

Abstract
In recent years, RNA interference (RNAi) therapeutics, most notably with lipid nanoparticle-based delivery systems, have advanced into human clinical trials. The results from these early clinical trials suggest that lipid nanoparticles (LNPs), and the novel ionizable lipids that comprise them, will be important materials in this emerging field of medicine. A persistent theme in the use of materials for biomedical applications has been the incorporation of biodegradability as a means to improve biocompatibility and/or to facilitate elimination. Therefore, the aim of this work was to further advance the LNP platform through the development of novel, next-generation lipids that combine the excellent potency of the most advanced lipids currently available with biodegradable functionality. As a representative example of this novel class of biodegradable lipids, the lipid evaluated in this work displays rapid elimination from plasma and tissues, substantially improved tolerability in preclinical studies, while maintaining in vivo potency on par with that of the most advanced lipids currently available.
Introduction
The key to enabling therapeutics based on RNA interference (RNAi) is the safe and efficacious delivery of short interfering RNAs (siRNAs), the mediators of RNAi, to the appropriate tissues, cells, and ultimately, intracellular compartments where the natural RNAi machinery may be engaged for target mRNA silencing resulting in protein knockdown. Currently, lipid nanoparticles (LNPs) represent the most advanced platform for the systemic delivery of siRNAs, and recent clinical data suggest that LNP-mediated RNAi therapeutics may soon be a reality. Clinical trials in liver cancer1,2 and transthyretin (TTR) amyloidosis,3,4 utilizing LNPs based on the lipid 1,2-dilinoleyloxy-3-dimethylaminopropane (DLin-DMA), have demonstrated initial proof of concept in humans. Even more recently, LNPs based on a more advanced novel lipid, DLin-MC3-DMA,5 demonstrated efficacy at even lower doses and were found to be well tolerated in two clinical trials, one in hypercholesterolemia6,7 and a second in TTR amyloidosis.8,9 These promising clinical developments have been enabled by a significant research effort in recent years focusing on both the elucidation of the mechanisms involved in LNP-mediated siRNA delivery,10 and the development of LNPs with improved potency.5,11,12 These potency improvements have been mainly attributable to the development of more efficacious delivery materials, utilizing both empirical combinatorial chemistry and screening approaches13,14 as well as rational design efforts to elucidate and utilize lipid structure-activity relationships.5,12 Particularly encouraging has been the finding that the potency improvements associated with more advanced LNPs have translated in humans, as evidenced by the recent hypercholesterolemia7 and TTR amyloidosis9 clinical trial results.
The results from these early clinical trials suggest that LNPs, and the novel ionizable lipids that comprise them, will be important materials in the field of medicine. A persistent theme in the development of biomaterials for therapeutic applications has been the incorporation of biodegradable design features as a means to improve biocompatibility and/or as a means to eliminate materials once they are no longer necessary.15 Common examples in routine medical use today include controlled release drug carriers, biodegradable coatings, tissue engineering scaffolds, as well as reabsorbable materials such as sutures, staples, screws, and pins. These considerations are similarly applicable to the ionizable lipids used in LNPs. Introducing biodegradability into these lipids may improve their biocompatibility and may facilitate their elimination once they have served their purpose to deliver siRNA to the appropriate intracellular compartments in vivo. In particular, unlike many biomaterial applications that may have a required service lifetime of weeks or months, functional LNP-mediated delivery of siRNA occurs within hours,16 obviating the need for persistent lipids. Therefore, the aim of this work was to further advance the LNP platform through the development of novel, next-generation lipids that combine the excellent potency of the most advanced lipids currently available with biodegradable functionality leading to rapid elimination in vivo. The strategy employed here was to build upon the extensive knowledge of lipid structure-activity relationships gained in recent years and to judiciously introduce biodegradability while maintaining the structural features required for lipid potency.
Results
General lipid design principles
Based on prior studies, we have correlated certain structural features with activity and arrived at structure-activity relationships that guide the design of lipids with good in vivo efficacy.5,12 Namely, the lipid should comprise an amphipathic structure with a hydrophilic headgroup region containing an ionizable amine and long hydrophobic dialkyl chains capable of promoting the self-assembly of formulation components into stable nanoparticles encapsulating the siRNA. We found that the acid dissociation constant (pKa) of the ionizable amino group strongly correlated with in vivo efficacy, with a pKa optimum between 6.2 and 6.5.5 Furthermore, the lipid should adopt an overall “cone” shape in acidic environments when paired with anionic endosomal membrane lipids such as phosphatidylserine. Lipids that adopt such a “cone” shape ion pair are hypothesized to promote the formation of non-bilayer phases, such as the hexagonal HII phase, that are associated with membrane disruption events.12,17 The structure-activity data indicate that modifications, such as double bonds, which alter the relative orientation of the alkyl chains from cylindrical to cone shape, increase the spacing between the alkyl chains, thereby enhancing the ability of the lipid to induce non-bilayer structures. The highly potent lipid DLin-MC3-DMA5 (Figure 1) appears to fulfill these criteria and was therefore used as a basis in the design of novel, highly potent, rapidly eliminated lipids.
Figure 1.

Design of lipids incorporating biocleavable ester functions within hydrophobic alkyl chains. positional variations of the ester linkage are indicated by numbers along the alkyl chain (see also Table 1).
We then turned our attention to define certain desirable features in order to further guide the lipid design. First, for ultimate therapeutic use in an LNP product, an ideal lipid would be rapidly metabolized in vivo, yet demonstrate good chemical stability in order to provide for sufficient shelf-life. Second, the resulting metabolites should be nontoxic and readily eliminated or further metabolized. Finally, in this respect, it may be particularly advantageous for the lipid to be rapidly metabolized into hydrophilic, water-soluble products with substantially reduced partitioning into biological membranes.
Earlier studies have shown that the ability of LNPs to effectively deliver siRNA is strongly influenced by the nature of the ionizable lipids that comprise them, with particular sensitivity to structural changes in the headgroup-linker region that affect the ionization behavior of the headgroup or the orientation of the lipid chains.5,12 Taken together, these design criteria pointed us toward the incorporation of ester linkages into the hydrocarbon chain region of the amino lipid, thereby preserving a headgroup-linker structure with demonstrated efficacy. Ester linkages were selected as biocleavable functionalities as they generally display good chemical stability at physiologic pH but can be hydrolyzed enzymatically by esterase or lipase activity present in tissues and intracellular compartments.18,19 Cleavage of an ester linkage within the hydrocarbon chain will result in products that are significantly more hydrophilic, liberating a carboxylic acid and an alcohol. We preferred the orientation of the ester linkage such that hydrolysis would generate a dimethylamino-dicarboxylic acid fragment and two relatively short and hydrophilic primary alcohols. The negatively charged carboxylates can thus neutralize or even reverse any positive charge associated with the amino headgroup and might therefore support the dissociation of the lipid–siRNA complex and the release of the siRNA from the carrier.20 We also hypothesized that the resulting hydrolysis products, in addition to being more readily eliminated directly, may be substrates for further metabolism by β-oxidation, the natural mechanism for the metabolism of free fatty acids.
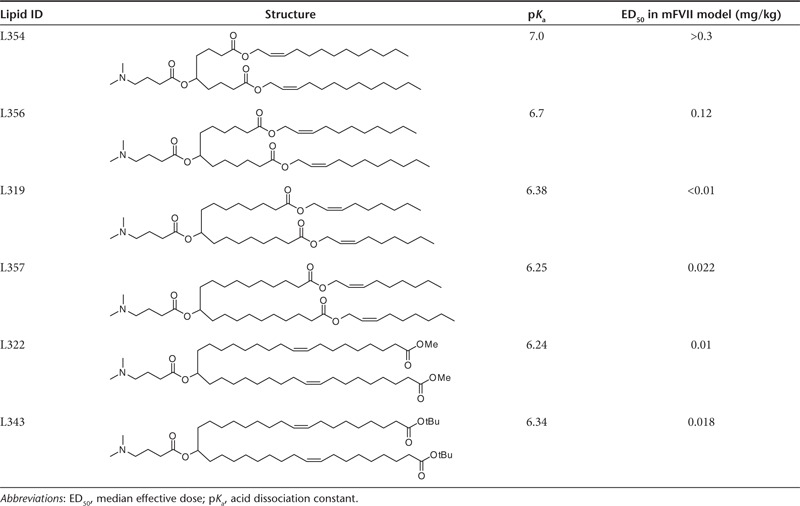
When considering the placement of the ester groups, we made note of the fact that the presence of the sp2-carbon of the ester would introduce a kink in the alkyl chain, similar to a double bond, thereby maintaining the spacing between the alkyl chains, which has been shown to be important for efficacy.21 Therefore, we rationalized that one of the double bonds of the linoleyl chain could be replaced with an ester group. For instance, replacing the 9,10-cis double bond with an ester (L319) places the degradable functionality centrally within the hydrocarbon chain, yielding hydrolysis products that would be predicted to be significantly more hydrophilic than the parent lipid (Figure 1). To gain further insight into the positional effect of the ester linkage, additional lipids containing esters at various positions along the alkyl chains (C4, C6, C10, and C18) were made and evaluated (Table 1). We rationalized that L343 containing sterically hindered tert-butyl esters should be substantially more stable against enzymatic degradation and may serve as a “non-degradable” control. The novel lipids were synthesized as summarized Supplementary Figures S1–S6 and described in Supplementary Materials and Methods.
Table 1. Structures and properties of novel lipids.
In vivo efficacy evaluation in mouse FVII model
LNPs were prepared using a spontaneous vesicle formation formulation procedure. The formulation resulted in small, uniform LNPs with a mean particle diameter of ~60 nm and greater than 90% siRNA encapsulation efficiency indicating that the presence of the ester group within the hydrophobic alkyl chains did not compromise the siRNA formulation properties of the lipids. Generally, the structural changes did not negatively impact the ionization behavior of the lipids as indicated by their apparent pKa values, which were determined by TNS (2-(p-toluidino)-6-naphthalene sulfonic acid) titration (Table 1). With the exception of L354 and L356 with ester positions shifted toward the headgroup-linker region (C4 and C6), the values were found to lie within the range of 6.2–6.5 reported as being optimal for in vivo activity.5
The in vivo activity of novel lipid-containing LNPs was determined using the mouse factor VII (FVII) gene silencing model.13 LNPs containing FVII-targeting siRNA (siFVII) were administered to mice at 0.01, 0.03, and 0.1 mg/kg via tail vein injection. In case of L319, this led to potent, dose-dependent knockdown of serum FVII protein, with ~75% silencing at the lowest dose tested (Figure 2). L319-containing LNPs were similarly efficacious in rats, with ~90% FVII silencing observed at 0.1 mg/kg (Supplementary Figure S7). Shifting the ester toward the headgroup-linker region led to a substantial decrease in potency as indicated in the more than tenfold and 30-fold higher median effective dose (ED50) levels for L356 and 354, respectively (Table 1). On the other hand, a shift in the opposite direction had only little effect on in vivo potency, with ED50 levels of L357, L322, and L343-containing LNPs found to be comparable with L319-LNPs. Potency for these more active lipids was found to be on par with that of the most potent lipids yet described in the literature, such as DLin-KC2-DMA12 and DLin-MC3-DMA.5 The in vivo activity data, as well as a preference for central placement of the degradable ester groups within alkyl chains to facilitate the generation of a smaller dimethylamino-dicarboxylic acid fragment, led us to focus the subsequent efforts on the novel lipid L319.
Figure 2.

Factor VII silencing activity of L319-lipid nanoparticle in mice after single intravenous administration. Serum was collected 48 hours postadministration and was analyzed for factor VII protein activity. Bars represent group mean ± SD (n = 3). PBS, phosphate-buffered saline.
Pharmacokinetics results demonstrate rapid elimination from plasma and tissues and confirm biodegradability
Having met two important criteria for siRNA delivery, good formulation characteristics and excellent in vivo activity, L319 was further evaluated to determine whether the lipid structure supports rapid metabolism and elimination from tissue. A pharmacokinetics (PK) study was performed in mice with an L319-based LNP formulation. The LNP was administered via tail vein injection and lipid levels were measured in blood, liver and spleen at various timepoints up to 168 hours postdose using liquid chromatography-mass spectrometry (LC-MS/MS) for lipid analysis and quantification. As additional controls, LNPs prepared with L343, containing a metabolically more stable, sterically hindered tert-butyl ester, as well as L322, its unhindered methyl ester analog, were also included in the PK study.
The L319 plasma PK profile indicated a very rapid distribution and/or elimination from plasma within the first few hours, with plasma levels decreasing more than 1,000-fold within the first 4 hours and dropping below the limit of quantification (= 0.8 pmol/ml) at 8 hours postdose (Figure 3a). The estimated plasma half-life was <30 minutes. A very rapid elimination rate was also observed for L319 in tissues. L319 accumulated in liver to a maximum level (Cmax) of ~4,600 pmol/g and levels rapidly decreased ~1,000-fold within the first 24 hours postdose (Figure 3b). At 48 hours, the lipid levels were below the limit of quantification (= 6 pmol/g). Similar observations were made in spleen with a Cmax of ~6,500 pmol/g and levels dropping ~100-fold within the first 24 hours postdose and below the limit of quantitation within 96 hours postdose (Supplementary Figure S8). L322, with its unhindered terminal methyl esters, displayed rapid elimination similar to L319, whereas L343, with its sterically hindered tert-butyl esters, exhibited slower elimination from plasma and higher and more persistent levels in liver (Figure 3a,b).
Figure 3.

Lipid pharmacokinetic profiles after single intravenous administration of lipid nanoparticle (0.3 mg/kg short interfering RNA) in mice. Concentration–time profiles of lipids L319, L343 and L322 in (a) plasma and (b) liver; concentration–time profiles of expected metabolites of L319 in (c) plasma and (d) liver; data points represent group mean ± SD (n = 2).
In addition to parent L319, the expected mono- as well as di-acid metabolites (Figure 1) could be detected in both plasma (Figure 3c) as well as tissues (Figure 3d and Supplementary Figure S8) confirming that the ester linkages were readily cleaved in vivo. Importantly, the metabolites were rapidly eliminated with rates similar to the parent compound and did not accumulate in plasma or tissues.
L319 is eliminated from intracellular compartments and is readily excreted in vivo
Considering the dramatic lipid elimination observed in vivo, we were interested to investigate the effect of biodegradability on intracellular LNP trafficking using fluorescently-labeled lipids. Previous studies have shown that LNPs enter cells via endocytosis with ionizable lipids accumulating in late endosome and lysosome compartments.16,22,23 To explore the intracellular trafficking and persistence in endo/lysosomal compartments, L319 was labeled with a fluorescent BODIPY dye (the details of the preparation are described in Supplementary Materials and Methods and Supplementary Figure S9). The labeled lipid was then incorporated into LNPs by replacing 10% of unlabeled lipid with labeled lipid. The 10% exchange with labeled lipid did not significantly impact in vitro or in vivo activity (Supplementary Figure S10). After 5-hour incubation with cells in vitro, the labeled formulation accumulated in LysoTracker Red positive compartments, i.e., primarily late endosomes and lysosomes (Figure 4a). Interestingly, however, 24 hours after the 5-hour incubation period, greater than eightfold decrease in vesicular fluorescent signal was observed in L319-BOD-treated cells (Figure 4b), indicative of rapid cellular clearance or degradation of L319-BOD.
Figure 4.

L319 is eliminated from intracellular compartments and is readily excreted in vivo. (a) Lipid nanoparticles (LNPs) containing BODIPY-labeled L319 were added at 20 nmol/l to HeLa cells co-stained with LysoTracker Red for 5 hours, green: BODIPY lipid, red: LysoTracker Red, blue: Hoechst. (b) Comparison of L319 lipid content per cell at 5 and 24 hours following a 5-hour pulse. (c) Excretion of L319 in urine and feces of rats after single intravenous administration of LNP-siRNA containing 14C-labeled cationic lipid. Urine and feces were collected 0–12, 12–24, 24–48, and 48–72 hours postadministration and analyzed for levels of 14C-label over each time period. Data show the cumulative amount of label detected over time and is expressed as % of injected dose and each data point indicates group mean ± SD (n = 4). RFU, relative fluorescence unit; siRNA, short interfering RNA.
To determine whether the rapid elimination of L319 from cells and tissues would translate into rapid and quantitative excretion, a 14C-label was introduced in the headgroup-linker region of L319 using 14C-labeled dimethylamino butyric acid (the details of the preparation are described in Supplementary Materials and Methods). Rats were dosed with LNPs containing 14C-labeled lipid and urine and feces were collected from each animal over 0–12, 12–24, 24–48, and 48–72-hour time periods. Blood and tissue were collected at 72 hours postdose. Consistent with the results of the lipid PK studies, L319 was rapidly eliminated with 30% of the injected dose detectable in the urine over the first 12 hours and 40% in the feces within 12–24 hours after administration (Figure 4c). Nearly, the entire injected dose of L319 was eliminated within 72 hours following intravenous administration. The vast majority was excreted within the first 24 hours with approximately half detected in each of the urine and the feces.
Safety studies in rat demonstrate that L319-LNP is well tolerated
One of the motivations for using biodegradable materials in medicine is to enhance biocompatibility; therefore, we evaluated the tolerability of L319-LNP in a dose escalation study in male Sprague-Dawley rats (n = 5/group). The formulation was administered as a single intravenous bolus injection at dose levels of 1, 3, 5, and 10 mg/kg. Potential test article-related effects were evaluated by clinical signs, body weight, serum chemistry (24 and 72 hours) and histopathology on select tissues (liver and spleen; 72 hours). L319-LNP was well tolerated up to 10 mg/kg. There were no adverse clinical signs or toxicologically significant alterations to body weight or serum chemistry parameters (Figure 5). Microscopic findings of single cell hepatocellular necrosis and/or vacuolation were observed in three of five animals at 10 mg/kg. Both findings were minimal to mild in severity and lacked any correlates to clinical chemistry parameters (Supplementary Table S1). There were no microscopic findings in the spleen of any dose group. These data represent a relative improvement in tolerability over the slowly eliminated control L343-LNP, which demonstrated signs of serum chemistry elevations (alanine transaminase, aspartate transaminase) at ≥3 mg/kg (Figure 5).
Figure 5.

Dose-dependent effect on rat serum chemistry (alanine transaminase (ALT) and aspartate transaminase (AST)) after intravenous infusion of L319-lipid nanoparticle (LNP) at 1, 3, 5, and 10 mg/kg dose and L343-LNP at 1, 3, and 5 mg/kg (based on short interfering RNA weight). Bars represent group mean ± SD (n = 5).
Efficacy and rapid elimination properties translate to higher species
Species-related differences in enzymatic activity could potentially impact the kinetics of lipid hydrolysis and metabolism, and therefore, diminish the benefits of this class of biodegradable lipids. Thus, it was important to confirm the translation of the favorable efficacy and elimination attributes of L319 in higher species. To address this issue, we conducted an exploratory efficacy and plasma PK study in nonhuman primates (NHPs). For this study, we chose to TTR, a hepatic gene of high therapeutic interest.24 Cynomolgus monkeys were treated with a single 15-minute intravenous infusion of TTR-targeting siRNA formulated in L319-based LNPs at a dose of 0.3 mg/kg. A liver biopsy was obtained from each animal at 48 hours postadministration and TTR liver mRNA levels were determined. Relative to control samples, ~70% silencing of TTR mRNA was observed in the treated animals (Figure 6a). In addition, plasma samples were collected from each animal at 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, and 168 hours after end-of-infusion. L319 levels were determined in plasma samples using LC-MS/MS analysis. Rapid elimination of lipid from plasma was observed, with plasma levels reduced nearly 100-fold within 4 hours, nearly 30,000-fold with 24 hours, and to undetectable levels by 48 hours (Figure 6b).
Figure 6.

Efficacy and lipid pharmacokinetics in nonhuman primates after single intravenous administration of 0.3 mg/kg TTR short interfering RNA formulated in L319-LNPs. (a) Liver TTR mRNA levels in biopsy samples collected 48 hours postadministration. Bars represent group mean ± SD (n = 3). (b) Lipid-concentration-time profile in plasma in L319-LNP–treated animals. Data points represent group mean (n = 3). LNP, lipid nanoparticle; TTR, transthyretin.
Discussion
Recent clinical trials have demonstrated the potential of RNAi as a therapeutic modality and LNPs as an RNAi delivery platform, in humans. In the present work, we have sought to further advance this platform by incorporating biodegradable design features into novel ionizable lipids used in LNPs. The key challenge was to introduce biodegradable functionality in such a way as to promote rapid in vivo metabolism into more hydrophilic, water-soluble products, while maintaining the excellent potency of the most advanced lipids currently available. This led us to design the lipid structure such that the biocleavable groups were positioned within the hydrophobic lipid tails. Further, while there are many available options for biodegradable linkers, ester linkages were selected due to their simple structure, good chemical stability and potential for rapid enzymatic cleavage in vivo. We rationalized that replacing one of the double bonds with the ester functionality, as in L319, would preserve the structural features of the lipid critical for in vivo activity. The loss of potency observed for L356 and L354-containing LNPs with the esters placed closer to the headgroup-linker region may at least in part be explained by an increase in apparent lipid pKa, found to be outside of the optimal range of 6.2–6.5. In contrast, moving the ester linkage toward the terminal ends of the alkyl chains had little effect on the lipid pKa and did not compromise the in vivo potency of the corresponding LNPs.
The novel lipids generally exhibited good formulation characteristics and, with the exception of L354 and L356, excellent activity in rodents, confirming that the presence of the ester linkage within the lipid tails was suitable for promoting siRNA encapsulation and for maintaining functional activity required for in vivo efficacy.
It has been demonstrated that LNPs enter cells via endocytosis and accumulate in endolysosomal compartments. The dramatic decrease in plasma and tissue levels observed for L319 as well as L322, together with the appearance of the expected mono- and di-acid metabolites, suggests that the lipids are susceptible to rapid enzymatic hydrolysis, conceivably mediated by lysosomal lipases with broad substrate specificity. The comparison with L343 indicates that the observed differences in elimination rates are mainly due to greater metabolic stability of the tert-butyl esters and further supports the role of enzymatic hydrolysis for the observed rapid elimination of L319 and L322. We rationalized that the polar structure of the primary hydrolysis products would support further metabolic processing and/or rapid excretion. In fact, PK profiles of the mono- and di-acid fragments indicated rapid elimination of these metabolites similar to that of the parent lipid.
Studies with labeled lipid confirmed the enhanced elimination of L319 from intracellular compartments in vitro and, importantly, the rapid and essentially quantitative excretion of radiolabel in vivo via both urine and feces. Collectively, these data highlight that the lipid structure supports rapid conversion into metabolites that can easily and quantitatively be removed from the body via excretion through urine and feces. Furthermore, the rapid elimination of L319 from target tissues and subsequent excretion translated into a favorable safety profile. L319-LNP was well tolerated up to 10 mg/kg in rats, representing a 100- to 1,000-fold multiple over efficacious dose levels and thereby demonstrating a substantial therapeutic index. Interestingly, these data are consistent with a proposed mechanism of action whereby functional LNP-mediated delivery of siRNA to the cytoplasm occurs rapidly, from early endosomal compartments, prior to transport to degradative late endosomes and lysosomes.
Finally, it was important to determine whether the favorable attributes of the novel L319-based LNPs translate into higher species. To address this, we conducted an exploratory efficacy and lipid PK study in NHPs, which confirmed the efficacy of L319-based LNPs with ~70% silencing of TTR mRNA after a single administration. It should be noted that the level of TTR gene silencing observed in the NHPs was less than the level of FVII gene silencing observed in mice (Figure 2). This difference may be in part attributable to the difference in siRNA and gene target, but the difference in activity may also be due in part to the fact that a composition optimized for mouse activity was used in the NHP study. Further optimization of the L319-LNP formulation composition will likely be required for optimal activity in the NHP. The rapid elimination of L319 observed in rodents was also confirmed in NHPs, with L319 in plasma reduced to undetectable levels by 48 hours postadministration.
In summary, we have described the development of a novel lipid that incorporates biodegradability into design principles elucidated in prior structure-activity relationship studies, resulting in rapid elimination and excretion, substantial tolerability and excellent potency in rodents and NHPs. To our knowledge, this is the first demonstration of a biodegradable lipid with an efficacy profile on par with that of the most advanced lipids currently available for siRNA delivery. With L319 as an exemplar, and with the possibility of further optimization, the class of biodegradable lipids described in this work shows great promise for use in future RNAi therapeutic programs.
Materials and Methods
General. PEG-DMG13 was synthesized as described previously, cholesterol was obtained from Sigma-Aldrich (St. Louis, MO), and DSPC was obtained from Avanti Polar Lipids (Alabaster, AL). All siRNAs were synthesized at Alnylam (Cambridge, MA) as described previously and characterized by electrospray MS and anion exchange high-performance liquid chromatography.25 The sequences for the sense and antisense strands of the siRNAs targeting FVII, TTR and the control siRNA targeting luciferase are as follows: siFVII sense: 5′-GGAUCAUCUCAAGUCUUACdTsdT, antisense: 5′-GUAAGACUUGAGAUGAUCCdTsdT; siTTR sense: 5′-GuAAccAAGAGuAuuccAudTdT-3′, antisense: 5′-AUGGAAuACUCUUGGUuACdTdT-3′ siLuc sense: 5′-cuuAcGcuGAGuAcuucGAdTsdT, antisense: 5′-UCGAAGuACUcAGCGuAAGdTsdT. 2′-O-methyl modified nucleotides are in lower case, 2′-fluoro modified nucleotides are underlined and phosphorothioate linkages are indicated as “s”. siRNAs were generated by annealing equimolar amounts of complementary sense and antisense strands. All animal studies were conducted in accordance with local, state and federal regulations as applicable and approved by the Institutional Animal Care and Use Committee and all study protocols were approved by the Institutional Animal Care and Use Committee.
Formulation and particle characterization. LNPs were prepared using ionizable lipid L319, distearoylphosphatidylcholine (DSPC), cholesterol and PEG-DMG at a molar ratio of 55:10:32.5:2.5 (L319:DSPC:cholesterol:PEG-DMG). The siRNA was introduced at a lipid nitrogen to siRNA phosphate ratio of 3, corresponding to a total lipid to siRNA weight ratio of ~10:1. A spontaneous vesicle formation process was used to prepare the LNPs. siRNA was diluted to ~1 mg/ml in 10 mmol/l citrate buffer, pH 4. The lipids were solubilized and mixed in the appropriate ratios in ethanol. Syringe pumps were utilized to deliver the siRNA solution and lipid solution at 15 and 5 ml/min, respectively. The syringes containing siRNA solution and lipid solution were connected to a union connector (0.05 in thru hole, #P-728; IDEX Health & Science, Oak Harbor, WA) with PEEK high-performance liquid chromatography tubing (0.02 in ID for siRNA solution and 0.01 in ID for lipid solution). A length of PEEK high-performance liquid chromatography tubing (0.04 in ID) was connected to the outlet of the union connector and led to a collection tube. The ethanol was then removed and the external buffer replaced with phosphate-buffered saline (155 mmol/l NaCl, 3 mmol/l Na2HPO4, 1 mmol/l KH2PO4, pH 7.5) by either dialysis or tangential flow diafiltration. Finally, the LNPs were filtered through a 0.2 μm sterile filter. Particle size was determined using a Malvern Zetasizer Nano ZS (Malvern, UK). siRNA content was determined by ultraviolet absorption at 260 nm and siRNA entrapment efficiency was determined by Quant-IT Ribogreen (Invitrogen, Carlsbad, CA) assay.21
Lipid PK in mice. Six to 8 weeks old male C57Bl/6 mice were obtained from Charles River Laboratories (Wilmington, MA). Mice were administered with LNP-siRNA systems containing nontargeting (luciferase) control siRNA (siLuc). Mice (n = 2/timepoint/group) were administered at 0.3 mg/kg by intravenous bolus injection via the lateral tail vein. Blood, liver, and spleen were collected at 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, and 168 hours postdose. Mice were perfused with saline before tissues collection. Blood samples were processed to obtain plasma. All samples were processed and analyzed by liquid chromatography with tandem LC-MS/MS as described in the Supplementary Materials and Methods.
Cellular trafficking studies. HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. LNPs were prepared using a BODIPY-labeled version of L319. Labeled lipid was incorporated into LNPs by replacing 10% of unlabeled lipid. Cells were exposed to LNPs diluted in regular media containing 1 μg/ml apolipoprotein E (Fitzgerald Industries, Acton, MA) for 5 hours. In some experiments, cells were counterstained with LysoTracker Red (Invitrogen) for the last 30 minutes of incubation, and in others, the media was changed and the uptake of the LNPs was allowed to proceed for another 24 hours. Post-LNP incubation the cells were counterstained with cell permeable Hoechst dye (Invitrogen) then imaged live using an Opera spinning disc confocal system (PerkinElmer, Waltham, MA). Image quantitation was done using Acapella software (PerkinElmer).
Excretion studies using radiolabeled lipid. Six to 8 weeks old Sprague-Dawley rats were obtained from Charles River Laboratories for the excretion studies. Animals were singly housed in metabolic cages (Techniplast, Philadelphia, PA) and provided with food and water ad libitum. LNP formulation encapsulating siFVII and containing 14C-labeled L319 was diluted to the appropriate concentration in sterile phosphate-buffered saline immediately before single intravenous bolus administration via the lateral tail vein at a lipid dose of 5.66 μmol/l/kg lipid (equivalent to 0.37 mg/kg siRNA) in a volume of 5 ml/kg. Urine and feces were collected from four animals per group over 0–12, 12–24, 24–48, and 48–72 hours time periods postadministration. These animals were then terminated and blood and tissues collected for determination of levels of radiolabel at 72 hours postadministration.
Rat toxicology studies. Male Sprague-Dawley rats were obtained from Charles River Laboratories and were 8 weeks old at initiation of dosing. L319-based LNP encapsulating a luciferase-targeting siRNA, was administered at 0, 1, 3, 5, and 10 mg/kg (5 animals/group) via single intravenous bolus injection at a dose volume of 5 ml/kg. Approximately, 1.0 ml of blood was obtained 24 hours postdose from the jugular vein of conscious animals using manual restraint and was processed to serum. All groups were terminated 72 hours postdose for postmortem evaluation. Potential test article-related effects were evaluated by the assessment of clinical signs, body weight, serum chemistry (complete panel, Olympus AU400 Serum Chemistry Analyzer; Beckman-Coulter, Brea, CA), macroscopic observations at necropsy, organ weights (liver and spleen) and histopathology on liver and spleen. Tissues were processed and evaluated by Experimental Pathology Laboratories (Sterling, VA).
NHP study. All procedures using cynomolgus monkeys were conducted by a certified contract research organization using protocols consistent with local, state and federal regulations, as applicable, and approved by the Institutional Animal Care and Use Committee. Cynomolgus monkeys (n = 3 per group) received 0.3 mg/kg of TTR siRNA formulated in L319-LNP as a 15-minute intravenous infusion (5 ml/kg) via the cephalic vein. A liver biopsy was obtaining from each animal at 48 hours postadministration and TTR liver mRNA levels, relative to GAPDH mRNA levels, were determined in liver samples using a branched DNA assay (QuantiGene Assay; Affymetrix, Santa Clara, CA). Plasma samples were collected from each animal at 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, and 168 hours after end-of-infusion. The samples were processed and analyzed by liquid chromatography with tandem LC-MS/MS as described in Supplementary Materials and Methods.
SUPPLEMENTARY MATERIAL Figure S1. Synthesis of lipid L319. Figure S2. Synthesis of lipid L354. Figure S3. Synthesis of lipid L356. Figure S4. Synthesis of lipid L357. Figure S5. Synthesis of lipid L322. Figure S6. Synthesis of lipid L343. Figure S7. Factor VII silencing activity of L319-LNP in rats. Figure S8. Concentration–time profiles of lipid L319 and its major metabolites in spleen after single IV administration of LNP (0.3 mg/kg siRNA) in mice. Figure S9. Synthesis of BODIPY-labeled lipid L319. Figure S10. Confirmation of silencing activity of LNPs containing 10% L319-BOD. Table S1. Histopathology incidence and severity of L319-LNPs in rats. Materials and Methods.
Acknowledgments
All the authors are employees of Alnylam Pharmaceuticals or have received research funding from Alnylam Pharmaceuticals.
Supplementary Material
References
- Dose escalation trial to evaluate the safety tolerability pharmacokinetics and pharmacodynamics of intravenous ALN-VSP02 In patients with advanced solid tumors with liver involvement.Registry of Federally and Privately Supported Clinical Trials, U.S. National Institutes of Health. < . < http://clinicaltrials.gov/ct2/show/NCT00882180 >.
- Gollob J.2011Phase I Dose Escalation Study of ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement.ASCO Annual Meeting, 48 June, Chicago, USA [Google Scholar]
- Trial to Evaluate Safety and Tolerability of ALN-TTR01 in Transthyretin (TTR) Amyloidosis Registry of Federally and Privately Supported Clinical Trials, U.S. National Institutes of Health . < http://clinicaltrials.gov/ct2/show/NCT01148953 >.
- Coelho T.2012Final Phase I Safety, Pharmacokinetic and Pharmacodynamic Results for ALN-TTR01, a novel rnai therapeutic for the treatment of transthyretin amyloidosis.XIII International Symposium on Amyloidosis, 10 May 2012, Groningen, Netherlands [Google Scholar]
- Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trial to Evaluate Safety and Tolerability of ALN-PCS02 in Subjects With Elevated LDL-Cholesterol (LDL-C)Registry of Federally and Privately Supported Clinical Trials, U.S. National Institutes of Health. < . < http://clinicaltrials.gov/ct2/show/NCT01437059 >.
- Fitzgerald K.2012Phase I Safety, Pharmacokinetic and Pharmacodynamic Results for ALN-PCS, A Novel RNAi Therapeutic for the Treatment of Hypercholesterolemia American Heart Association's Arteriosclerosis, Thrombosis and Vascular Biology 2012 Scientific Sessions19 April 2012, Chicago, USA [Google Scholar]
- Trial to Evaluate Safety, Tolerability, and Pharmacokinetics of ALN-TTR02 in Healthy Volunteer SubjectsRegistry of Federally and Privately Supported Clinical Trials, U.S. National Institutes of Health. < . < http://clinicaltrials.gov/ct2/show/NCT01559077 >.
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Timothy S, Mant T, et al. N Engl J Med. in press; 2013. Safety and efficacy of an RNAi therapeutic targeting transthyretin (TTR) for TTR amyloidosis. [DOI] [PubMed] [Google Scholar]
- Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol. 2008;26:561–569. doi: 10.1038/nbt1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon KP, Love KT, Whitehead KA, Qin J, Akinc A, Leshchiner E, et al. Combinatorial approach to determine functional group effects on lipidoid-mediated siRNA delivery. Bioconjug Chem. 2010;21:1448–1454. doi: 10.1021/bc100041r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner BD, Hoffman AS, Schoen FJ, Lemons JE. Biomaterials Science, Third Edition: An Introduction to Materials in Medicine Waltham. Academic Press, Oxford; 2013. [Google Scholar]
- Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, et al. Nat Biotechnol. in press; 2013. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. [DOI] [PubMed] [Google Scholar]
- Hafez IM, Maurer N, Cullis PR. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001;8:1188–1196. doi: 10.1038/sj.gt.3301506. [DOI] [PubMed] [Google Scholar]
- Gilham D, Lehner R. Techniques to measure lipase and esterase activity in vitro. Methods. 2005;36:139–147. doi: 10.1016/j.ymeth.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Wong H, Schotz MC. The lipase gene family. J Lipid Res. 2002;43:993–999. doi: 10.1194/jlr.r200007-jlr200. [DOI] [PubMed] [Google Scholar]
- Prata CA, Zhao Y, Barthelemy P, Li Y, Luo D, McIntosh TJ, et al. Charge-reversal amphiphiles for gene delivery. J Am Chem Soc. 2004;126:12196–12197. doi: 10.1021/ja0474906. [DOI] [PubMed] [Google Scholar]
- Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release. 2005;107:276–287. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Akinc A.2012Strategies for delivery of RNAi therapeutics Asia Tides28 February–1 March 1, 2012Tokyo, Japan [Google Scholar]
- Maier M.2012Recent advances in lipid nanoparticle-mediated delivery of RNAi therapeutics. 8th Annual Meeting of the Oligonucleotide Therapeutics Society 2831 October, Boston [Google Scholar]
- Kurosawa T, Igarashi S, Nishizawa M, Onodera O. Selective silencing of a mutant transthyretin allele by small interfering RNAs. Biochem Biophys Res Commun. 2005;337:1012–1018. doi: 10.1016/j.bbrc.2005.09.142. [DOI] [PubMed] [Google Scholar]
- Addepalli H., Meena, , Peng CG, Wang G, Fan Y, Charisse K, et al. Modulation of thermal stability can enhance the potency of siRNA. Nucleic Acids Res. 2010;38:7320–7331. doi: 10.1093/nar/gkq568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.