity ratio measured in K-Ras/Cdc42 activated versus pLPC control samples (both with PAK4 knockout) was required to be converse (minimum twofold). To highlight different categories of identified phosphopeptides, hierarchical clustering was performed on log2-transformed raw intensity levels using Ward's method and Pearson correlation coefficient as the distance metric.
Using the ‘GOstats’ R package significantly overrepresented biological processes were identified on the basis of hypergeometric tests. All identified phosphorylated proteins were used as gene universe, whereas phosphoproteins with minimum threefold intensity changes were selected as subsets. In the hypergeometric model, each GO term is treated as independent classification and the resulting probability reflects whether the number of selected phosphoproteins associated with the specified GO term is larger than expected.
RESULTS
Depth of the Detected Phosphoproteomes
To measure quantitative changes in the levels of phosphorylation sites induced by perturbing K-Ras, Cdc42, or PAK4 signaling, we employed the label-free based PTMScan® method from Cell Signaling Technology (50) in wild-type and PAK4 knockout NIH3T3 cell lines expressing oncogenic K-Ras, activated Cdc42, or an empty vector control (Fig. 1B) (Materials and Methods). These cell lines have previously been used to show a critical role for PAK4 in K-Ras- and Cdc42-driven tumor formation (32), and provide a novel platform to explore signaling downstream of PAK4, as PAK4-selective small molecule inhibitors are currently unavailable.
Prior to mass spectrometric analysis, a kinome-wide view of cellular phosphorylation was obtained by probing NIH3T3 protein lysates with 17 phospho-motif antibodies (KinomeView® Service, Cell Signaling Technology, (54), Materials and Methods). Phospho-motif antibodies associated with kinase substrate recognition of CDK, CK, or PKD yielded the most evident up- as well as down-regulation of phosphorylation based on immunoblot profiling, and were therefore selected for phosphopeptide enrichment (supplemental Fig. S1). The corresponding substrate sequence motifs are [K R]-pS-P-X-[K R], pT-[D E]-X-[D E], and L-X-R-X-X-p[S T], respectively, where “X” represents any amino acid, and “p” reflects the phosphorylated site. In the subsequent PTMScan® method, proteins were extracted from each cell line, digested with trypsin, and separated from non-peptide material by solid-phase extraction with Sep-Pak C18 cartridges (50). Phosphorylation motif antibodies were used to isolate phosphorylated peptides in serial immunoaffinity purifications, in the following order: CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], PKD substrate motif l-X-R-X-X-p[S T]. The samples were run in duplicate and tandem mass spectra were acquired with an LTQ-Orbitrap VELOS hybrid mass spectrometer.
With this approach, we detected 2359 phosphorylation sites matching the intended phosphorylation motifs from 1062 proteins with an estimated false positive rate of less than 1% (Fig. 2). In total, 99% of the 2152 reported phosphorylated peptides with unique m/z values could be quantified throughout all conditions. With an average coefficient of determination R2 value of 0.94, phosphopeptide intensities correlated very well between technical replicates (supplemental Fig. S2). Median coefficients of variation (CV) between analytical replicate injections of the same samples ranged from 13.5% to 16.4% (supplemental Table S1) indicating reproducible quantitation. All identified phosphorylated peptides are listed in supplemental Table S1. Phosphorylated peptides with a minimum fold change of 2.5 were manually reviewed by inspecting their corresponding extracted ion chromatograms and are highlighted in bold (supplemental Table S1). Individually, CDK, CK, and PKD motif antibody enrichment led to the identification of 617, 742, and 827 phosphopeptides with unique m/z values, respectively. Some of the phosphopeptides were detected in more than one phosphorylation state. Removing this redundancy results in the mapping of 1682 unique peptides. Different sequence motif enrichments proved to be complementary, as 98%, 94% and 93% of the 1682 peptides were detected exclusively with CDK, CK, and PKD phospho-motif enriched experiments, respectively. The majority of the identified peptides were singly phosphorylated (68%), but multiply phosphorylated peptides were also observed. This explains the occurrence of identified phosphorylation sites that do not match with the sequence motif of the corresponding antibody as illustrated by the sequence logos in Fig. 2. Furthermore, sequence motif analysis of the CDK phosphomotif antibody set revealed high specificity for proline on the +1 position relative to the phosphorylation site, but low specificity for arginine and lysine on the −1 and +3 positions.
Fig. 2.

Depth of the detected phosphoproteomes. The number of identifications was similar between different phospho-motif antibody enrichment methods ranging from 617 to 827 phosphopeptides after filtering out duplicates. Around 67% of the mapped phosphosites were novel in reference to the PhosphoSitePlus database. Overall, 68% of the identified peptides were singly phosphorylated, whereas 32% of the peptides carried multiple phosphorylation sites.
Overall, 1581 phosphosites (67%) identified were not recorded in the PhosphoSitePlus database (www.phosphosite.org). A similar range of previously unreported phosphorylation events has been described in prior studies (42) supporting the hypothesis that the identification of the eukaryotic phosphoproteome is far from complete. This trend also suggests that protein kinases phosphorylate more than the estimated one-third of the proteome.
Global Phosphoproteome Response to K-Ras, Cdc42, and PAK4 Perturbation
Quantitation of identified phosphorylated peptides revealed that K-Ras, Cdc42, and PAK4 perturbations yielded both up- as well as down-regulation of phosphorylation. However, the overall phosphorylation levels varied only minimally between samples, so that the median phosphorylation intensities were almost identical (supplemental Fig. S3). Consequently, normalization factors were only minimal (supplemental Table S1) as evident from the similarity between distributions of normalized versus raw phosphorylation changes (supplemental Fig. S4) (Materials and Methods). In the rare case of phosphopeptides that were detected across multiple antibody enrichments, only their first identification in the sequential enrichment series was considered in the quantitative analysis. To examine the signaling networks regulated by oncogenic K-Ras, we compared the phosphoproteome of NIH3T3 cells expressing K-Ras V12 with that of NIH3T3 cells expressing an empty vector control. The G12V mutation in K-Ras renders the protein constitutively active and is commonly observed in human tumors (55). We used a threefold cutoff to define up- or down-regulation, as 90% of the phosphopeptide intensity changes fall within the ± threefold change interval for most comparisons. The relative abundance of 298 phosphopeptides representing 192 proteins was at least threefold different between the control and K-Ras V12 expressing samples (Fig. 3A). Of these, 152 phosphopeptides (102 proteins) were up-regulated by expression of K-Ras V12, whereas the remaining 146 phosphopeptides (93 proteins) were down-regulated. Phosphorylated residues positively modulated by K-Ras V12 include known regulatory phosphorylation sites on ERK1 (extracellular-signal-regulated kinase 1) and ERK2 (extracellular-signal-regulated kinase 2) as well as sites on the signaling proteins GAB2 (GRB2-associated-binding protein 2) and afadin (supplemental Table S1). Phosphorylated residues negatively modulated by K-Ras V12 include sites on the adaptor protein DAB2 (Disabled homolog 2), the Rap effector RADIL (Ras-associating and dilute domain-containing protein), and the tumor suppressor protein MTUS1 (Microtubule-associated tumor suppressor homolog 1).
Fig. 3.

Quantitative phosphoproteomic analysis of K-Ras, Cdc42, and PAK4 perturbation. A, The numbers of phosphorylated peptides (-proteins) with threefold intensity changes induced by K-Ras activation, Cdc42 activation, or PAK4 knockout were in a comparable range. The number of regulated phosphoproteins is given in parentheses. Up-regulated phosphopeptides are boxed in red, whereas down-regulated phosphopeptides are boxed in green. Density plots illustrate the distributions of logarithmized phosphopeptide intensity ratios for given comparisons. Venn diagrams depict the overlap in regulated phosphopeptides (upper diagram) and phosphoproteins (lower diagram). WT: wild-type; KO: knockout; pLPC: empty vector control. B, Clustering (using Ward's method) of the 100 most variable phosphorylated peptides based on measured intensities elucidated different regulation patterns. C, Biological processes that were significantly overrepresented in the set of regulated proteins based on Gene Ontology annotation. The expected number of proteins associated with a certain cellular process was derived from the total set of identified phosphoproteins. Enrichment of GO terms among the targets of K-Ras, Cdc42 and PAK4 was determined using the R package GOstats.
Activation of Cdc42 signaling by expression of the constitutively active Cdc42 V12 mutant affected the relative abundance of 194 phosphopeptides representing 142 proteins (Fig. 3A). Comparatively, Cdc42 V12 regulated 104 fewer phosphopeptides than K-Ras V12. The ratio of phosphopeptide changes positively and negatively regulated by Cdc42 V12 was around 1:1, as was the case for K-Ras V12. Phosphopeptides regulated by Cdc42 V12 include those derived from small GTPase RHEB and the Rho GTPase activating protein RALBP1 (RalA-binding protein 1), indicating additional possible roles for Cdc42 in modulating GTPase signaling (56, 57).
To identify PAK4-dependent signaling changes, we compared the phosphoproteome of NIH3T3 PAK4 knockout cells with that of NIH3T3 wild-type cells. The relative abundance of 283 phosphopeptides representing 211 proteins was at least threefold different between the two conditions (Fig. 3A). The total number of significant phosphopeptide changes induced by PAK4 loss was similar to those induced by K-Ras V12 expression. However, the majority of phosphopeptides affected by PAK4 loss were reduced in relative abundance (57% phosphopeptides down-regulated compared with 43% up-regulated). Loss of PAK4 reduced the abundance of phosphopeptides derived from the scaffold proteins IQGAP1 and ZO-2 and DNA damage response proteins GTSE1 (G2 and S phase-expressed protein 1) and BRCA1 (Breast cancer type 1 susceptibility protein homolog) (supplemental Table S1). In contrast, PAK4 loss increased the abundance of phosphopeptides derived from the Rho guanine nucleotide exchange factor DOCK6 (Dedicator of cytokinesis protein 6) and the lipid kinases PIP5K (1-phosphatidylinositol 3-phosphate 5-kinase) and PIK3CB (Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform), indicating a potential role for PAK4 in modulating the phosphorylation status of upstream regulators. Phosphopeptides derived from the microtubule-associated cytoskeletal protein MAP1A (Microtubule-associated protein 1A) also increased by PAK4 loss.
The greatest overlap in regulation was observed for phosphopeptides regulated by K-Ras and Cdc42. Phosphoproteins co-regulated by K-Ras and Cdc42 include the actin binding proteins Filamin A, Filamin B, and Filamin C. Despite the fact that Cdc42 regulates 104 fewer total phosphopeptides than K-Ras, the overlap of phosphopeptides regulated by Cdc42 and PAK4 (58 peptides) is quite similar to the overlap between K-Ras and PAK4 regulation (59 peptides). Phosphopeptides derived from the microtubule-associated CLASP (Cytoplasmic linker associated protein) family proteins were among those showing a pattern of co-regulation. Interestingly, K-Ras and PAK4 regulate the abundance of the CLASP1 S598 phosphopeptide, whereas Cdc42 and PAK4 regulate the abundance of the CLASP2 S1021 phosphopeptide. In addition, the top 100 differentially regulated phosphopeptides were clustered based on measured intensities (Fig. 3B). The resulting heatmap shows distinct regulation patterns induced by perturbing K-Ras, Cdc42, or PAK4 signaling. The most dramatic changes in phosphopeptide abundance are induced by K-Ras V12 expression, consistent with its known role as a master regulator of several downstream effector pathways.
As highlighted above, several novel phosphoprotein targets of K-Ras, Cdc42 and PAK4 signaling regulate microtubule dynamics, including phosphoproteins belonging to the CLASP and MAP1 protein families. Indeed, Gene Ontology (GO) enrichment analysis shows that microtubule depolymerization is an overrepresented biological process in PAK4 knockout cells (supplemental Table S2; Fig. 3C) underlying the observation that all associated phosphorylated proteins (Mtap1a, Mtap1b, Clasp1, Clasp2) show changes in phosphopeptide intensities (p < 0.001). These findings are aligned with the reported role of PAK4 in regulating microtubule-dependent cell morphological changes and metaphase spindle assembly (34, 58). Phosphoproteins involved in cell adhesion, cytoskeletal organization, cell cycle regulation and intracellular transport were also significantly overrepresented in the set of regulated phosphoproteins, consistent with established roles for Ras, Cdc42, and PAK4 signaling.
Technical Validation by Immunoblot
Although the screen identified several interesting phosphopeptides, validation of phosphorylation regulation is limited by the availability of phosphosite-specific antibodies. We chose to focus our validation efforts on the S6 ribosomal protein and the eEF-2 (Eukaryotic translation elongation factor 2) translation elongation factor, as phospho-specific antibodies for these proteins are readily available. Loss of PAK4 reduced the abundance of phosphopeptides derived from S6; however, identical phosphopeptides derived from eEF-2 show divergent regulation patterns in the CK and PKD substrate motif immunoaffinity purifications (supplemental Table S1), thereby warranting further validation.
Immunoblot analysis shows that PAK4 knockout NIH3T3 cells show a reduction in S6 phosphorylation and an increase in eEF-2 phosphorylation, with respect to wild-type cells (Fig. 4). These results are consistent with eEF-2 phosphopeptide abundances observed in the CK substrate motif immunoaffinity purification. Due to low quantities of starting material, phosphopeptides were enriched by serial immunoaffinity purifications, using immobilized motif antibodies in the following order: CDK substrate motif [K R]-pS-P-X-[K R], CK substrate motif pT-[D E]-X-[D E], PKD substrate motif L-X-R-X-X-p[S T]. Thus, the results from the immunoblot analysis indicate that in cases where the same peptide is identified across multiple serial immunoaffinity purifications, the quantitation from the first immunoaffinity purification will be most accurate. Consequently, quantitations from subsequent purifications were excluded from our global quantitative analysis.
Fig. 4.
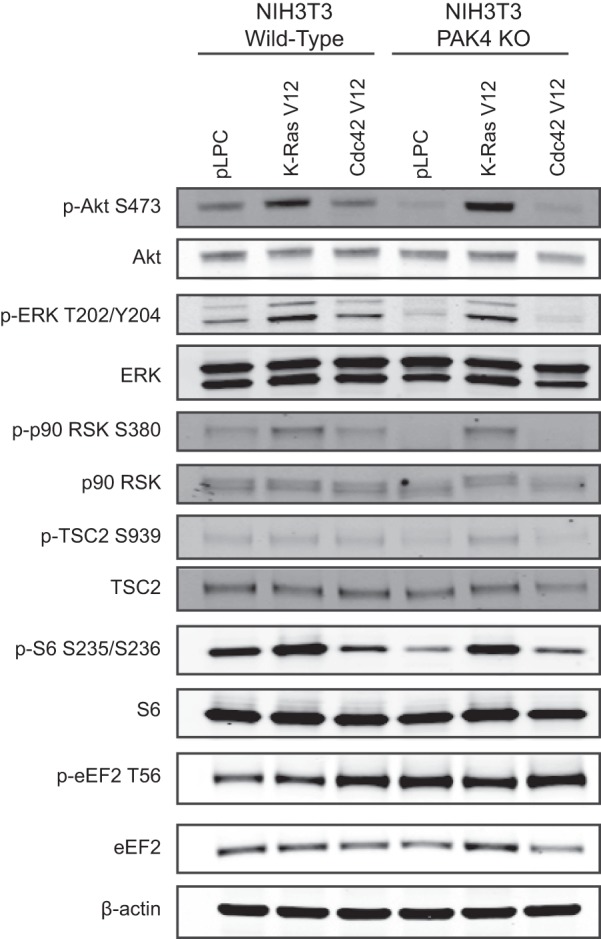
Validation of PAK4-mediated phosphorylation events. Lysates derived from NIH3T3 wild-type (WT) or PAK4 knockout (KO) cells expressing K-Ras V12, Cdc42 V12, or a pLPC empty vector control were subjected to immunoblot analysis.
Upstream Regulation of PAK4-mediated Phosphorylation Events
We next generated a plot to directly compare the K-Ras, Cdc42, and PAK4-mediated regulation of selected phosphoproteins involved in diverse biological processes including microtubule polymerization, cytoskeletal organization, as well as GTPase, lipid kinase, and DNA damage signaling (Fig. 5A). Interestingly, expression of K-Ras V12 or Cdc42 V12 in PAK4 knockout NIH3T3 cells reverses phosphopeptide intensity changes induced by PAK4 knockout, indicating K-Ras and/or Cdc42 can serve as compensatory regulators of these particular phosphosites in the absence of PAK4.
Fig. 5.

Upstream regulation of PAK4-mediated phosphorylation events. A, The regulation of select phosphoproteins by K-Ras activation, Cdc42 activation, or PAK4 knockout is depicted. “Regulation observed in distinct phosphopeptides” indicates that the abundances of multiple distinct phosphopeptides of a particular phosphoprotein are modulated by pathway perturbation. “Diverse regulation” indicates that the abundances of distinct phosphopeptides are differentially regulated by pathway perturbation. “Reverse regulation” depicts phosphoproteins for which phosphopeptide intensity changes induced by PAK4 knockout are reversed by K-Ras V12 or Cdc42 V12 expression. B, Phosphorylated peptides with minimum threefold intensity changes induced by PAK4 knockout in pLPC control samples were defined as ‘reversed’ by K-Ras V12 or Cdc42 V12 expression, if there was no intensity change (less than threefold) or opposite changes between K-Ras/Cdc42 activated PAK4 knockout and pLPC treated wild-type samples. In addition the intensity ratio measured in K-Ras/Cdc42 V12 versus pLPC control samples (both with PAK4 knockout) was required to be converse (minimum twofold).
We next examined the number of PAK4-dependent phosphopeptide intensity changes that are reversed by K-Ras V12 or Cdc42 V12 expression (Fig. 5B). Expression of K-Ras V12 reverses the regulation of more PAK4-dependent phosphopeptides than Cdc42 V12. Additionally, the regulation of several PAK4-dependent phosphopeptides can be reversed by both K-Ras V12 and Cdc42 V12. These findings suggest that several PAK4-mediated phosphorylation events can also be independently regulated by K-Ras and/or Cdc42.
DISCUSSION
Although K-Ras, Cdc42 and PAK4 are known to function in the same signaling cascade, an in depth investigation into the regulation of independent and overlapping signaling events mediated by these proteins had not yet been performed. To the best of our knowledge, our study is the first to comprehensively characterize the spectrum of phosphorylation signaling downstream of each of these key signaling proteins. We find that phosphoproteins affected by K-Ras, Cdc42, and PAK4 perturbation are involved in numerous biological processes, including cell cycle regulation, cell adhesion, and microtubule depolymerization. Although there is significant overlap in the phosphoproteins regulated by K-Ras, Cdc42, and PAK4, we also identified several novel targets of PAK4 signaling that are regulated independently from upstream K-Ras or Cdc42 signaling. These findings are consistent with recent reports demonstrating that PAK4 can regulate cellular proliferation in K-Ras mutant cancer cells via pathways independent from Raf/MEK/ERK and PI3K/Akt signaling (59).
Our results indicate that many actin- and microtubule-binding phosphoproteins are regulated by K-Ras, Cdc42 and PAK4 signaling, including those belonging to the Filamin and CLASP protein families. Filamin proteins cross-link actin into orthogonal networks and can serve as scaffolds for cytoplasmic signaling proteins (60). Several unique phosphopeptides derived from Filamin A, Filamin B, and Filamin C were identified in our phosphoproteomic screen. Previous studies show that PAK1 phosphorylates Filamin A at S2152 to mediate actin cytoskeleton reorganization (61), whereas we find that loss of PAK4 increases the abundance of the Filamin A Y1632 phosphopeptide. Though future studies are required to delineate the consequence of Y1632 phosphorylation, these findings suggest that multiple PAK kinases might converge on Filamin A to regulate its activity. In addition, we find that Cdc42 and PAK4 regulate the abundance of the CLASP2 S1021 phosphopeptide. CLASP family members are microtubule plus-end tracking proteins that promote microtubule stability (62). The S1021 phosphorylation site resides within a region of CLASP2 that mediates association to the cell cortex via LL5β/PHLDB2 (63). In light of our findings, it would be interesting to test whether phosphorylation of CLASP2 at S1021 modulates its ability to interact with LL5β. Collectively these findings support a critical role for the Ras/Cdc42/PAK4 signaling network in regulating cytoskeletal dynamics.
Our findings indicate that several PAK4-mediated phosphorylation events can be compensated by activated K-Ras or Cdc42 in the absence of PAK4. We find that in many cases, overexpression of oncogenic K-Ras or activated Cdc42 can reverse the changes in phosphopeptide abundance induced by PAK4 loss. This suggests that some PAK4-mediated phosphorylation events can also be independently regulated by K-Ras or Cdc42. Expression of K-Ras reversed more PAK4-regulated phosphopeptides than Cdc42, consistent with its role further upstream. These findings are also consistent with xenograft experiments demonstrating that PAK4 loss more significantly attenuates tumor formation induced by Cdc42 V12 than K-Ras V12 (32).
PAK4 has recently drawn attention as a potential therapeutic target, and thus there is significant interest in characterizing novel downstream signaling pathways. Our findings indicate that PAK4 modulates the phosphorylation status of key regulators of protein translation. S6 is a component of the 40S ribosome and phosphorylation on residues S235/S236 correlates with an increase in mRNA translation. Phosphorylation of eEF-2 at T56 inhibits its ability to catalyze the ribosomal translocation step during translation elongation, which is required for protein synthesis. The decrease in S6 phosphorylation at S235/S236 and increase eEF-2 phosphorylation at T56 observed in PAK4 knockout cells suggests that PAK4 positively regulates protein translation. Additional studies are required to determine the molecular mechanism by which PAK4 impinges on this signaling cascade, although we also found that loss of PAK4 deregulates upstream signaling at the level of Akt, ERK, RSK (Ribosomal protein S6 kinase alpha-1), and TSC2 (Tuberous sclerosis protein 2) (Fig. 4). Expression of oncogenic K-Ras in PAK4 knockout cells increases the phosphorylation levels of these upstream markers and rescues the defect in S6 phosphorylation, indicating that PAK4 might modulate S6 signaling via the Raf/MEK/ERK and PI3K/Akt signaling pathways.
We also found that K-Ras, Cdc42 and PAK4 regulate several novel phosphoproteins involved in GTPase signaling, lipid kinase signaling, cytoskeletal organization, and the DNA damage response. Future studies are required to characterize the functional role of novel phosphorylation events identified in this study. Collectively, our findings broaden our understanding of this critical oncogenic signaling network and lay the groundwork for future studies to characterize novel targets of oncogenic K-Ras signaling and validate biomarkers of PAK4 inhibition.
Supplementary Material
Acknowledgments
We thank Adrian Jubb and Yingying Liu for insightful discussions and technical assistance.
Footnotes
* The costs for this article was covered by Genentech Inc.
 This article contains supplemental Figs. S1 to S4 and Tables S1 and S2.
This article contains supplemental Figs. S1 to S4 and Tables S1 and S2.
1 The abbreviations used are:
- BCA
- bicinchoninic acid
- BRCA1
- Breast cancer type 1 susceptibility protein homolog
- CLASP
- Cytoplasmic linker associated protein
- DAB2
- Disabled homolog 2
- DMEM
- Dulbecco's Modified Eagle Medium
- DOCK
- Dedicator of cytokinesis protein
- DTT
- dithiothreitol
- eEF-2
- Eukaryotic translation elongation factor 2
- ERK
- extracellular-signal-regulated kinase
- FBS
- fetal bovine serum
- GAB2
- GRB2-associated-binding protein 2
- GO
- Gene Ontology
- GTSE1
- G2 and S phase-expressed protein 1
- LC
- liquid chromatography
- LDS
- lithium dodecyl sulfate
- LTQ
- linear trap quadrupole
- MAP1
- Microtubule-associated protein 1
- MS
- mass spectrometry
- MTUS1
- Microtubule-associated tumor suppressor homolog 1
- PAK
- p21-activated kinase
- PBS
- phosphate buffered saline
- PHLDB2
- pleckstrin homology-like domain family B member 2
- PI3K
- phosphoinositide 3-kinase
- PIK3CB
- Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform
- PIP5K
- 1-phosphatidylinositol 3-phosphate 5-kinase
- PMSF
- phenylmethylsulfonyl fluoride
- RADIL
- Ras-associating and dilute domain-containing protein
- RALBP1
- RalA-binding protein 1
- RSK
- Ribosomal protein S6 kinase alpha-1
- RTK
- receptor tyrosine kinase
- SDS
- sodium dodecyl sulfate
- TFA
- trifluoroacetic acid
- TSC2
- Tuberous sclerosis protein 2.
REFERENCES
- 1. Schubbert S., Shannon K., Bollag G. (2007) Hyperactive Ras in developmental disorders and cancer. Nat. Rev. 7, 295–308 [DOI] [PubMed] [Google Scholar]
- 2. Baines A. T., Xu D., Der C. J. (2011) Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3, 1787–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gysin S., Salt M., Young A., McCormick F. (2011) Therapeutic strategies for targeting ras proteins. Genes Cancer 2, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Courtney K. D., Corcoran R. B., Engelman J. A. (2010) The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 28, 1075–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pratilas C. A., Solit D. B. (2010) Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin. Cancer Res. 16, 3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai Y., Edamatsu H., Maeda S., Saito H., Suzuki N., Satoh T., Kataoka T. (2004) Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res. 64, 8808–8810 [DOI] [PubMed] [Google Scholar]
- 7. Gonzalez-Garcia A., Pritchard C. A., Paterson H. F., Mavria G., Stamp G., Marshall C. J. (2005) RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 7, 219–226 [DOI] [PubMed] [Google Scholar]
- 8. Khosravi-Far R., Solski P. A., Clark G. J., Kinch M. S., Der C. J. (1995) Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 15, 6443–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malliri A., van der Kammen R. A., Clark K., van der Valk M., Michiels F., Collard J. G. (2002) Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 417, 867–871 [DOI] [PubMed] [Google Scholar]
- 10. Qiu R. G., Chen J., Kirn D., McCormick F., Symons M. (1995) An essential role for Rac in Ras transformation. Nature 374, 457–459 [DOI] [PubMed] [Google Scholar]
- 11. White M. A., Nicolette C., Minden A., Polverino A., Van Aelst L., Karin M., Wigler M. H. (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell 80, 533–541 [DOI] [PubMed] [Google Scholar]
- 12. Cheng C. M., Li H., Gasman S., Huang J., Schiff R., Chang E. C. (2011) Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol. Cell. Biol. 31, 983–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stengel K. R., Zheng Y. (2012) Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PloS One 7, e37317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng Y., Xia Y., Hawke D., Halle M., Tremblay M. L., Gao X., Zhou X. Z., Aldape K., Cobb M. H., Xie K., He J., Lu Z. (2009) FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol. Cell 35, 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stengel K., Zheng Y. (2011) Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 23, 1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Wang Y., Zhang Y., Miao Y., Zhao Y., Zhang P. X., Jiang G. Y., Zhang J. Y., Han Y., Lin X. Y., Yang L. H., Li Q. C., Zhao C., Wang E. H. (2009) Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer 63, 375–382 [DOI] [PubMed] [Google Scholar]
- 17. Gómez Del Pulgar T., Valdés-Mora F., Bandres E., Perez-Palacios R., Espina C., Cejas P., Garcia-Cabezas M. A., Nistal M., Casado E., Gonzalez-Baron M., Garcia-Foncillas J., Lacal J. C. (2008) Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int. J. Oncol. 33, 185–193 [PubMed] [Google Scholar]
- 18. Tucci M. G., Lucarini G., Brancorsini D., Zizzi A., Pugnaloni A., Giacchetti A., Ricotti G., Biagini G. (2007) Involvement of E-cadherin, beta-catenin, Cdc42 and CXCR4 in the progression and prognosis of cutaneous melanoma. Br. J. Dermatol. 157, 1212–1216 [DOI] [PubMed] [Google Scholar]
- 19. Kamai T., Yamanishi T., Shirataki H., Takagi K., Asami H., Ito Y., Yoshida K. (2004) Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 10, 4799–4805 [DOI] [PubMed] [Google Scholar]
- 20. Fritz G., Just I., Kaina B. (1999) Rho GTPases are over-expressed in human tumors. Int. J. Cancer 81, 682–687 [DOI] [PubMed] [Google Scholar]
- 21. Appledorn D. M., Dao K. H., O'Reilly S., Maher V. M., McCormick J. J. (2010) Rac1 and Cdc42 are regulators of HRasV12-transformation and angiogenic factors in human fibroblasts. BMC Cancer 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu R. G., Abo A., McCormick F., Symons M. (1997) Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell. Biol. 17, 3449–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crawford J. J., Hoeflich K. P., Rudolph J. (2012) p21-Activated kinase inhibitors: a patent review. Expert Opin. Ther. Pat. 22, 293–310 [DOI] [PubMed] [Google Scholar]
- 24. Ong C. C., Jubb A. M., Zhou W., Haverty P. M., Harris A. L., Belvin M., Friedman L. S., Koeppen H., Hoeflich K. P. (2011) p21-activated kinase 1: PAK'ed with potential. Oncotarget 2, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar R., Gururaj A. E., Barnes C. J. (2006) p21-activated kinases in cancer. Nat. Rev. Cancer 6, 459–471 [DOI] [PubMed] [Google Scholar]
- 26. Whale A., Hashim F. N., Fram S., Jones G. E., Wells C. M. (2011) Signalling to cancer cell invasion through PAK family kinases. Front. Biosci. 16, 849–864 [DOI] [PubMed] [Google Scholar]
- 27. Callow M. G., Clairvoyant F., Zhu S., Schryver B., Whyte D. B., Bischoff J. R., Jallal B., Smeal T. (2002) Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J. Biol. Chem. 277, 550–558 [DOI] [PubMed] [Google Scholar]
- 28. Chen S., Auletta T., Dovirak O., Hutter C., Kuntz K., El-ftesi S., Kendall J., Han H., Von Hoff D. D., Ashfaq R., Maitra A., Iacobuzio-Donahue C. A., Hruban R. H., Lucito R. (2008) Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol. Ther. 7, 1793–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kimmelman A. C., Hezel A. F., Aguirre A. J., Zheng H., Paik J. H., Ying H., Chu G. C., Zhang J. X., Sahin E., Yeo G., Ponugoti A., Nabioullin R., Deroo S., Yang S., Wang X., McGrath J. P., Protopopova M., Ivanova E., Zhang J., Feng B., Tsao M. S., Redston M., Protopopov A., Xiao Y., Futreal P. A., Hahn W. C., Klimstra D. S., Chin L., DePinho R. A. (2008) Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 19372–19377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parsons D. W., Wang T. L., Samuels Y., Bardelli A., Cummins J. M., DeLong L., Silliman N., Ptak J., Szabo S., Willson J. K., Markowitz S., Kinzler K. W., Vogelstein B., Lengauer C., Velculescu V. E. (2005) Colorectal cancer: mutations in a signalling pathway. Nature 436, 792. [DOI] [PubMed] [Google Scholar]
- 31. Paliouras G. N., Naujokas M. A., Park M. (2009) Pak4, a novel Gab1 binding partner, modulates cell migration and invasion by the Met receptor. Mol. Cell. Biol. 29, 3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y., Xiao H., Tian Y., Nekrasova T., Hao X., Lee H. J., Suh N., Yang C. S., Minden A. (2008) The pak4 protein kinase plays a key role in cell survival and tumorigenesis in athymic mice. Mol. Cancer Res. 6, 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dan C., Kelly A., Bernard O., Minden A. (2001) Cytoskeletal changes regulated by the PAK4 serine/threonine kinase are mediated by LIM kinase 1 and cofilin. J. Biol. Chem. 276, 32115–32121 [DOI] [PubMed] [Google Scholar]
- 34. Callow M. G., Zozulya S., Gishizky M. L., Jallal B., Smeal T. (2005) PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 118, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 35. Cammarano M. S., Nekrasova T., Noel B., Minden A. (2005) Pak4 induces premature senescence via a pathway requiring p16INK4/p19ARF and mitogen-activated protein kinase signaling. Mol. Cell. Biol. 25, 9532–9542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gnesutta N., Qu J., Minden A. (2001) The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J. Biol. Chem. 276, 14414–14419 [DOI] [PubMed] [Google Scholar]
- 37. Rennefahrt U. E., Deacon S. W., Parker S. A., Devarajan K., Beeser A., Chernoff J., Knapp S., Turk B. E., Peterson J. R. (2007) Specificity profiling of Pak kinases allows identification of novel phosphorylation sites. J. Biol. Chem. 282, 15667–15678 [DOI] [PubMed] [Google Scholar]
- 38. Guha U., Chaerkady R., Marimuthu A., Patterson A. S., Kashyap M. K., Harsha H. C., Sato M., Bader J. S., Lash A. E., Minna J. D., Pandey A., Varmus H. E. (2008) Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proc. Natl. Acad. Sci. U.S.A. 105, 14112–14117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sudhir P. R., Hsu C. L., Wang M. J., Wang Y. T., Chen Y. J., Sung T. Y., Hsu W. L., Yang U. C., Chen J. Y. (2011) Phosphoproteomics identifies oncogenic Ras signaling targets and their involvement in lung adenocarcinomas. PLoS One 6, e20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 41. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 42. Pan C., Gnad F., Olsen J. V., Mann M. (2008) Quantitative phosphoproteome analysis of a mouse liver cell line reveals specificity of phosphatase inhibitors. Proteomics 8, 4534–4546 [DOI] [PubMed] [Google Scholar]
- 43. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 44. Gnad F., de Godoy L. M., Cox J., Neuhauser N., Ren S., Olsen J. V., Mann M. (2009) High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 9, 4642–4652 [DOI] [PubMed] [Google Scholar]
- 45. Zielinska D. F., Gnad F., Jedrusik-Bode M., Wiśniewski J. R., Mann M. (2009) Caenorhabditis elegans has a phosphoproteome atypical for metazoans that is enriched in developmental and sex determination proteins. J. Proteome Res. 8, 4039–4049 [DOI] [PubMed] [Google Scholar]
- 46. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amanchy R., Zhong J., Hong R., Kim J. H., Gucek M., Cole R. N., Molina H., Pandey A. (2009) Identification of c-Src tyrosine kinase substrates in platelet-derived growth factor receptor signaling. Mol. Oncol. 3, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moritz A., Li Y., Guo A., Villén J., Wang Y., MacNeill J., Kornhauser J., Sprott K., Zhou J., Possemato A., Ren J. M., Hornbeck P., Cantley L. C., Gygi S. P., Rush J., Comb M. J. (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., Wetzel R., Macneill J., Ren J. M., Yuan J., Bakalarski C. E., Villen J., Kornhauser J. M., Smith B., Li D., Zhou X., Gygi S. P., Gu T. L., Polakiewicz R. D., Rush J., Comb M. J. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 [DOI] [PubMed] [Google Scholar]
- 50. Stokes M. P., Farnsworth C. L., Moritz A., Silva J. C., Jia X., Lee K. A., Guo A., Polakiewicz R. D., Comb M. J. (2012) PTMScan direct: identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol. Cell. Proteomics 11, 187–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stokes M. P., Silva J. C., Jia X., Lee K. A., Polakiewicz R. D., Comb M. J. (2012) Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct. Int. J. Mol. Sci. 14, 286–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 53. Lundgren D. H., Martinez H., Wright M. E., Han D. K. (2009) Protein identification using Sorcerer 2 and SEQUEST. Curr. Protoc. Bioinformatics Chapter 13, Unit 13 13 [DOI] [PubMed] [Google Scholar]
- 54. Zhang H., Zha X., Tan Y., Hornbeck P. V., Mastrangelo A. J., Alessi D. R., Polakiewicz R. D., Comb M. J. (2002) Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 277, 39379–39387 [DOI] [PubMed] [Google Scholar]
- 55. Prior I. A., Lewis P. D., Mattos C. (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cau J., Hall A. (2005) Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J. Cell Sci. 118, 2579–2587 [DOI] [PubMed] [Google Scholar]
- 57. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 58. Bompard G., Rabeharivelo G., Cau J., Abrieu A., Delsert C., Morin N. (2012) P21-activated kinase 4 (PAK4) is required for metaphase spindle positioning and anchoring. Oncogene. 32, 910–919 [DOI] [PubMed] [Google Scholar]
- 59. Tabusa H., Brooks T., Massey A. J. (2012) Knockdown of PAK4 or PAK1 Inhibits the Proliferation of Mutant KRAS Colon Cancer Cells Independently of RAF/MEK/ERK and PI3K/AKT Signaling. Mol. Cancer Res. 11, 109–121 [DOI] [PubMed] [Google Scholar]
- 60. Zhou A. X., Hartwig J. H., Akyürek L. M. (2010) Filamins in cell signaling, transcription and organ development. Trends Cell. Biol. 20, 113–123 [DOI] [PubMed] [Google Scholar]
- 61. Vadlamudi R. K., Li F., Adam L., Nguyen D., Ohta Y., Stossel T. P., Kumar R. (2002) Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 4, 681–690 [DOI] [PubMed] [Google Scholar]
- 62. Al-Bassam J., Chang F. (2011) Regulation of microtubule dynamics by TOG-domain proteins XMAP215/Dis1 and CLASP. Trends Cell Biol. 21, 604–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lansbergen G., Grigoriev I., Mimori-Kiyosue Y., Ohtsuka T., Higa S., Kitajima I., Demmers J., Galjart N., Houtsmuller A. B., Grosveld F., Akhmanova A. (2006) CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev. Cell 11, 21–32 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.