

Abstract
Saposin C-dioleoylphosphatidylserine (SapC-DOPS) nanovesicles are a nanotherapeutic which effectively target and destroy cancer cells. Here, we explore the systemic use of SapC-DOPS in several models of brain cancer, including glioblastoma multiforme (GBM), and the molecular mechanism behind its tumor-selective targeting specificity. Using two validated spontaneous brain tumor models, we demonstrate the ability of SapC-DOPS to selectively and effectively cross the blood–brain tumor barrier (BBTB) to target brain tumors in vivo and reveal the targeting to be contingent on the exposure of the anionic phospholipid phosphatidylserine (PtdSer). Increased cell surface expression of PtdSer levels was found to correlate with SapC-DOPS–induced killing efficacy, and tumor targeting in vivo was inhibited by blocking PtdSer exposed on cells. Apart from cancer cell killing, SapC-DOPS also exerted a strong antiangiogenic activity in vitro and in vivo. Interestingly, unlike traditional chemotherapy, hypoxic cells were sensitized to SapC-DOPS–mediated killing. This study emphasizes the importance of PtdSer exposure for SapC-DOPS targeting and supports the further development of SapC-DOPS as a novel antitumor and antiangiogenic agent for brain tumors.
Introduction
Glioblastoma multiforme (GBM) is the most common and most destructive form of primary brain tumor. The aggressive and drug-resistant nature of this tumor is compounded by protection behind the blood–brain tumor barrier (BBTB).1,2,3 Despite decades of improvements in radiation and chemotherapy, the median survival remains <15 months.4,5 A significant obstacle and bottleneck in the development of therapeutics for brain tumors is the inability of systemic therapies to efficiently cross the BBTB.3 Currently, drug delivery has centered on transiently permeabilizing the BBTB or directly bypassing the BBTB through intratumoral or intraventricular administration.1,2 Utilizing nanotechnology to deliver chemotherapy is an emerging paradigm; however, dualistic nanocarriers which can cross the BBTB and also exert intrinsic antitumor effects are lacking.6,7
Saposin C (SapC) is a sphingolipid-activating protein found ubiquitously throughout the body that functions to catabolize glycosphingolipids.8 When SapC is coupled with dioleoylphosphatidylserine (DOPS), stable nanovesicles are formed which selectively fuse with cancer cells, leading to ceramide accumulation, caspase activation, and eventual apoptosis.9 Interestingly, intravenously administered SapC-DOPS nanovesicles are capable of delivering fluorescent probes and magnetic resonance contrast agents directly into the tumor tissue, attesting to the specificity of the systemic administration.10 Although the SapC component was required for in vivo tumor-selective targeting, the molecular mechanism behind this targeting has not been elucidated.
The main objective of this study was to evaluate the ability of SapC-DOPS to cross the BBTB and target GBMs in vivo. We also sought to elucidate the molecular mechanism behind its targeting to develop novel strategies to increase overall therapeutic efficacy. Here, we show that tumor-specific targeting of SapC-DOPS is reliant on phosphatidylserine (PtdSer) externalization and that high levels of PtdSer on the surface of GBM and the tumor-associated endothelium allow for the specific antitumor and antiangiogenic effects of SapC-DOPS treatment.
Results
SapC-DOPS nanovesicles target human xenografts and spontaneous mouse brain tumors in vivo
To investigate if SapC-DOPS could effectively target glioma cells in vitro, we utilized SapC-DOPS nanovesicles labeled with the lipophilic fluorescent probe CellVue Maroon (CVM). Initial targeting was tested using U87ΔEGFR cells, human glioma cells harboring EGFRvIII: a truncated, constitutively active, mutant epidermal growth factor receptor (ΔEGFR). EGFR amplification is the most common genetic alteration in GBM and many of these tumors overexpressing EGFR also harbor the constitutively active form EGFRvIII, a strong prognostic indicator of poor survival.11 Following treatment of U87ΔEGFR-Luc cells with SapC-DOPS-CVM, accumulation of CVM within the cell membrane was evident by fluorescent microscopy. Quantification of the targeting by flow cytometry revealed that SapC-DOPS was incorporated into glioma cell membranes within minutes and remained stably incorporated in the cell membrane for up to an hour (Figure 1a). To determine if SapC-DOPS could target human GBM cells in vivo, we implanted green fluorescent protein-expressing primary GBM-derived X12v2 cells (Figure 1b) or U87ΔEGFR-Luc cells (Supplementary Figure S1a) intracranially into mice. Ten days following tumor cell implantation, mice were treated intravenously with SapC-DOPS-CVM by tail vein injection and its localization was evaluated by monitoring fluorescence by the IVIS 200 imaging system (Figure 1b and Supplementary Figure S1a). Consistent with the rapid targeting observed in vitro, SapC-DOPS-CVM was found to localize to intracranial tumors within minutes of intravenous injection and persisted for up to 48 hours post-treatment. Fluorescent microscopy of frozen sections of tumor-bearing brains from these mice revealed that SapC-DOPS-CVM colocalized with green fluorescent protein-expressing GBM cells in vivo (Supplementary Figure S1b). Importantly, no fluorescence signal was detected in normal non-neoplastic brain parenchyma.
Figure 1.
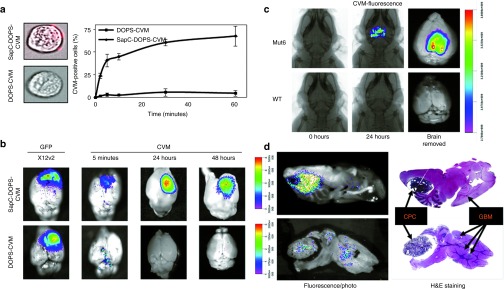
SapC-DOPS efficiently targets glioblastoma multiforme (GBM) cells in vitro and in vivo. (a) Fluorescent microscopy images of U87ΔEGFR-Luc cells treated with CellVue Maroon (CVM)-labeled SapC-DOPS (top) and control CVM-labeled DOPS (bottom panel) showing ability of SapC-DOPS to target GBM cells in vitro. Right panel is quantification of targeting by imaging flow cytometry ± SD. (b) Fluorescence images of brains (superimposed on bright field) of mice bearing intracranial glioma (X12v2 cells) treated with intravenous SapC-DOPS-CVM (top) or DOPS-CVM 10 days post-tumor cell implantation. (c,d) Fluorescent IVIS images of spontaneous tumor bearing (c) Mut6 and (d) cKO mice treated with a single dose of SapC-DOPS-CVM. Hematoxylin and eosin (H&E) staining of tumor-bearing brain sections imaged in d. CPC, choroid plexus carcinoma; GFP, green fluorescent protein; SapC-DOPS, Saposin C-dioleoylphosphatidylserine.
To further confirm the ability of SapC-DOPS to cross the BBTB, we utilized two genetically engineered mouse models which develop spontaneous brain tumors. Mut6 mice (GFAP-cre; Nf1loxP/+; p53−/loxP; PtenloxP/+)12 and quadruple cKO mice (GFAP-CreER; PtenloxP/loxP; p53loxP/loxP; Rb1loxP/loxP; p107−/−) (L.M.L. Chow and S.J. Baker, unpublished data) were obtained and monitored daily for development of neurological symptoms (seizures, paralysis, etc.). Once symptoms of tumor burden (mild hemiparesis, lack of grooming, or lethargy) were observed (~15 weeks for Mut6 and 10 weeks for quadruple cKO), mice were treated with a single intravenous dose of SapC-DOPS-CVM and imaged 24 hours later. Tumor-specific CVM fluorescence was observed in both Mut6 and quadruple cKO spontaneous glioma-bearing mice (Figure 1c,d). cKO mice occasionally develop choroid plexus carcinoma, a rare brain tumor which occurs mainly in young children. Histological analysis of the brains from these mice revealed efficient targeting of choroid plexus carcinoma as well as small foci of spontaneous glioma by SapC-DOPS-CVM (Figure 1d). These data demonstrate that SapC-DOPS can effectively cross the BBTB to selectively target brain tumors in vivo.
PtdSer exposure on cell surface is essential for SapC-DOPS targeting
Collectively these results displayed a very high specificity of SapC-DOPS for neoplastic brain tumor cells with minimal effects on non-neoplastic normal brain. We further explored the specificity of SapC-DOPS for glioma by investigating its targeting mechanism. SapC is known to associate and fuse to negatively charged phospholipids including PtdSer.13,14 Normally sequestered on the inner leaflet of the cell membrane, PtdSer is externalized to the outer leaflet of plasma membrane of neoplastic cells.15,16,17 To test if cancer cell specificity of SapC-DOPS was dependent on surface exposure of PtdSer, we compared sensitivity of GBM cells with low or high surface exposure of PtdSer to SapC-DOPS–induced cytotoxicity. GBM cells expressing high surface levels of PtdSer were significantly more sensitive to SapC-DOPS treatment compared with GBM cells with low PtdSer exposure levels (Figure 2a,b).
Figure 2.

SapC-DOPS targets exposed phosphatidylserine on glioma cells in vitro and in vivo. (a) Quantification of the mean fluorescence of exposed PtdSer in a panel of glioblastoma multiforme (GBM) cell lines. (b) Percent survival of GBM cells 72 hours after treatment with SapC-DOPS (SapC 50 μmol/l). Data shown are mean values of percent surviving cells for low and high PtdSer-expressing cell lines. **P = 0.01. (c) Representative luminescent and fluorescence IVIS images of mice implanted subcutaneously with U87ΔEGFR-Luc cells incubated with lactadherin (Lact-C2), β-2-glycoprotein-1 (β2GP1), or PBS, after intravenous treatment with SapC-DOPS-CVM, respectively. CVM, CellVue Maroon; SapC-DOPS, Saposin C-dioleoylphosphatidylserine; PBS, phosphate-buffered saline; PtdSer, phosphatidylserine.
To further corroborate this finding in vivo, we tested the ability of the PtdSer-specific binding proteins lactadherin C2 (Lact-C2) and β-2-glycoprotein-1 (β2GP1) to block in vivo targeting of SapC-DOPS to GBM cells. U87ΔEGFR-Luc cells were incubated with Lact-C2 or β2GP1 before subcutaneous implantation into nude mice. Cells were implanted under the skin and not intracranially to permit visualization of signal without allowing cells to proliferate and loose cell surface bound Lact-C2 or β2GP1. One hour following tumor cell implantation, mice were treated with SapC-DOPS-CVM intravenously and imaged for CVM fluorescence. Luciferase imaging confirmed viable tumor cell implantation in all mice. Fluorescent imaging for SapC-DOPS revealed efficient localization of CVM-labeled SapC-DOPS in control mice, whereas no labeling was observed in mice implanted with GBM cells treated with PtdSer blocking Lact-C2 or β2GP1 proteins (Figure 2c). Because many tumors contain high levels of dead and dying cells which expose PtdSer, we next investigated whether SapC-DOPS preferentially targeted areas of necrosis containing high levels dead/dying cells. To do this we established subcutaneous U87ΔEGFR-Luc tumors in mice and let them grow to a large size of 1,500 mm3, a size in which we know there to be large areas of necrosis and then treated with a single dose of SapC-DOPS-CVM. Tumors were then harvested 24 hours later for immunofluorescence. Fluorescent imaging of CVM shows SapC-DOPS localization in both necrotic and non-necrotic tumor tissue (Supplementary Figure S1c). Collectively, these results provide evidence that PtdSer exposure on GBM cells is necessary for SapC-DOPS targeting in vivo.
SapC-DOPS exerts potent antiangiogenic effects in vitro and in vivo
Similar to cancer cells, high levels of PtdSer exposure have also been detected on tumor blood vessels in comparison to normal endothelium and have even been referred to as a universal marker for tumor vasculature.18,19,20 In light of the specificity of SapC-DOPS for PtdSer, we investigated SapC-DOPS's impact on angiogenesis in brain tumors. We first determined that proliferating endothelial cells succumbed to SapC-DOPS–induced cytotoxicity at comparable doses to GBM cells in vitro (Supplementary Figure S2a). Treatment of human dermal microvascular endothelial cells (HDMECs) (Figure 3b) and human umbilical vein endothelial cells (HUVECs) (Supplementary Figure S2b) with SapC-DOPS caused a significant inhibition of cell migration in a standard Boyden chamber assay and nearly abolished the viable tube formations of HDMECs (Figure 3a) and HUVECs (Supplementary Figure S2c) on Matrigel. SapC-DOPS also substantially inhibited vessel sprouting in the ex vivo rat aortic ring assay (Figure 3c).
Figure 3.
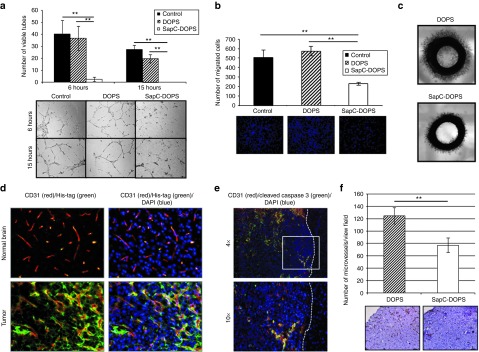
Antiangiogenic effects of SapC-DOPS in vitro and in vivo. (a) Inhibition of endothelial cell tube formation by SapC-DOPS. Human dermal microvascular endothelial cells (HDMECs) were incubated with SapC-DOPS, DOPS, or control (media alone). Cells were then plated onto polymerized Matrigel. Six and 15 hours post-plating, the number of tubes/view field were quantified. Data shown are mean number of tubes/view field ± SD, **P < 0.01. Bottom panel shows representative images of endothelial cell tubes from each group. (b) Reduced migration of HDMECs treated with SapC-DOPS. HDMECs treated with SapC-DOPS, DOPS, or control (media alone) were allowed to migrate in a standard Boyden chamber assay, and the number of cells that migrated to the other side of the membrane were quantified. Data shown are mean number of cells/view field ± SD. Representative fluorescent images of migrated cells stained with Hoechst, **P < 0.001. (c) Reduced ex vivo sprouting of rat aorta rings treated with SapC-DOPS. One millimeter thick rings of rat aorta rings treated as indicated were plated in Matrigel, and the amount of endothelial sprouting was analyzed 48 hours later. Shown are representative images of sprouting aorta from n = 4/group. (d) SapC-DOPS targets tumor vasculature in vivo. Immunofluorescent images (20x) from the brains of mice bearing intracranial U87ΔEGFR tumors treated with SapC-DOPS: His-tag (green), CD31 (red), and DAPI (blue). (e) Immunofluorescent images from brains in d stained with CD31 (red), cleaved caspase-3 (green), and DAPI (blue). Dotted white line divides tumor (left) from normal brain parenchyma (right). (f) SapC-DOPS reduced angiogenesis in vivo. Subcutaneous Gli36ΔEGFR tumor bearing mice (100–200 mm3) were treated with five consecutive daily doses of SapC-DOPS or DOPS control, and then analyzed for microvessel density as described in Materials and Methods. Data shown are mean MVD ± SD for each group, n = 2–4 sections/tumor and n = 4 tumors/group, **P = 0.001. Bottom shows representative images of tumor sections analyzed by immunohistochemistry for CD31 to highlight blood vessels. Bars = 100 μmol/l. DAPI, 4′,6-diamidino-2-phenylindole; MVD, microvessel density; SapC-DOPS, Saposin C-dioleoylphosphatidylserine.
To investigate whether SapC-DOPS could target the tumor vasculature in vivo, we implanted U87ΔEGFR glioma cells intracranially in nude mice. Mice were treated 11 days later with a single dose of His-tagged SapC-DOPS or DOPS control and animals were killed and brains were harvested for immunofluorescence analysis 3 hours later. We observed colocalization of SapC-DOPS (His-tag) with the tumor vasculature (CD31) and glioma cells (DAPI (4′,6-diamidino-2-phenylindole)) within the tumor (Figure 3d). Importantly, His-tag staining was found to be absent in the normal brain parenchyma including both the normal blood vessels and normal neuronal cells. Furthermore, we observed colocalization (yellow) of CD31 (red) and cleaved caspase-3 (green) within the tumor but not in the normal brain (Figure 3e). These results attest to the specificity of SapC-DOPS targeting of glioma cells and the tumor vasculature as opposed to normal brain tissue.
To elucidate the effect of SapC-DOPS on the tumor vasculature in vivo, nude mice with established subcutaneous GBMs (100–200 mm3) were treated with five consecutive daily doses of SapC-DOPS or DOPS control. Twenty-four hours following the final treatment, animals were killed and the tumors were harvested for immunohistochemistry analysis. Quantification of CD31-positive microvessels21 demonstrated a remarkable reduction in the microvessel density of GBMs treated with SapC-DOPS compared with DOPS control (Figure 3f). Overall, these results reveal novel antiangiogenic effects of SapC-DOPS.
SapC-DOPS sensitization in hypoxia
Antiangiogenic therapy for high grade GBM has been associated with tumor vessel destruction resulting in elevated tumoral hypoxia.22 Tumoral hypoxia is recognized for its influence in resistance to standard therapeutics including ionizing radiation and chemotherapy.23 Since SapC-DOPS possesses significant antiangiogenic effects, we tested the efficacy of SapC-DOPS in a hypoxic environment. Cell viability of Gli36ΔEGFR and X12v2 cells treated with effective SapC-DOPS doses in hypoxia (1% O2) or normoxia (20% O2) revealed increased sensitivity to SapC-DOPS–induced cytotoxicity in hypoxia (Figure 4a). Interestingly, exposure to hypoxia has been shown to increase PtdSer exposure on the outer membrane of colon carcinoma cells.24 In an effort to understand increased sensitivity of hypoxic GBM cells to SapC-DOPS, we investigated the impact of hypoxia on cell surface PtdSer exposure. Fluorescence-activated cell sorting analysis of GBM cells following 72 hours incubation in hypoxia compared with normoxia revealed a significant increase in exposed PtdSer on the outer membrane (Figure 4b).
Figure 4.

Glioblastoma multiforme (GBM) cells show higher sensitivity to SapC-DOPS and increased levels of PtdSer in hypoxia. (a) X12v2 or Gli36ΔEGFR cells were treated with the indicated doses of SapC-DOPS in normoxia (20% O2) or hypoxia (1% O2) for 72 hours and cell viability was measured by MTT. All values were normalized to untreated control cells in normoxia or hypoxia. Data shown are mean ± SD of percentage viable cells after treatment with SapC-DOPS in normoxia and hypoxia, **P < 0.01. (b) PtdSer exposure was measured by flow cytometry using Annexin V-Pacific Blue following 72 hours of normoxia or hypoxia. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PtdSer, phosphatidylserine; SapC-DOPS, Saposin C-dioleoylphosphatidylserine.
Systemic treatment with SapC-DOPS yields antitumor efficacy in vivo
The ability of SapC-DOPS to effectively cross the BBTB to target the tumor-associated vasculature and both normoxic and hypoxic GBM cells led us to assess its in vivo antitumor efficacy in mice bearing intracranial tumors. Mice with intracranial U87ΔEGFR-Luc tumors were treated with SapC-DOPS or control DOPS intravenously, and tumor progression was followed by in vivo imaging of tumor cell expressed luminescence as indicated (Figure 5a). Tumor burden could be visualized by luminescence imaging as early as 3 days postimplantation, and while control DOPS-treated animals showed progressive tumor growth until day 17, no detectable tumor could be found in mice treated with SapC-DOPS (Figure 5a). To elucidate the antitumor efficacy of SapC-DOPS, we compared survival of mice with intracranial tumors in two different tumor models: U87ΔEGFR-Luc and X12v2 with low and high PtdSer exposure respectively (Figure 5b,c). While SapC-DOPS treatment resulted in a significant increase in survival in both models, it is interesting to note that only 25% of the mice implanted with PtdSer low U87ΔEGFR-Luc tumors were long-time survivors, whereas 75% of the mice implanted with high PtdSer-expressing X12v2 tumors were long-term survivors. In conclusion, systemic treatment with SapC-DOPS is able to target GBM tumors with both low and high levels of exposed PtdSer in vivo yielding a significant increase in survival in both models.
Figure 5.

Intravenous administration of SapC-DOPS yields significant antitumor efficacy in vivo. (a) U87ΔEGFR-Luc cells were implanted intracranially and treated with SapC-DOPS. Images were taken for tumor luminescence at 3, 12, and 17 days post-tumor implantation. (b,c) Kaplan–Meier survival curve for mice with intracranial (b) U87ΔEGFR-Luc and (c) X12v2 glioma treated with intravenous injections of DOPS or SapC-DOPS. SapC-DOPS, Saposin C-dioleoylphosphatidylserine.
Discussion
A significant complication for effective chemotherapy in GBM patients is the inability of drugs and small molecules to cross the BBTB. Formation and regulation of the BBB is thought to be dependent on the interactions of endothelial cell tight junctions, astrocytes, and pericytes.25,26 While the BBB can be partially disrupted within the tumor, the BBTB is still thought to have significant impact on the systemic delivery of chemotherapeutics.1 Here, we investigated the ability of SapC-DOPS nanovesicles to cross the BBTB and target intracranial tumors. Utilizing two distinct spontaneous mouse glioma models, we were able to demonstrate the ability of SapC-DOPS nanovesicles to cross the BBTB and specifically target GBM cells in vivo. Although SapC-DOPS has been previously shown to be able to deliver fluorescent probes and magnetic resonance contrast agents to subcutaneous tumors in vivo, this is the first report showing its ability to cross the BBTB to target orthotopic and spontaneous brain tumors in vivo.10 The ability of SapC-DOPS to efficiently target GBM in vivo coupled with the ability to load it with small molecule inhibitors or other chemotherapeutics has broad implications on future treatment paradigms.
Our results demonstrate that PtdSer exposure on the surface of tumor cells plays a significant role in the brain tumor-selective targeting of SapC-DOPS. Recently, several groups have used liposomes containing PtdSer-binding ligands such as Annexin A5 to target PtdSer-rich membranes27 and radiolabeled Annexin 5 has even been used in the clinic as a means to assess therapeutic efficacy of chemotherapy and radiation.28,29 While great strides have been made using nanocarriers for drug delivery and diagnostic imaging,6,30 what distinguishes SapC-DOPS is the ability to not only cross the BBTB to target GBM, but to destroy these cells as well. Consistent with the PtdSer targeting, we found a correlation between GBM PtdSer exposure levels and sensitivity to SapC-DOPS in vitro and in vivo.
PtdSer has been shown to be elevated on the surface of tumor-associated endothelium.18,19,20 Several studies have shown vascular targeting anti-PtdSer antibodies to possess antitumor efficacy in several tumor types including GBM.31,32 Bavituximab, a chimeric monoclonal PtdSer-targeting antibody, is currently under investigation in clinical trials for numerous solid tumor malignancies (http://www.clinicaltrials.gov). In light of the specificity of SapC-DOPS for PtdSer, we chose to investigate the effects on angiogenesis. Here, we have shown that SapC-DOPS can specifically target brain tumor vasculature and possesses significant antiangiogenic effects in vitro and in vivo, demonstrating a dualistic mechanism for the antitumor effects of SapC-DOPS against GBM. In addition to the tumor cells and the tumor-associated vasculature, PtdSer has also been shown to be highly expressed on the surface of microvesicles.33 Tumor microvesicles have been shown to be involved in extracellular communication in tumorigenesis in numerous cancers including GBM.34 It is interesting to speculate as to whether SapC-DOPS can bind to these microvesicles and future studies will be necessary to examine these possible interactions and the effects on cancer progression and response to therapy.
Interestingly, while both chemotherapy and radiation treatment show reduced efficacy in hypoxic conditions, we found increased sensitivity of GBM cells to SapC-DOPS in hypoxia compared with normoxia. Thus, its antiangiogenic effects combined with tumor sensitization in hypoxia orchestrate a very significant antiglioma response in vivo. The increased sensitization of hypoxic glioma cells is likely due to the increased PtdSer exposure in low oxygen conditions. Therefore, it is interesting to speculate that SapC-DOPS will likely synergize when combined with chemotherapy and irradiation both of which are known to increase PtdSer exposure on cancer cells.31,35
Taken together, the data presented here highlight the ability of systemic treatment of SapC-DOPS to efficiently cross the BBTB, to target and destroy intracranial GBM in mice. The efficient ability of SapC-DOPS nanovesicles to cross the BBTB could be further exploited as a vehicle to deliver a host of therapeutics such as small molecular inhibitors, cytotoxic drugs, oncolytic viruses, etc. These findings support the further development of SapC-DOPS as a novel therapeutic for GBM.
Materials and Methods
Cell culture and reagents. Human GBM cell lines were obtained from ATCC (Manassas, VA), Gli36 cells subcloned to express a truncated, constitutively active, mutant epidermal growth factor receptor (Gli36ΔEGFR), U87ΔEGFR, and U87ΔEGFR-Luc, were obtained from Dr Webster Cavenee (Ludwig Cancer Institute, San Diego, CA), X12 primary tumor-derived cells were obtained from Dr Sarkaria, and were subcloned to express green fluorescent protein to generate X12v2 (Mayo Clinic, Rochester, MN). All cells are routinely checked for mycoplasma contamination. Cells were cultured with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units of penicillin/ml, and 10 mg of streptomycin/ml. HDMECs and HUVECs were purchased from ScienCell Research Laboratories (San Diego, CA) and cultured with endothelial cell medium supplemented with 2% fetal bovine serum, 100 units of penicillin/ml, and 10 mg of streptomycin/ml. All cells were cultured at 37 °C in an atmosphere containing 5% CO2 and 20% O2 for normoxia and 1% O2 for hypoxia. For cytotoxicity assays, cells were plated at 10,000 cells/well in 96-well dishes overnight and treated with SapC-DOPS for 72 hours. Cell viability was determined as described previously9 using a standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. PtdSer-specific binding proteins, Lact-C2 and β2GP1 were obtained from Gary E. Gilbert (Department of Medicine, Department of Veterans Affairs, VA Boston Healthcare System, Brigham and Women's Hospital, Harvard Medical School, Boston, MA) and Haematologic Technologies (Essex Junction, VT). Exposed PtdSer on cells was measured using binding of Annexin V-Pacific Blue purchased from Invitrogen (Carlsbad, CA) and was used according to the manufacturer's instructions.
Preparation of SapC and SapC-DOPS nanovesicles. Recombinant SapC with and without His-tag were expressed using the pET system in Escherichia coli cells as previously described with minor modifications.9 Expressed SapC was purified by ethanol precipitation and ion-exchange high-performance liquid chromatography. After lyophilization using a tertiary butyl alcohol/water co-solvent system, protein powder was used and its concentration was determined by its weight. All phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL). SapC-DOPS nanovesicles were prepared by two methods. (i) Bath sonication as previous described:9,10 after solvent removal under nitrogen gas, DOPS (Avanti Polar Lipids) was mixed with weighed dry amounts of SapC in an acid buffer (pH 5) and quickly diluted in phosphate-buffered saline or media. The mixture was then sonicated to assemble into nanovesicles. The sonicated samples were stored at 4 °C. (ii) Lypholization with a co-solvent solution: dry DOPS was suspended in 80% tert-butanol. SapC and sucrose (10 mg/ml) were dissolved in water. The mixture of DOPS and SapC + sucrose (1:0.6, vol:vol) was lyophilized to a stable powder cake in a freeze dryer (VirTis Unitop 1000L linked o a Freezemobile 25XL; The VIRTIS, Gardiner, NY). The cake was resuspended in phosphate-buffered saline or media to form SapC-DOPS nanovesicles. Once formed, the vesicles were monitored by a N4 plus subsize particle size analyzer. SapC-DOPS for in vitro experiments was formulated at a 1:3 molar ratio of SapC:DOPS.
In vivo xenografts. For intracranial tumor studies, anesthetized mice were fixed in a stereotactic apparatus, and U87ΔEGFR-Luc (1 × 105 cells) or X12v2 (green fluorescent protein) (2 × 105 cells) were implanted at 2 mm lateral to bregma, at a depth of 3 mm. For survival studies, mice were treated with SapC-DOPS (SapC 12 mg/kg, DOPS 4.6 mg/kg) or DOPS (4.6 mg/kg) intravenously on the following days post-tumor implantation: 4–11, 13, 15, 17, 19, 22, 25, 28, and 31 days for U87ΔEGFR-Luc tumors and 5–9, 11, 13, and 15 days for X12v2 tumors. For studies involving CD31 and cleaved caspase-3 staining, mice were treated on days 6–10 following implantation and brains were harvested 24 hours later. For subcutaneous tumor studies, 1.5 × 106 Gli36ΔEGFR or 1 × 106 U87ΔEGFR-Luc cells were implanted in the rear flanks of athymic nude mice. When Gli36ΔEGFR tumors reached 100–200 mm3, they were treated with five consecutive daily doses of SapC-DOPS (SapC 13 mg/kg, DOPS 8 mg/kg) or DOPS (8 mg/kg) by tail vein injection. When U87ΔEGFR-Luc tumors reached 1,500 mm3, mice were treated with a single dose of SapC-DOPS-CVM (SapC 3.2 mg/kg, DOPS 1.8 mg/kg, CVM 1.6 μmol/l). Tumors were excised 24 hours following the final treatment and harvested for immunohistochemistry analysis.
Immunohistochemistry/immunofluorescence analysis. Subcutaneous tumors and mouse brains were fixed in 4% buffered paraformaldehyde followed by 30% sucrose at 4 °C, embedded in optimal-cutting temperature, and frozen at −80 °C. Subcutaneous tumors were divided into 2–4 pieces, and 10-μm sections from each piece were stained with anti-CD31 (BD Pharmingen, San Jose, CA). The three most vascularized areas within the tumor were chosen at low magnification, and vessels were counted in a representative high magnification field in each view with n = 4 tumors/group. Mouse brains bearing intracranial tumors were sectioned at 5 μm and stained using the following antibodies: anti-CD31 (BD Pharmingen), anti-His (GenScript, Piscataway, NJ), anti-cleaved caspase 3 (Cell Signaling, Danvers, MA), Alexa Fluor 594 and 488 (Invitrogen).
Genetically engineered mouse models. All mouse experiments and care were approved by the Institutional Animal Care and Use Committee of The Ohio State University. We bred Mut3 (GFAP-cre; Nf1loxP/+; Trp53−/+) male mice with Trp53loxP/loxP; PtenloxP/loxP females to generate Mut6 mice (GFAP-cre; Nf1loxP/+; Trp53−/loxP; PtenloxP/+).12 We maintained Mut3 mice in B6CBAF1/J strain by breeding male Mut3 mice with female B6CBAF1/J females (The Jackson Laboratory, Bar Harbor, ME). We genotyped the mice between P9 and P12 as described12 and confirmed the genotypes after harvesting their tissues.
Quadruple conditional knockout mice were derived from mice previously described.36 Briefly, GFAP-CreER; PtenloxP/loxP; Trp53loxP/loxP; Rb1loxP/loxP mice were bred with Rb1−/− mice (p107-null)37 to generate the four gene-targeted strain. Tumors were induced by intraperitoneal tamoxifen injections and mice were monitored as previously described.36
In vivo bioluminescence and fluorescence imaging. CVM (PTI Research, Exton, PA) in ethanol was mixed with phospholipid solvent for bath sonication preparation by the procedure as previously described.9,10 CVM-labeled SapC-DOPS nanovesicles were separated from free CVM dye using a Sephadex G25 column (PD-10; Amersham Pharmacia Biotech, Piscataway, NJ). SapC-DOPS-CVM (SapC 3.2 mg/kg, DOPS .656 mg/kg, CVM 320 μmol/l) or CVM-labeled DOPS (DOPS .656 mg/kg, CVM 320 μmol/l), was administrated by tail vein injection into orthotropic and transgenic brain tumor-bearing mice. Real-time images were taken using an IVIS 200 Series (Clipper, Alameda, CA) or a Kodak FX (Carestream Health, Toronto, Ontario, Canada) imaging system. For in vitro targeting experiments, cells were treated with SapC-DOPS-CVM (SapC 33 μmol/l, DOPS 100.5 μmol/l, CVM 1.6 μmol/l) or DOPS-CVM (DOPS 100.5 μmol/l, CVM 1.6 μmol/l) and evaluated by fluorescence-activated cell sorting analysis. For PtdSer-blocking experiments, cells were incubated in 0.4 mg/ml of Lact-C2 or β2GP1 for 30 minutes at 37 °C before injecting subcutaneously (100,000 cells) above the skull of nude mice. One hour later, mice were treated with CVM-labeled SapC-DOPS (SapC 3.2 mg/kg, DOPS .656 mg/kg, CVM 320 μmol/l) and imaged 1 hour later as described above (n = 3).
In vitro angiogenesis assays. For the endothelial cell migration assays, HDMECs or HUVECS were cultured in 0% serum-containing media with SapC-DOPS (SapC 50 μmol/l, DOPS 152 μmol/l), DOPS (152 μmol/l), or media alone for 30 minutes at 37 °C. Cells (1 × 106) were plated in the upper chamber of transwell chambers (ISC BioExpress, Kaysville, UT) with an 8 μm pore size, and complete endothelial cell media was used as a chemoattractant in the bottom chamber. The cells were allowed to migrate for 6 hours, and were then fixed and stained with 0.5% crystal violet. The migrated cells were quantified as number of cells/view field (n = 3 view fields/well and four wells/group). For the tube formation assay, 40,000 HDMECs or HUVECs were cultured as above and plated on 250 μl of polymerized Matrigel (BD Biosciences, Bedford, MA) diluted to 75% in complete endothelial cell medium, and incubated at 37 °C. Pictures of formed tubes were taken at 6 and 15 hours and viable tubes (>200 μm) were quantified by counting one ×10 microscopic view/well, and the data presented as means of four wells. Ex vivo aortic ring assay was completed by removing the full-length aorta from a Fisher-344 rat and sectioned into 1-mm long rings and polymerized in Matrigel. Rings were treated with SapC-DOPS (SapC 50 μmol/l, DOPS 152 μmol/l), DOPS (152 μmol/l) or media alone and pictures were taken 48 hours later, n = 4.
Statistical analysis. Student's t-test was used to analyze in vitro experiments. A P value <0.05 was considered statistically significant in Student's t-test. Kaplan–Meier curves were compared using the log-rank test using GraphPad Prism S/W (GraphPad Software, La Jolla, CA). All error bars represent SD.
SUPPLEMENTARY MATERIAL Figure S1. SapC-DOPS targeting in vivo. Figure S2. Antiangiogenic effect of SapC-DOPS.
Acknowledgments
We thank Ray Takigiku, Charlie Cruze, and Ellen Monson for comments on the manuscript. Patents are pending for the intellectual property disclosed in this manuscript. We thank Luis Parada for Mut3 mice, Hong Wu for loxP-Pten mice, and Eva Lee for loxP-Trp53 mice. We thank Suzanne J. Baker for the use of quadruple cKO mice in this study. L.M.L.C. is a St. Baldrick's Foundation Scholar and is supported by a Distinguished Scientist Award from the Sontag Foundation. We thank Amanda N. White for flow cytometric experiment. This work was supported in part by 1R01CA158372 and New Drug State Key Project grant number 009ZX09102-205 (to X.Qi), 1R43 CA136017 and 2R44 CA136017 grants from National Institutes of Health-National Cancer Institute (to X.Qi and Kevin Xu). X.Qi is listed as an inventor on the patent for SapC-DOPS technology that is the subject of this research. Consistent with current Cincinnati Children's Hospital Medical Center policies, the development and commercialization of this technology has been licensed to Bexion Pharmaceuticals, LLC, in which Dr Qi, holds a minor (<5%) equity interest. The other authors declared no conflict of interest.
Supplementary Material
References
- de Vries NA, Beijnen JH, Boogerd W, van Tellingen O. Blood-brain barrier and chemotherapeutic treatment of brain tumors. Expert Rev Neurother. 2006;6:1199–1209. doi: 10.1586/14737175.6.8.1199. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12:495–508. doi: 10.1038/nrn3060. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- Silva GA. Neuroscience nanotechnology: progress, opportunities and challenges. Nat Rev Neurosci. 2006;7:65–74. doi: 10.1038/nrn1827. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584:1700–1712. doi: 10.1016/j.febslet.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Qi X, Chu Z, Mahller YY, Stringer KF, Witte DP, Cripe TP. Cancer-selective targeting and cytotoxicity by liposomal-coupled lysosomal saposin C protein. Clin Cancer Res. 2009;15:5840–5851. doi: 10.1158/1078-0432.CCR-08-3285. [DOI] [PubMed] [Google Scholar]
- Kaimal V, Chu Z, Mahller YY, Papahadjopoulos-Sternberg B, Cripe TP, Holland SK, et al. Saposin C coupled lipid nanovesicles enable cancer-selective optical and magnetic resonance imaging. Mol Imaging Biol. 2011;13:886–897. doi: 10.1007/s11307-010-0417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Grabowski GA. Differential membrane interactions of saposins A and C: implications for the functional specificity. J Biol Chem. 2001;276:27010–27017. doi: 10.1074/jbc.M101075200. [DOI] [PubMed] [Google Scholar]
- Vaccaro AM, Tatti M, Ciaffoni F, Salvioli R, Serafino A, Barca A. Saposin C induces pH-dependent destabilization and fusion of phosphatidylserine-containing vesicles. FEBS Lett. 1994;349:181–186. doi: 10.1016/0014-5793(94)00659-8. [DOI] [PubMed] [Google Scholar]
- Fernandes RS, Kirszberg C, Rumjanek VM, Monteiro RQ. On the molecular mechanisms for the highly procoagulant pattern of C6 glioma cells. J Thromb Haemost. 2006;4:1546–1552. doi: 10.1111/j.1538-7836.2006.01985.x. [DOI] [PubMed] [Google Scholar]
- Riedl S, Rinner B, Asslaber M, Schaider H, Walzer S, Novak A, et al. In search of a novel target - phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim Biophys Acta. 2011;1808:2638–2645. doi: 10.1016/j.bbamem.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51:3062–3066. [PubMed] [Google Scholar]
- Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62:6132–6140. [PubMed] [Google Scholar]
- Ran S, Thorpe PE. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiat Oncol Biol Phys. 2002;54:1479–1484. doi: 10.1016/s0360-3016(02)03928-7. [DOI] [PubMed] [Google Scholar]
- Thorpe PE. Targeting anionic phospholipids on tumor blood vessels and tumor cells. Thromb Res. 2010;125 suppl. 2:S134–S137. doi: 10.1016/S0049-3848(10)70031-1. [DOI] [PubMed] [Google Scholar]
- Uzzan B, Nicolas P, Cucherat M, Perret GY. Microvessel density as a prognostic factor in women with breast cancer: a systematic review of the literature and meta-analysis. Cancer Res. 2004;64:2941–2955. doi: 10.1158/0008-5472.can-03-1957. [DOI] [PubMed] [Google Scholar]
- Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. 2009;5:610–620. doi: 10.1038/nrneurol.2009.159. [DOI] [PubMed] [Google Scholar]
- Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- Schilling D, Gehrmann M, Steinem C, De Maio A, Pockley AG, Abend M, et al. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. 2009;23:2467–2477. doi: 10.1096/fj.08-125229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Garnier B, Bouter A, Gounou C, Petry KG, Brisson AR. Annexin A5-functionalized liposomes for targeting phosphatidylserine-exposing membranes. Bioconjug Chem. 2009;20:2114–2122. doi: 10.1021/bc9002579. [DOI] [PubMed] [Google Scholar]
- Rottey S, Loose D, Vakaet L, Lahorte C, Vermeersch H, Van Belle S, et al. 99mTc-HYNIC Annexin-V imaging of tumors and its relationship to response to radiotherapy and/or chemotherapy. Q J Nucl Med Mol Imaging. 2007;51:182–188. [PubMed] [Google Scholar]
- van de Wiele C, Lahorte C, Vermeersch H, Loose D, Mervillie K, Steinmetz ND, et al. Quantitative tumor apoptosis imaging using technetium-99m-HYNIC annexin V single photon emission computed tomography. J Clin Oncol. 2003;21:3483–3487. doi: 10.1200/JCO.2003.12.096. [DOI] [PubMed] [Google Scholar]
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15:6871–6880. doi: 10.1158/1078-0432.CCR-09-1499. [DOI] [PubMed] [Google Scholar]
- Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res. 2005;11:1551–1562. doi: 10.1158/1078-0432.CCR-04-1645. [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123 Pt 10:1603–1611. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer Res. 2005;65:4408–4416. doi: 10.1158/0008-5472.CAN-05-0031. [DOI] [PubMed] [Google Scholar]
- Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19:305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, et al. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.