


Abstract
The range of heterogeneous approaches available for quantifying protein abundance via mass spectrometry (MS)1 leads to considerable challenges in modeling, archiving, exchanging, or submitting experimental data sets as supplemental material to journals. To date, there has been no widely accepted format for capturing the evidence trail of how quantitative analysis has been performed by software, for transferring data between software packages, or for submitting to public databases. In the context of the Proteomics Standards Initiative, we have developed the mzQuantML data standard. The standard can represent quantitative data about regions in two-dimensional retention time versus mass/charge space (called features), peptides, and proteins and protein groups (where there is ambiguity regarding peptide-to-protein inference), and it offers limited support for small molecule (metabolomic) data. The format has structures for representing replicate MS runs, grouping of replicates (for example, as study variables), and capturing the parameters used by software packages to arrive at these values. The format has the capability to reference other standards such as mzML and mzIdentML, and thus the evidence trail for the MS workflow as a whole can now be described. Several software implementations are available, and we encourage other bioinformatics groups to use mzQuantML as an input, internal, or output format for quantitative software and for structuring local repositories. All project resources are available in the public domain from the HUPO Proteomics Standards Initiative http://www.psidev.info/mzquantml.
The Proteomics Standards Initiative (PSI) has been working for ten years to improve the reporting and standardization of proteomics data. The PSI has published minimum reporting guidelines, called MIAPE (Minimum Information about a Proteomics Experiment) documents, for MS-based proteomics (1) and molecular interactions (2), as well as data standards for raw/processed MS data in mzML (3), peptide and protein identifications in mzIdentML (4), transitions for selected reaction monitoring analysis in TraML (5), and molecular interactions in PSI-MI format (6). Standards are particularly important for quantitative proteomics research, because the associated bioinformatics analysis is highly challenging as a result of the range of different experimental techniques for deriving abundance values for proteins using MS. The techniques can be broadly divided into those based on (i) differential labeling, in which a metabolic label or chemical tag is applied to cells, peptides, or proteins, samples are mixed, and intensity signals for peptide ions are compared within single MS runs; or (ii) label-free methods in which MS runs occur in parallel and bioinformatics methods are used to extract intensity signals, ensuring that like-for-like signals are compared between runs (7). In most label-based and label-free approaches, peptide ratios or abundance values must be summarized in order for one to arrive at relative protein abundance values, taking into account ambiguity in peptide-to-protein inference. Absolute protein abundance values can typically be derived only using internal standards spiked into samples of known abundance (8, 9). The PSI has recently developed a MIAPE-Quant document defining and describing the minimal information necessary in order to judge or repeat a quantitative proteomics experiment.
Software packages tend to report peptide or protein abundance values in a bespoke format, often as tab or comma separated values, for import into spreadsheet software. In complementary work, the PSI has developed a standard format for capturing these final results in a standardized tab separated value format, called mzTab, suitable for post-processing and visualization in end-user tools such as Microsoft Excel or the R programming language. The final results of a quantitative analysis are sufficient for many purposes, such as performing statistical analysis to determine differential expression or cluster analysis to find co-expressed proteins. However, mzTab (or similar bespoke formats) was not designed to hold a trace of how the peptide and protein abundance values were calculated from MS data (i.e. metadata is lost that might be crucial for other tasks). For example, most quantitative software packages detect and quantify so-called “features” (representing all ions collected for a given peptide) in two-dimensional MS data, where the two dimensions are retention time from liquid chromatography (LC) and mass over charge (m/z). Without capturing the two-dimensional coordinates of the features, it is not possible to write visualization software showing exactly what the software has quantified; researchers have to trust that the software has accurately quantified all ions from isotopes of a given peptide, excluding any overlapping ions derived from other peptides. The history of proteomics research has been one in which studies of highly variable quality have been published. There is also little quality control or benchmarking performed on quantitative software (10), meaning it is difficult to make quality judgments on a set of peptide and protein abundance values. The PSI has recently developed mzML, which can capture raw or processed MS data in a vendor neutral format, and the mzIdentML standard, to capture search engine results and the important metadata (such as software parameters), such that peptide and protein identification data can be interpreted consistently. These two standards are now being used for data sharing and to support open source software development, so that informatics groups can focus on algorithmic development rather than file format conversions. Until now, there has been no widely used open source format or data standard for capturing metadata and data relating to the quantitation step of analysis pipelines. In this work, we report the mzQuantML standard from the PSI, which has recently completed the PSI standardization process (11), from which version 1.0 was released. We believe that quantitative proteomics research will benefit from improved capabilities for tracing what manipulations have happened to data at each stage of the analysis process. The mzQuantML standard has been designed to store quantitative values calculated for features, peptides, proteins, and/or protein groups (where there is ambiguity in protein inference), plus associated software parameters. It has also been designed to accommodate small molecule data to improve interoperability with metabolomics investigations. The format can represent experimental replicates and grouping of replicates, and it has been designed via an open and transparent process.
EXPERIMENTAL PROCEDURES
The mzQuantML model was developed over several years at dedicated workshops, annual PSI meetings (12–14), and regular conference calls between contributors around the world. The primary use cases and guiding principles for the development of mzQuantML are as follows (these are edited extracts from the formal specification document).
• General principles that the format should support: journal requirements for the reporting of quantitative proteomic data from MS; reporting according to MIAPE guidelines; submission of quantitative data to public databases; data exchange between software tools; import of data into statistical processing tools; and the ability to reprocess or recreate the analysis workflow using the same parameters, assuming no manual steps have taken place.
• Use cases that the format should capture: final abundance values (relative or absolute) for peptides, proteins, and protein groups; quantitation values about peptide/protein modifications; abundance values at the level of a single run and logical groupings of runs; the evidence trail for how final abundance values were calculated, such as the features used for quantifying peptides and proteins; relationships between features either on different regions of the same MS run or on different MS runs that report on the same peptide or small molecule; and details about pre-fractionation sufficient to describe the combination of multiple input data files.
All development meetings have been advertised and open to any interested parties to ensure that the process is transparent and the widest possible input can be obtained. The model has been developed as an XML Schema Definition (XSD) file accompanied by controlled vocabulary terms and definitions as part of the PSI-MS controlled vocabulary (CV) (15), also used in mzML, mzIdentML, TraML, and mzTab. To cope with the heterogeneity of different quantitative methods, additional semantic validation rules have been defined as part of the version 1.0 release and implemented in software. These rules are required to differentiate between the four techniques included in the first release: (i) intensity-based label-free, (ii) MS label-based, (iii) MS2 tag-based, and (iv) spectral counting sub-types of mzQuantML files. Additional rules are under development to support selected reaction monitoring techniques that will be released in 2013. The semantic encoding rules are difficult to encode in a single XSD file but are required to ensure that software exporting mzQuantML files encode data from a particular technique consistently (see the section “Semantic Validation and Controlled Vocabularies”). All development resources have been maintained in the public domain under subversion repository since the inception of the project.
RESULTS
In the following sections, we describe different aspects of the mzQuantML model, which is summarized in Fig. 1. The model captures metadata about how a quantitative analysis was performed by software and, importantly, a description of the experimental design in terms of biological or technical replication and grouping of replicates into so-called study variables. These aspects are important to capture, as many quantitative software packages use such information for reporting data averaged over replicates. The format defines matrix structures for capturing data values at various levels from individual MS runs, inferred peptide ion signals derived originally from different samples (within or between MS runs), and inferred proteins or protein groups quantified by the software. The format also has basic structures for capturing data about small molecules, as the PSI looks to build links with the metabolomics community and develop shared standards. Further details about the structures reported in the following sections can be obtained from the Twenty Minute Guide to mzQuantML or the formal specification document (available from the PSI website).
Fig. 1.

A diagrammatic representation of the data model for mzQuantML. Dashed boxes indicate non-mandatory elements. RT, retention time.
Metadata, Software, and Parameters
As shown in Fig. 1, the file captures metadata about the CVs used in the element <CvList> (angle brackets denote an element in an mzQuantML XML file), the provider of the document (<Provider>), and their contact details (<AuditCollection>). A valid file must contain particular CV terms within <AnalysisSummary> describing the type of data represented in the file (e.g. MS label-based, MS intensity-based label free, MS2 tag-based, spectral counting) and whether the software is reporting values for features, peptides, proteins, and/or protein groups. The <InputFiles> element captures references to the data files used for analysis including raw MS data files (e.g. in mzML format), identification data (e.g. in mzIdentML format), the protein database from which proteins have been identified (e.g. in FASTA format), and configuration or methods files required for the analysis—for example, input transitions for a selected reaction monitoring analysis (e.g. in TraML format). There is no dependence on any particular input format, so long as the format(s) used can be referenced by a Uniform Resource Identifier and, in the case of identification file formats, contains unique identifiers for peptide-spectrum matches and/or detected proteins. The format captures a description of the software and version used in <SoftwareList>, the analysis steps performed with parameters captured as CV terms in <DataProcessingList>, and any bibliographic references associated with the data represented in the file in <BibliographicReference>.
The Experimental Design
The concept of an <Assay> in mzQuantML typically represents analysis of a single biological sample. Additional replicate analyses of the same sample are modeled as extra <Assay> elements. For techniques in which multiple samples have been compared within a single MS run, multiple <Assay> elements are defined that all refer to the same raw MS data file(s) (specified within <InputFiles>). For label-free techniques, there is typically a one-to-one mapping from an <Assay> to a raw file. In label or tag-based techniques, the <Assay> must also capture the label or tag used to differentiate the peptide ion, such as the iTRAQ or SILAC reagent.
<StudyVariable> elements are used to apply logical groupings to sets of <Assay> elements, for which quantitative values may be reported. A typical study variable might be a collection of biological replicates for which the analysis software has calculated average quantitative values from <Assay> elements (e.g. “disease group” or “non-affected individual group”).
During the development of mzQuantML, it was observed that many quantitative software packages report ratios (say, of peptide or protein abundance values) rather than intensity values, and thus an important use case for mzQuantML is the ability to report this type of data. The <RatioList> can capture definitions of <Ratio> elements, where each ratio has a numerator and denominator referencing <StudyVariable> or <Assay> elements.
Reporting Data Values in mzQuantML
The format has a matrix-based structure designed to be both flexible and economical in storage space called a <QuantLayer>, which holds a two-dimensional matrix of data values. The various sub-types of <QuantLayer> elements are named according to the part of the experimental design for which data values are exported—assays, study variables, ratios, global values, and so on—and these elements form the columns of the data matrix. The location of the <QuantLayer> within the file defines the type of object for which values are reported—protein groups, proteins, peptides, features, or small molecules—and is used to form the rows of the data matrix. For example, an <AssayQuantLayer> within the <ProteinList> contains a <DataMatrix> in which the columns reference <Assay> elements and the rows reference <Protein> elements, as further exemplified below. The formal specification document describes how missing values, zeros, infinite values (e.g. in ratios), and calculation errors (“not a number” errors) can be encoded in the <DataMatrix> element.
Taking the <ProteinList> as a representative example (Fig. 2), data values can be captured for each protein identified in the file for each <Assay> in an <AssayQuantLayer>, for each <StudyVariable> in a <StudyVariableQuantLayer>, for any <Ratio> elements defined in <RatioQuantLayer>, or for the entire experiment, such as global counts, scores, or statistics, in a <GlobalQuantLayer>. With the exception of a <GlobalQuantLayer> (which can store multiple different types of data if required), each <QuantLayer> can store only one type of data value within its <DataMatrix>; for example, in Fig. 2 the <DataMatrix> contains only normalized protein abundance values. If the software wished to export raw protein abundance values additionally, a second <QuantLayer> would be required. The <ColumnIndex> of an <AssayQuantLayer> specifies <Assay> elements for which corresponding data values are reported for each <Protein> in the <Row> elements of the <DataMatrix>. For example, the <Row> specifying “prot_0” in Fig. 2 references the definition of the (yeast) <Protein> with accession YDL081C, followed by 12 data values, one per <Assay>. It is thus straightforward to process files to retrieve data values for all <Assay> elements or for a specific <Assay> element as required. This design has been employed to ensure that files are not overly verbose, as the data type has to be specified only once per <AssayQuantLayer>, and files are easily interpretable.
Fig. 2.

An example from a partial mzQuantML file for a label-free analysis. Data values for proteins in which 12 samples were analyzed are shown: <ColumnIndex> references 12 <Assay> elements. Each <Row> contains 12 quantitative values about a single protein (as defined by the <ColumnIndex>). The data type within the <AssayQuantLayer> is defined using a CV term under <DataType>.
Each <Protein> element can reference <PeptideConsensus> elements from which the protein-level quantitative values were derived. A <PeptideConsensus> element represents a peptide that has been quantified in one or more <Assay>s. It can have a peptide sequence and modifications (or it can be an unidentified peptide that has been quantified), and it can reference <Feature> elements in the <FeatureList> (Fig. 3). Additionally, references can be provided to elements in external files, such as mzIdentML, containing detailed evidence for the identification of the peptide via a set of peptide-spectrum matches and scores (described in “Linkage from mzQuantML to mzIdentML and mzML” in the Twenty Minute Guide). Similar to the <ProteinList>, data values can be stored for peptides in <QuantLayer> elements for each <Assay>, <StudyVariable>, or <Ratio> defined in the file (not shown).
Fig. 3.

Partial examples from a label-free analysis in mzQuantML, showing the associations from <Protein> to <PeptideConsensus> to <Feature>.
For each analysis of a raw file (or group of raw files where sample pre-fractionation has occurred), a <FeatureList> will be produced. A <FeatureList> contains a list of positions in two-dimensional LC-MS space that have been quantified, called <Feature> elements. A minimal <Feature> definition includes the m/z value, the predicted charge, the retention time (if LC has been performed), and a unique identifier. The specifications optionally allow for a <MassTrace> element to be included whereby the precise regions in two-dimensional space that have been quantified by the software can be specified (details in the specification document), supporting the development of visualization and validation software. Each <FeatureList> can contain a <FeatureQuantLayer> in which values can be reported that are not appropriate at the <PeptideConsensus> level, such as descriptors of the quality of the feature's isotope profile. An <AssayQuantLayer> or <StudyVariableQuantLayer> cannot be provided within a <FeatureList>, because a <Feature> is by definition a region within one raw file (prior to features being matched that report on the same peptide) and thus cannot have different values for each <Assay> or <StudyVariable>. One exception to this rule is MS2 quantification techniques, such as iTRAQ or TMT, in which multiple assays are quantified from the same MS1 feature (further discussed in the “MS2 Tag-based” section of the Twenty Minute Guide).
Small Molecule Data
The model has an extension for use with metabolite data, via the inclusion of the <SmallMoleculeList> element. Each <SmallMolecule> can have references to external databases for formally identifying the molecule, and to <Feature> elements (as for <PeptideConsensus>) and associated quantitative data stored in <QuantLayer> elements for assays, study variables, and ratios as for peptides, proteins, or protein groups. This part of the model was developed to encourage software developers working with such data to use mzQuantML and join this development effort rather than develop a separate format. However, it has not been tested to the same level as the proteomics examples, and thus it stands as a placeholder for additional development in future versions.
Semantic Validation and Controlled Vocabularies
The associated tutorial document and example files accessible from the project home page explain how different types of experimental approaches should be encoded within the general structures described above. Clearly, there is considerable difference between the ways in which data from an MS2 tag-based approach, such as iTRAQ, should be represented and the presentation of data from a spectral counting label-free approach. To ensure that software packages export data consistently, a set of semantic validation rules have been defined alongside the XSD, written in natural language. These rules have been encoded in validation software that checks (i) whether an mzQuantML file is valid against the XSD, (ii) whether CV terms have been used appropriately, and (iii) whether the additional rules have been fulfilled. CV terms are stored in the PSI-MS CV, which contains around 2000 terms and definitions used in mzML, mzIdentML, mzQuantML, mzTab, and TraML describing a wide range of aspects of MS-based protein analysis (instrument and software parameters, enzymes, data formats, software scores, etc.). As one example of the type of validation performed in a duplex SILAC experiment (one replicate only), a valid mzQuantML file must contain two <Assay> elements, each describing one sample analyzed (e.g. one flagged as unlabeled, the other flagged as heavy labeled) but both referencing the same raw MS data file (rules can be found at the mzQuantML project website). The <Assay> element for the heavy labeled sample must contain CV terms describing the SILAC reagent(s) and the mass shift.
<AssayList id = “assaylist1”>
<Assay id = “a_887303905526135715” rawFilesGroup_ref = “rfg_11416597224957566492”>
<Label>
<Modification massDelta = “0”>
<cvParam cvRef = “PSI-MS” accession = “MS:1002038” name = “unlabeled sample”/>
</Modification>
</Label>
</Assay>
<Assay id = “a_5154939017891837577” rawFilesGroup_ref = “rfg_11416597224957566492”>
<Label>
<Modification massDelta = “8.0141988132”>
<cvParam cvRef = “UNIMOD” accession = “259” name = “Label:13C(6)15N(2)” value = “Lys8”/>
</Modification>
<Modification massDelta = “10.0082686”>
<cvParam cvRef = “UNIMOD” accession = “267” name = “Label:13C(6)15N(4)” value = “Arg10”/>
</Modification>
</Label>
</Assay>
</AssayList>
Where replicates are performed with the labels switched across samples, the corresponding <Assay> elements are created with relevant labels and grouped under a common <StudyVariable> element to indicate that replicate samples have been analyzed.
Software Implementations
A number of software tools are currently available that support mzQuantML (see details linked from the PSI mzQuantML home page). A Java application programming interface for mzQuantML called jmzQuantML is available that provides a bidirectional mapping from XML to Java objects, with methods for reading and writing valid files (available from the mzQuantML project website). The application programming interface is used in the semantic validation software, conversion software for exporting files from Progenesis LC-MS (16) and MaxQuant (17), and in a library of Java routines (currently under development) for manipulating and viewing mzQuantML files and performing conversions to mzTab (for further details, consult the PSI mzQuantML home page).
There is a prototype Microsoft Excel to mzQuantML converter that reads in spectral count values of time series data of two samples under different experimental conditions represented in a tab-delimited format and generates an mzQuantML output file containing the relative abundance value ratios for the peptides and proteins. The mzQuantML data model is also being used as the backbone for quantitative analyses in the open-source Proteosuite toolkit, which writes output in and visualizes mzQuantML files.
The open-source framework OpenMS (18) implements support for reading and writing mzQuantML files in C++. Based on OpenMS, TOPP (19) can import/export mzQuantML from the quantitation TOPP tools SILACAnalyzer and ITRAQAnalyzer. It can also import mzQuantML files for further internal use. The XMLValidator TOPPtool can check files for XML schema consistency, and the SemanticValidator TOPP tool is capable of reading mzQuantML files to verify the schema semantics and the proper use of the CV. TOPP also provides tools for the conversion/export of mzQuantML data to mzTab.
In the context of MS proteomics repositories, the storage of quantitative data has been limited (20) because of the lack of data standardization and the wide variety of existing experimental approaches. It is expected that mzQuantML will be used in the ProteomeXchange consortium data workflow. The ProteomeXchange consortium aims to promote standard submission and data sharing policies among the main MS-based proteomics data repositories, including PRIDE (21) and PeptideAtlas (22). At present, the first implementation of the workflow for qualitative data has been set up, and a number of data submissions have already been done (for an updated list of public datasets, see the Proteome Central website). At present, quantitative information can be uploaded in any format as additional files (not mandatory) that are stored and available for download by users. The formalization of the data workflow for quantification information is due in 2013. The formalization and wide acceptance of new formats like mzQuantML and mzTab are essential for the success of these efforts, and this is why the development of mzQuantML is a formal deliverable in the “ProteomeXchange” grant, funded by the EU FP7 program.
Example Files
Files are available from the project website that exemplify the four types of experimental techniques covered by the version 1.0 release, from which selected parts are described below.
The file “MS1Label/oms-data-silacanalyzer.mzq” contains a single MS run from a SILAC analysis (light versus heavy using Lys8 and Arg10 labels), performed in the SILACAnalyzer tool in OpenMS, from which quantitative values are reported for features (raw intensity) and peptide ratios. “MS2Tag/oms-data-itraqanalyzer-id.mzq” contains a single MS run from an iTRAQ analysis using four reagents (114, 115, 116, and 117 Da reporter ions); the reporter ion intensity is exported for each <Assay> from within each <Feature>, referenced by <PeptideConsensus> elements for each identified peptide. “label-free/CPTAC-Progenesis.mzq.gz” contains 12 MS runs in a label-free analysis, performed using Progenesis LC-MS. Two data types are reported for both proteins and peptides (normalized and raw abundance). Each <Feature> identified also has a specification of the region quantified by the software, using the <MassTrace> element (note: a single rectangle is encoded encompassing the entire region quantified, as Progenesis LC-MS does not currently export the coordinates of the individual isotopes quantified). Each <PeptideConsensus> has references to all peptide-spectrum matches made by the search engine (Mascot, Matrix Science, London, UK in a separate file, encoded in mzIdentML “CPTAC_Progenesis_Identifications.mzid.gz.” The CPTAC mzQuantML and mzIdentML example files have been created to demonstrate a complete analysis trace for a genuine experimental data set, and thus these files are significantly larger than other example files. It is intended that mzQuantML files will be always be stored, transferred, and interpreted by software as zipped versions (gzip is recommended in the specification document). The mzQuantML file is 26 MB zipped and 158 MB unzipped. We believe such file sizes are acceptable, given that the raw data for this analysis totalled ∼7 GB (.raw files) or ∼15 GB (.mzml files). The file “spectral-count/mzQuantML_draft_spectralCount_from_Excel_MPC.mzq” contains a spectral counting example file in which two biological samples were quantified in a time series analysis at five successive time points after a treatment took place, making ten <Assay> elements in total and two <StudyVariable> elements to summarize the replicate analyses of the same original sample.
Relationship with mzTab
The mzQuantML and mzTab specifications have been developed in a coordinated effort by the PSI to serve different user groups. Primarily mzQuantML has been designed as a format for tool developers for import into visualization or advanced post-processing software and to ensure that a full trace of analysis steps is maintained in a standardized format, which might be particularly useful for proteomics users in clinical domains. In contrast, mzTab has been developed as a lightweight layer for the simple transfer of final results, allowing end-user visualization in spreadsheets or statistical software. There is considerable overlap between the two formats for reporting final abundance values for proteins or peptides and to demonstrate how the formats map onto each other. For this part, the “label-free/CPTAC-Progenesis.mzq” file has been converted to mzTab “label-free/CPTAC_Progenesis_label_free_mzq.mzTab.” We also provide a table detailing the features and use cases covered by mzTab, mzQuantML, and mzIdentML (Table I).
Table I. Feature comparison among the file formats mzTab, mzIdentML, and mzQuantML.
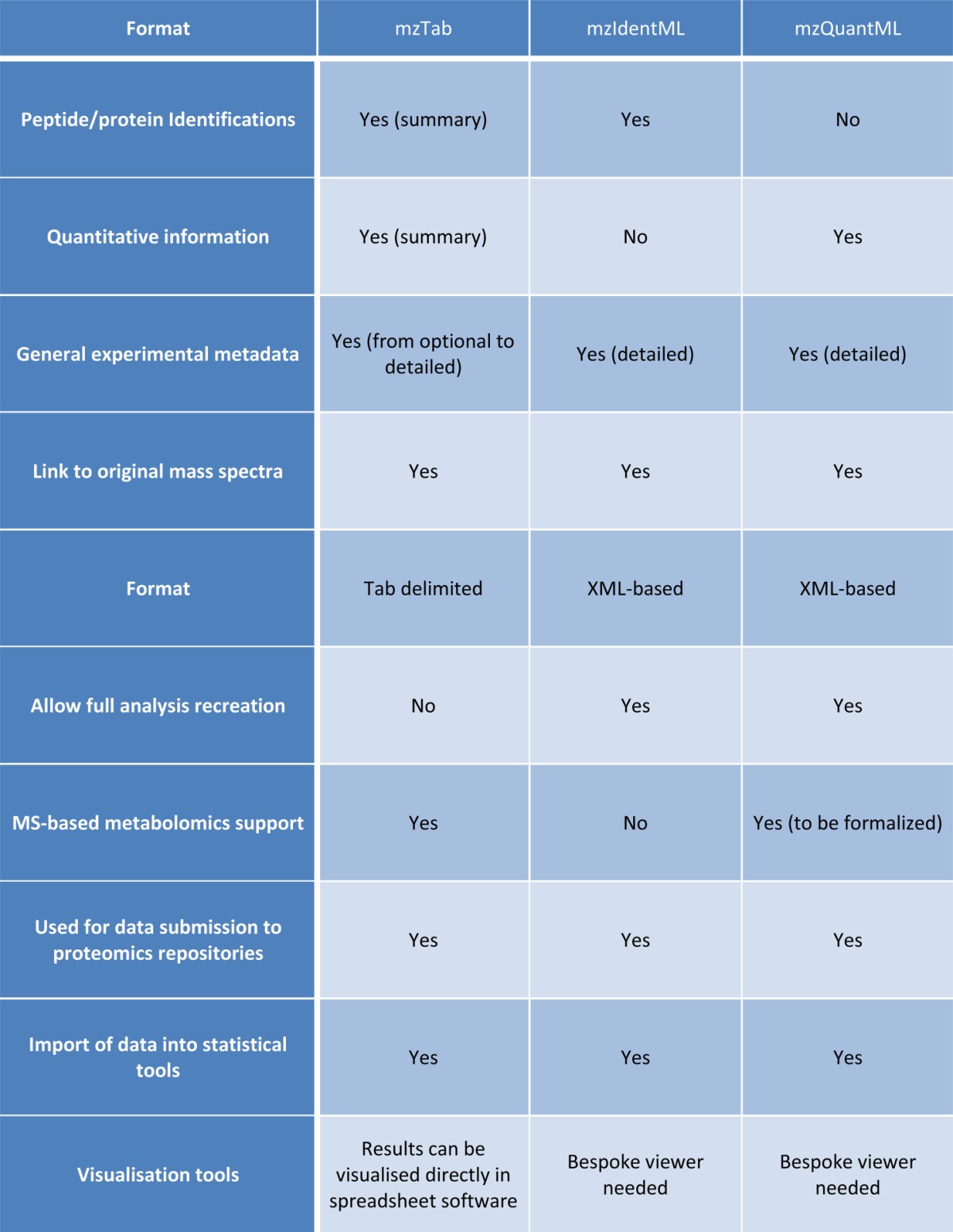
Relationship with MIAPE Quant and Publication Guidelines
The PSI has recently developed and released a minimum information guideline document for quantification studies called MIAPE Quant (version 1.0 is available from the PSI website). The MIAPE Quant document describes what information is essential to report in order to allow the study to be critically appraised, including a description of the labeling protocol employed, correction factors, software and parameters employed, normalization, grouping of replicates, and so on. An mzQuantML file has the capacity to represent a fully MIAPE Quant–compliant analysis, as detailed in the supplementary material associated with the version 1.0 specification document. It should be noted that a valid mzQuantML file might not be MIAPE compliant, as particular details might not be available to the exporting software. Conversely, mzQuantML also has the capacity to represent more information than requested by MIAPE Quant. For example, mzQuantML can capture a detailed trace of a software package's internal data types and parameters, which might not be requested by MIAPE Quant. In parallel with MIAPE efforts, several journals, including Molecular & Cellular Proteomics, have written and adopted guidelines on the protocol information, metadata, and data that should be reported alongside a proteomics publication (23). mzQuantML has been designed to support such guidelines for quantitative data, as exemplified in the supplementary material associated with the version 1.0 specification document.
DISCUSSION
The mzQuantML standard has been designed to improve the capabilities for open-source software development in proteomics, including re-analysis and visualization of data sets, and, importantly, to ensure that data sets submitted to public repositories contain a trace of how protein values were calculated via peptide intermediates, back to regions in two-dimensional LC-MS space. The project is supported by validation software to ensure that the stable generic core of mzQuantML can be used to cover different experimental methods currently widely used in proteomics and adapt to new scenarios and techniques as they are published. Several different open-source projects are now using mzQuantML as the backbone of their pipeline infrastructure, and the ongoing development and maintenance are supported by an active mailing list of developers around the world. With the release of the standard format, there is hope that public repositories for proteomics data will start to incorporate quantitative data sets for community re-use. We welcome further input and contributions to the project through attendance at a PSI meeting, conference calls, and/or contributions via the mailing list or the Google code repository.
Acknowledgments
We acknowledge numerous colleagues from the Proteomics Standards Initiative for helpful discussions and feedback at meetings.
Footnotes
* M.W. is funded by MedSys (BMBF grant number 0315450). G.M. and D.Q. are funded by the EU FP7 “ProteomeXchange” grant (grant number 260558). J.U. is funded by CLIB (“Cluster Industrielle Biotechnologie”) within the QProM project (contract number 616 40003 0315413B). M.E. is funded by P.U.R.E. (Protein Unit for Research in Europe), a project of Nordrhein-Westfalen, a federal state of Germany. F.F.G.-G. is funded by BBSRC (BB/I00095X/1). J.F. and C.B. are funded by BBSRC (BB/I001131/1). E.W.D. is funded by NIGMS grant GM087221, NHGRI grant HG005805, EU FP7 grant “ProteomeXchange” (grant number 260558), and the Systems Biology Initiative of the State of Luxembourg. F.R. is supported by the Wellcome Trust (grant number WT085949MA). J.A.V. is supported by the EU FP7 grants LipidomicNet (grant number 202272) and ProteomeXchange (grant number 260558). J.A.M.-A. and J.P.A. are supported by the Spanish Research Council (CSIC) and the Spanish National Proteomics Institute (ProteoRed-ISC III) (grant number 2005X747_3). O.K. is supported by grants from EU FP7 PRIME-XS (grant number 262067) and MARINA (grant number 236215), as well as by BMBF (SARA - FKZ 0315395F and BIOMARKERS - FKZ 01GI1104A). A.R.J. is supported by ProteomeXchange (grant number 260558) and the BBSRC (BB/I00095X/1, BB/H024654/1).
1 The abbreviations used are:
- CV
- controlled vocabulary
- iTRAQ
- isobaric tag for relative and absolute quantitation
- LC
- liquid chromatography
- MIAPE
- Minimum Information about a Proteomics Experiment
- MS
- mass spectrometry
- PSI
- Proteomics Standards Initiative
- SILAC
- stable isotope labeling by amino acids in cell culture
- XSD
- XML Schema Definition.
REFERENCES
- 1. Taylor C. F., Paton N. W., Lilley K. S., Binz P.-A., Julian R. K., Jones A. R., Zhu W., Apweiler R., Aebersold R., Deutsch E. W., Dunn M. J., Heck A. J. R., Leitner A., Macht M., Mann M., Martens L., Neubert T. A., Patterson S. D., Ping P., Seymour S. L., Souda P., Tsugita A., Vandekerckhove J., Vondriska T. M., Whitelegge J. P., Wilkins M. R., Xenarios I., Yates J. R., Hermjakob H. (2007) The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 25, 887–893 [DOI] [PubMed] [Google Scholar]
- 2. Orchard S., Salwinski L., Kerrien S., Montecchi-Palazzi L., Oesterheld M., Stumpflen V., Ceol A., Chatr-aryamontri A., Armstrong J., Woollard P., Salama J. J., Moore S., Wojcik J., Bader G. D., Vidal M., Cusick M. E., Gerstein M., Gavin A.-C., Superti-Furga G., Greenblatt J., Bader J., Uetz P., Tyers M., Legrain P., Fields S., Mulder N., Gilson M., Niepmann M., Burgoon L., Rivas J. D. L., Prieto C., Perreau V. M., Hogue C., Mewes H.-W., Apweiler R., Xenarios I., Eisenberg D., Cesareni G., Hermjakob H. (2007) The minimum information required for reporting a molecular interaction experiment (MIMIx). Nat. Biotechnol. 25, 894–898 [DOI] [PubMed] [Google Scholar]
- 3. Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., Tang W. H., Römpp A., Neumann S., Pizarro A. D., Montecchi-Palazzi L., Tasman N., Coleman M., Reisinger F., Souda P., Hermjakob H., Binz P.-A., Deutsch E. W. (2011) mzML—a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones A. R., Eisenacher M., Mayer G., Kohlbacher O., Siepen J., Hubbard S., Selley J., Searle B., Shofstahl J., Seymour S., Julian R., Binz P.-A., Deutsch E. W., Hermjakob H., Reisinger F., Griss J., Vizcaino J. A., Chambers M., Pizarro A., Creasy D. (2012) The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 11, M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deutsch E. W., Chambers M., Neumann S., Levander F., Binz P.-A., Shofstahl J., Campbell D. S., Mendoza L., Ovelleiro D., Helsens K., Martens L., Aebersold R., Moritz R. L., Brusniak M.-Y. (2012) TraML—a standard format for exchange of selected reaction monitoring transition lists. Mol. Cell. Proteomics 11, R111.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hermjakob H., Montecchi-Palazzi L., Bader G., Wojcik J., Salwinski L., Ceol A., Moore S., Orchard S., Sarkans U., von Mering C., Roechert B., Poux S., Jung E., Mersch H., Kersey P., Lappe M., Li Y., Zeng R., Rana D., Nikolski M., Husi H., Brun C., Shanker K., Grant S. G. N., Sander C., Bork P., Zhu W., Pandey A., Brazma A., Jacq B., Vidal M., Sherman D., Legrain P., Cesareni G., Xenarios I., Eisenberg D., Steipe B., Hogue C., Apweiler R. (2004) The HUPO PSI's molecular interaction format—a community standard for the representation of protein interaction data. Nat. Biotechnol. 22, 177–183 [DOI] [PubMed] [Google Scholar]
- 7. Gonzalez-Galarza F. F., Lawless C., Hubbard S. J., Hermjakob H., Jones A. R. (2012) A critical appraisal of techniques, software packages and standards for quantitative proteomic analysis. OMICS 16, 431–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 9. Pratt J. M., Simpson D. M., Doherty M. K., Rivers J., Gaskell S. J., Beynon R. J. (2006) Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043 [DOI] [PubMed] [Google Scholar]
- 10. Eisenacher M., Schnabel A., Stephan C. (2011) Quality meets quantity—quality control, data standards and repositories. Proteomics 11, 1031–1036 [DOI] [PubMed] [Google Scholar]
- 11. Vizcaíno J. A., Martens L., Hermjakob H., Julian R. K., Paton N. W. (2007) The PSI formal document process and its implementation on the PSI website. Proteomics 7, 2355–2357 [DOI] [PubMed] [Google Scholar]
- 12. Orchard S., Jones A., Albar J.-P., Cho S. Y., Kwon K.-H., Lee C., Hermjakob H. (2010) Tackling quantitation: a report on the Annual Spring Workshop of the HUPO-PSI 28–30 March 2010, Seoul, South Korea. Proteomics 10, 3062–3066 [DOI] [PubMed] [Google Scholar]
- 13. Orchard S., Albar J. P., Deutsch E. W., Eisenacher M., Vizcaíno J. A., Hermjakob H. (2011) Enabling BioSharing—a report on the Annual Spring Workshop of the HUPO-PSI April 11–13, 2011, EMBL-Heidelberg, Germany. Proteomics 11, 4284–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orchard S., Binz P.-A., Borchers C., Gilson M. K., Jones A. R., Nicola G., Vizcaino J. A., Deutsch E. W., Hermjakob H. (2012) Ten years of standardizing proteomic data: a report on the HUPO-PSI Spring Workshop. Proteomics 12, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer G., Montecchi-Palazzi L., Ovelleiro D., Jones A. R., Binz P.-A., Deutsch E., Orchard S., Vizcaíno J. A., Hermjakob H., Stephan C., Meyer H. E., Eisenacher M. (2013) The HUPO Proteomics Standards Initiative—mass spectrometry controlled vocabulary. Database (Oxford) Vol. 2013, doi:10.1093/database/bat009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qi D., Brownridge P., Beynon R. J., Xia D., Mackay K., Gonzalez F., Kenyani J., Jones A. R. (2012) A software toolkit and interface for performing stable isotope labelling and top3 quantification using Progenesis LC-MS. OMICS 16, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 18. Sturm M., Bertsch A., Gropl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohlbacher O., Reinert K., Gropl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP—the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 [DOI] [PubMed] [Google Scholar]
- 20. Vizcaíno J. A., Foster J. M., Martens L. (2010) Proteomics data repositories: providing a safe haven for your data and acting as a springboard for further research. J. Proteomics 73, 2136–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vizcaíno J. A., Côté R., Reisinger F., Barsnes H., Foster J. M., Rameseder J., Hermjakob H., Martens L. (2010) The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 38, D736–D742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Desiere F., Deutsch E. W., King N. L., Nesvizhskii A. I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S. N., Aebersold R. (2006) The PeptideAtlas project. Nucleic Acids Res. 34, D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bradshaw R. A., Burlingame A. L., Carr S., Aebersold R. (2006) Reporting protein identification data: the next generation of guidelines. Mol. Cell. Proteomics 5, 787–788 [DOI] [PubMed] [Google Scholar]