

Abstract
Both genetic inactivation and pharmacological inhibition of the cholesteryl ester synthetic enzyme acyl-CoA:cholesterol acyltransferase 1 (ACAT1) have shown benefit in mouse models of Alzheimer's disease (AD). In this study, we aimed to test the potential therapeutic applications of adeno-associated virus (AAV)-mediated Acat1 gene knockdown in AD mice. We constructed recombinant AAVs expressing artificial microRNA (miRNA) sequences, which targeted Acat1 for knockdown. We demonstrated that our AAVs could infect cultured mouse neurons and glia and effectively knockdown ACAT activity in vitro. We next delivered the AAVs to mouse brains neurosurgically, and demonstrated that Acat1-targeting AAVs could express viral proteins and effectively diminish ACAT activity in vivo, without inducing appreciable inflammation. We delivered the AAVs to the brains of 10-month-old AD mice and analyzed the effects on the AD phenotype at 12 months of age. Acat1-targeting AAV delivered to the brains of AD mice decreased the levels of brain amyloid-β and full-length human amyloid precursor protein (hAPP), to levels similar to complete genetic ablation of Acat1. This study provides support for the potential therapeutic use of Acat1 knockdown gene therapy in AD.
Introduction
Alzheimer's disease (AD) is a progressive neurological disease that causes deterioration of cognitive processes in afflicted individuals. There is currently no cure for AD and only symptomatic treatments are available.1,2 One biochemical characteristic of AD is the accumulation of plaques in the brain composed of amyloid-β (Aβ), an insoluble cleavage product of the amyloid precursor protein (APP).3,4
Acyl-CoA:cholesterol acyltransferase (ACAT) enzymes (also known as sterol-O-acyltransferases, abbreviated as SOAT; referred to herein by the name ACAT) catalyze the formation of cholesteryl esters and perform diverse functions in the body, including mediating storage of cholesterol and cellular cholesterol homeostasis.5 There are two known isoforms of ACAT in mammals: ACAT1 and ACAT2.
Multiple previous studies have linked the pathogenesis of AD with lipid metabolism in the brain.6,7,8 Several have indicated that diminishing the activity or protein levels of ACAT1 benefited mouse models of AD (see ref. 9 for review). One group used pharmacological inhibition of ACAT on an AD mice and found that it diminished the amyloid plaque burden in their brains and improved cognitive function.10,11 In a previous study in our own laboratory, Bryleva and colleagues genetically ablated Acat1 in AD mice and found that it diminished the levels of Aβ42, decreased the amyloid plaque burden and full-length human APP (hAPP), and also improved the cognitive function of the mice.12 Here, we asked whether Acat1 inhibition could have therapeutic potential for AD, and aimed to test whether a specific genetic knockdown of Acat1 in the mouse brain, administered late in life after the onset of the disease phenotype, could benefit AD.
We used the Triple Transgenic (3xTg-AD) mouse model, originally created by Oddo and colleagues, which manifests amyloid plaques and neurofibrillary tangles, creating a phenocopy of human AD.13 The 3xTg-AD mouse manifests cognitive deficits by 9 months of age12 and develops significant Aβ42 accumulation beginning at approximately 10 months of age.13,14 The 3xTg-AD mice with or without Acat112 were used in the current study, and are referred to herein as AD or AD/Acat1−/−, respectively.
We chose to administer Acat1 gene knockdown using a recombinant adeno-associated virus (AAV) mediated gene therapy approach. AAVs are popular gene therapy vectors15 because they produce long-term (at least 1 year),16 stable, episomal expression of the viral transgene, provoke only a minimal inflammatory response, have minimal oncogenic potential, can infect and transduce postmitotic cells (for instance, neurons),17,18 and have been used successfully in many studies to transduce central nervous system tissues19,20,21 and other tissues.22 Recombinant AAVs with hybrid serotype 1/2 efficiently target and transduce neurons.23 AAV vectors have been used successfully in a wide variety of gene therapy studies in mouse models of neurodegenerative diseases, including AD,24,25,26,27 and have demonstrated safety in human trials. One phase II human clinical trial that utilizes an AAV gene therapy approach to treat AD is currently ongoing.28 In the current study, we administered AAVs expressing artificial microRNAs (miRNA) that targeted Acat1 for knockdown to AD mice to test the effect on the AD phenotype. We utilized miRNAs because they were found to be less inflammatory in mouse brain tissue than were similar short hairpin RNAs.29
Results
Engineering of AAVs for Acat1 knockdown
To test the potential therapeutic use of ACAT1-targeting AAVs, we designed artificial miRNAs targeting four different regions within the Acat1 (Soat1) gene (NCBI reference sequence: NM_009230.3). We also selected a scrambled control sequence that was not shown to match the sequence of any mouse gene (miRNA sequences are available in Supplementary Table S1).
Each miRNA was cloned into the pcDNA 6.2-GW/miR plasmid, a mammalian expression vector, where the cloned miRNA insert is expressed co-cistronically with emerald green fluorescent protein (GFP), enabling selection of GFP-positive cells. Mouse 3T3 fibroblast cells were transfected with the miRNA expression constructs. Five days after transfection, GFP-positive cells were selected by cell sorting, lysed, and the lysates subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis. Western blot results showed that miRNAs #54 and #55 most effectively diminished levels of ACAT1 protein in the cells relative to the loading control glyceraldehyde 3-phosphate dehydrogenase (Figure 1a). Subsequently, miRNAs #54, #55, and negative control were used for AAV production.
Figure 1.

Engineering of AAVs for Acat1 knockdown gene therapy. (a) Testing of miR sequences in mouse 3T3 fibroblasts. DNA sequences encoding artificial miRNAs targeting Acat1 were inserted into the mammalian expression plasmid pcDNA 6.2-GW/miR and used to transfect mouse 3T3 fibroblasts grown in culture. The cells were grown for 5 days after transfection, then lysed and subjected to SDS-PAGE and western blotting for ACAT1 and GAPDH as a loading control. Cells in the “mock transfected” lane were not transfected with any plasmid. (b) AAV-miR-GFP vector map. The viral construct consists of an AAV2 backbone with AAV2 inverted terminal repeats, co-cistronically expressed pre-miRNA and enhanced green fluorescent protein under control of the hybrid cytomegalovirus early enhancer element and chicken β-actin promoter, the woodchuck hepatitis virus post-transcriptional regulatory element, and bovine growth hormone polyadenylation sequence. (c) Cloning of miRNA inserts into viral backbone and verification of inverted terminal repeats integrity. Quantification of DNA, verification of the correct size of AAV vector + insert by BglII digestion, and verification of integrity of inverted terminal repeats by SmaI digestion. (d) AAV production and purification. SDS-PAGE of pure AAV stocks produced for this study. CAG, chicken β-actin; eGFP, enhanced green fluorescent protein; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ITR, inverted terminal repeat; pA, polyadenylation sequence; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element.
Next, DNA fragments containing emerald GFP plus the miRNA (negative control miRNA, or Acat1-targeting miRNA #54 or #55) were subcloned into the pAM/CAG-pL-WPRE-bGHpA AAV vector, creating an AAV2 backbone with GFP and miRNA expression driven by the strong universal CAG promoter (Figure 1b). Three DNA constructs—AAV-Acat1(54), AAV-Acat1(55), and AAV-NC—were created (Figure 1c) and used for production of hybrid serotype 1/2 AAVs (Figure 1d).
In Vitro testing of AAVs
We next sought to test the AAVs for their ability to infect mouse neurons and glia, and for their effect on ACAT activity in these cells in vitro. Since GFP and the miRNA are expressed co-cistronically, GFP expression in a cell after AAV infection indicates that miRNAs are also being expressed. The results (Figure 2a) showed thatGFP expression could be detected in the majority of AAV-treated cells by fluorescence microscopy. All AAV stocks—AAV-NC, AAV-Acat1(54), and AAV-Acat1(55)—were tested and found to be equivalent in their ability to infect cultured cells (a representative image from one of the AAV stocks is shown in Figure 2a).
Figure 2.
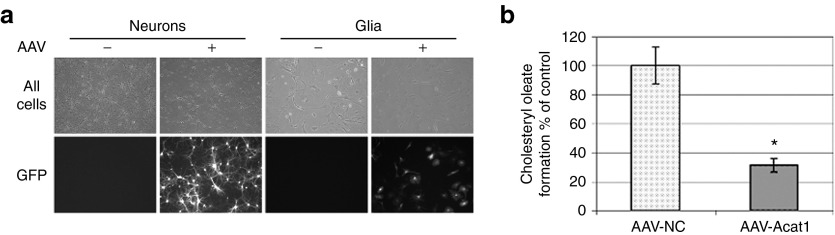
Expressions of AAVs in cell culture and in vivo. (a) Primary cultures of mouse neurons or mixed glia were transfected with AAV on the day of plating. Live cells were observed for GFP expression 14 days later. All AAV stocks were tested (AAV-NC, AAV-Acat1) and a representative picture is shown. (b) ACAT activity assay in primary cultured mouse neurons treated with AAV. Error bars represent mean ± SEM. Average of two experiments with samples run in duplicate or triplicate for each point. *P < 0.01. AAV, adeno-associated virus; GFP, green fluorescent protein.
Oleate pulse is an assay that can be used to measure ACAT1 activity in intact cells.30 Briefly, cells are treated with 3H-oleate for a period of time. The 3H-oleate containing metabolites, including cholesteryl esters synthesized by ACAT, are subsequently analyzed by thin layer chromatography and quantified relative to the amount of protein present in the sample. Oleate pulse assay was performed on cultured AD mouse neurons treated with AAV, to measure ACAT1 activity. Neurons were isolated, plated in 6-well dishes, and treated with 0.5 μl/well of AAV-NC or a mixture of AAV-Acat1(54) and AAV-Acat1(55); the mixture is referred to as AAV-Acat1. As an additional control, we isolated neurons from AD/Acat1−/− mice, which lack any functional Acat1, and used the values as a baseline. Compared to AAV-NC treatment, AAV-Acat1 treatment could suppress ACAT1 activity in cultured AD mouse neurons by ~70% (Figure 2b).
In Vivo testing of AAVs and AAV delivery
We next moved to in vivo testing of the AAVs we had engineered, and delivered them neurosurgically to mouse brains. Each mouse received injections of ~1.0 µl of virus, bilaterally in the hippocampi. The mice were housed postoperatively for one month and then used for experiments. Upon isolation and dissection of the mouse brains 1 month after brain injection, macroscopic GFP expression was visible in the hippocampus, cortex, and along the midline of the brain (Supplementary Figure S1). Western blot analysis of several brain regions showed widespread and robust GFP expression, with the most robust GFP immunoreactivity evident in the hippocampi, followed by less robust but still apparent GFP signal in the cortex and basal ganglia, and a smaller amount of GFP expression in the cerebellum of some individuals. The strength and distribution of GFP expression were comparable between individual mice and AAV stocks (Figure 3a).
Figure 3.

In vivo testing of adeno-associated virus (AAV). (a) Western blot of brain regions from mice treated with AAV. Cb, cerebellum; BG basal ganglia; Hp hippocampus; Cx cortex. (b) ACAT activity assay in hemibrains of wild-type, Acat1−/−, and Acat2−/− mice treated with sham surgery or PBS brain injection. Mice underwent surgery at 4 months of age and were subjected to sham surgery, or injected bilaterally in the dorsal hippocampi with 1.0 µl of sterile PBS. One month later, the animals were euthanized and their brain homogenates used for ACAT assay. N = 3 mice per group. Error bars represent SEM. (c) Real-time PCR for inflammatory transcripts on mouse brains after sham surgery, PBS or AAV brain injection. At 6 months of age, mice underwent sham surgery or were injected bilaterally in the dorsal hippocampi with 1.0 µl of sterile PBS or AAV solution. One month later, the animals were euthanized and their brain RNA was used for real time PCR. Samples were run in duplicate. N = 3 mice per group. Error bars represent mean ± SEM. GFP, green fluorescent protein; PBS, phosphate-buffered saline.
We next undertook to address concerns about brain inflammation. ACAT2 activity can be induced in monocyte-derived macrophages under certain pathophysiological conditions.31 To address concerns that the trauma of the surgical procedure could induce ACAT2, we tested brain ACAT activity in vitro in mixed micelles,32 in brain homogenates from 4-month-old wild-type, Acat1−/−,33 and Acat2−/−34 mice, which had either received sham surgery or brain injection with sterile phosphate-buffered saline (PBS) (Figure 3b). We included Acat1−/− and Acat2−/− controls because this ACAT assay does not differentiate between ACAT1 and ACAT2 activity. The results of this ACAT assay indicated that Acat1−/− mice had virtually no ACAT activity, while wild-type and Acat2−/− mice had very similar levels of ACAT activity. There were no significant difference in the levels of ACAT activity between sham surgery mice and PBS brain injected mice in any of the genotypes tested. This result was consistent with previous data showing that ACAT1 is the primary isoform responsible for ACAT activity in adult mouse brains.12
To monitor brain inflammation, we performed quantitative PCR for various inflammatory mRNA transcripts in 6-month-old mice that had received sham surgery, or brain injections of PBS or AAV solution. We did not observe any significant change in any of the transcripts measured between any of the treatment groups (Figure 3c). In this experiment, we also attempted to perform quantitative PCR for Acat2 transcript, and found that the signal was close to the lower limit of detection in brain tissue in all of the samples (Acat2, data not shown).
The measurement of ACAT1 protein levels in mouse brains by western blot is complicated by the presence of a cross-reacting band of the same molecular weight as ACAT1, which appears on western blots of Acat1−/− mouse brain tissue, but not other mouse body tissues (unpublished observations).12 For this reason, we opted to assay mouse brain ACAT activity in vitro rather than perform western blot for ACAT1 to test the effectiveness of our AAV at diminishing ACAT1 activity in mouse brains. We used the same ACAT activity assay as in Figure 3b, which measures ACAT assay in mixed micelles.32 We used homogenates from the hemibrains of 6-month-old sex-matched male and female mice that had received brain injections in the hippocampi of 1 µl of PBS or AAV solution, 1 month after injection. Mice received sham surgery, PBS injection, or injection with AAV-NC or AAV-Acat1. Each treatment was tested in AD and AD/Acat1−/− mice (Figure 4). In AD/Acat1−/− mice, consistent with previous results (Figure 3b),12 virtually no ACAT activity was present under any of the conditions tested. In AD mice, sham, PBS, and AAV-NC groups had similar levels of brain ACAT activity, whereas in the AAV-Acat1 treated group, there was a statistically significant, ~45% decrease in whole brain ACAT activity.
Figure 4.

ACAT activity assay in mouse hemibrains. At 6 months of age, AD or AD/Acat1−/− mice underwent sham surgery or were injected bilaterally in the dorsal hippocampi with 1.0 µl of sterile PBS or AAV solution. One month later, the animals were euthanized and their brain homogenates used for ACAT activity assay. The number of mice in each group is indicated as N. Error bars represent mean ± SEM. **P < 0.005. AAV, adeno-associated virus; ACAT, acyl-CoA:cholesterol acyltransferase; AD, Alzheimer's disease; PBS, phosphate-buffered saline.
Testing of AAV gene therapy effects on AD mice
To clarify the temporal profile of the AD-like phenotype in the particular line of 3xTg-AD mice used in our lab in this study, we compared several different age groups of 3xTg-AD mice with and without Acat1 (Supplementary Figure S2). We performed ELISA assay for total Aβ42 (Supplementary Figure S2a) and Aβ40 (Supplementary Figure S2b) in formic acid extracts of pooled samples from groups of 6-, 9-, or 12-month-old AD or AD/Acat1−/− mice. Our data showed that the mice had a surge in the levels of total Aβ42, and to a lesser extent Aβ40, between 9 and 12 months of age. The surge in total Aβ levels was less robust in AD/Acat1−/− mice than in AD mice.
We next undertook a larger study (schematized in Figure 5). To test the effect of AAV-Acat1 on the AD phenotype, AD mice received brain injections of either PBS (control), AAV-NC (control), or AAV-Acat1. As an additional set of controls, we administered all three of these treatments (PBS, AAV-NC, or AAV-Acat1) to AD/Acat1−/− mice. The brain injections were administered to 10-month-old, sex-matched groups of mice. At 12 months of age, the mice were euthanized and their brains were used for analysis of AD markers. We performed western blot analysis for GFP and confirmed that viral gene expression in these mice was similar between groups (data not shown).
Figure 5.

Experimental design. AD or AD/Acat1−/− mice underwent surgery at 10 months of age to deliver PBS, AAV-NC, or AAV-Acat1 solution to their brains. Each mouse received bilateral injections of 1.0 µl of solution in the dorsal hippocampus. The mice were housed for 2 months postoperatively and then euthanized for brain analysis. The number of mice in each treatment group is indicated as N. AAV, adeno-associated virus; ACAT, acyl-CoA:cholesterol acyltransferase; AD, Alzheimer's disease; PBS, phosphate-buffered saline.
In a previous study, HMG-CoA reductase (HMGR), the rate-limiting enzyme in cholesterol synthesis, was linked with the decrease in full-length hAPP and the decrease in amyloid-β (1-42) (Aβ42) with genetic ablation of Acat1 in AD mice.12 That study demonstrated that AD/Acat1−/− male mice had lower levels of HMGR than AD mice at 1–4 months of age. In our current study, we tested whether 12-month-old mice, which had received brain injections showed any changes in HMGR protein levels. We first attempted to reproduce the phenomenon of decreased HMGR in male AD/Acat1−/− mice of various ages. We found that in up to 4-month-old male mice which had not received any brain injections, there was a statistically significant, ~50% decrease in HMGR levels in AD/Acat1−/− mice compared to AD mice (Figure 6a). However, we observed that older male mice (>4 up to 22 months) that had not received any brain injections have significant variation in HMGR protein levels between individuals. Although a decrease in HMGR levels in AD/Acat1−/− compared to AD mice was still observable at the age of 22 months, it was less obvious and less robust (Figure 6b). Next, we measured HMGR protein levels in individual 12-month-old male AD or AD/Acat1−/− mice, which had received brain injections with PBS, AAV-NC, or AAV-Acat1. We found a high degree of interindividual variability in HMGR protein levels. When we quantified HMGR protein levels in the groups of mice that had multiple males, we found no significant differences in HMGR protein levels between treatment groups (Figure 6c).
Figure 6.
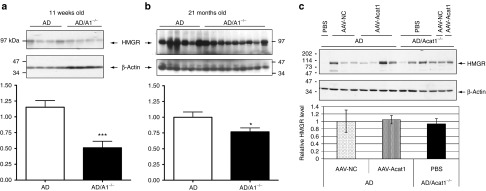
Analysis of HMGR in AD and AD/Acat1−/− mouse brains. (a) Eleven weeks old male AD or AD/Acat1−/− mouse brain homogenates were analyzed by tricine SDS-PAGE followed by western blot for HMGR (top) and β-actin (bottom; loading control). Western blot pictured is representative of N = 3 experiments. With quantification. ***P < 0.0002. (b) Twenty-one months old male AD or AD/Acat1−/− mouse brain homogenates were analyzed as in a. *P < 0.05. (c) Individual 12-month-old male AD or AD/Acat1−/− male mice treated with PBS, AAV-NC, or AAV-Acat1 at 10 months of age. Analyzed by SDS-PAGE followed by western blot for HMGR (top) and β-actin (bottom; loading control). With quantification of the groups in which there were multiple samples. All quantifications in this figure were performed with ImageJ software by measuring the intensity of the HMGR band, with background value subtracted, and measuring the intensity of the loading control band, with background value subtracted, and then calculating the ratio of the HMGR signal to loading control signal. The bars represent the mean relative HMGR intensity with error bars ± SEM. In a and b, the quantifications represent data from three separate western blots and a representative blot is shown. In c, the quantification represents data from one western blot. AAV, adeno-associated virus; ACAT, acyl-CoA:cholesterol acyltransferase; AD, Alzheimer's disease; HMGR, HMG-CoA reductase; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Next, homogenates from the hemibrains of 12-month-old AD or AD/Acat1−/− mice that had received brain injections with 1.0 µl of PBS, AAV-NC, or AAV-Acat1 in the hippocampi at 10 months of age were examined. Equal amounts of brain homogenate from individual mice were combined to create sample pools representative of each group. The sample pools were subjected to formic acid extraction, followed by ELISA assay for Aβ42 (Figure 7a). Analysis of variance analysis showed that there was a difference among the means and was followed by post-hoc t-tests to compare groups to each other. Consistent with previous results12 Aβ42 levels were significantly decreased in all groups of AD/Acat1−/− mice, compared to AD mice treated with PBS or AAV-NC. Aβ42 levels were significantly decreased in AD mice treated with AAV-Acat1, compared with AD mice treated with AAV-NC or PBS. There was no significant difference in Aβ42 levels in AD mice treated with AAV-Acat1 versus AD/Acat1−/− mice; also, there were no significant differences in Aβ42 levels between the AD/Acat1−/− mice treated with PBS, AAV-NC, or AAV-Acat1. Unexpectedly, there was a statistically significant decrease in Aβ42 in AD mice treated with AAV-NC, compared with AD mice treated with PBS. However, the effect of AAV-Acat1 on Aβ42 levels in AD mice was more robust than that of AAV-NC. Amyloid-β (1-40) (Aβ40) levels were also assayed by ELISA (Supplementary Figure S3). Aβ40 levels in these mice were near the lower limit of detection in our assay, and analysis of variance analysis did not detect a statistically significant difference in the group means for Aβ40.
Figure 7.

Analysis of hAPP and amyloid-β (1-42) in brain injected mice. (a) Amyloid β (1-42) ELISA assay on mouse brain homogenates after brain injection. Sample pools of mouse brain homogenates in sucrose buffer were subjected to formic acid extraction. The neutralized extracts were assayed for Aβ42 by ELISA. Formic acid extraction followed by ELISA was performed N = 3 separate times on different sample pools; each time the ELISA assay was run in duplicate or triplicate for each sample. Error bars represent SEM. *P < 0.05; **P < 0.005. (b) Representative Western blot of full-length hAPP in brain homogenate sample pools from 12-month-old AD or AD/Acat1−/− mice treated with either PBS, AAV-NC, or AAV-Acat1 brain injection at 10 months of age. Top panel: hAPP. Bottom: Ponceau S stain (loading control). (c) Quantification of N = 6 western blots of total (mature + immature) full length hAPP in brain homogenate sample pools from 12-month-old AD or AD/Acat1−/− mice treated with either PBS, AAV-NC, or AAV-Acat1 that received brain injection at 10 months of age. Total hAPP band intensity was normalized to Ponceau S staining in the lane; data is shown relative to AD mice treated with PBS. Error bars represent mean ± SEM. *P < 0.05. AAV, adeno-associated virus; ACAT, acyl-CoA:cholesterol acyltransferase; AD, Alzheimer's disease; hAPP, human amyloid precursor protein; PBS, phosphate-buffered saline.
In a previous study, full-length hAPP was decreased with genetic ablation of Acat1 in AD mice.12 To investigate the mechanism for the decrease in Aβ42 observed in our current study, we measured the levels of full-length hAPP by isolation of the membrane fraction of mouse brain homogenates (from the mice described above and used in Figure 7a), followed by western blotting using the 6E10 antibody, which recognizes an N-terminal sequence in human APP and does not recognize mouse APP. A representative western blot result is shown in Figure 7b. Quantification of full-length hAPP levels by western blot showed that its levels essentially paralleled those of Aβ42 in each group of mice (Figure 7c). There was a statistically significant decrease in full-length hAPP in AD mice treated with AAV-Acat1, compared with AD mice treated with PBS. There was also a significant decrease in full-length hAPP levels in all treatment groups of AD/Acat1−/− mice, compared to AD mice treated with PBS.
We next tested whether the levels of oligomeric Aβ, which is widely regarded to be the most toxic form of Aβ, were affected by treatment with AAV-Acat1 in AD mice. We performed dot blot analysis with the A11 antibody, which recognizes oligomeric Aβ and has previously been used to monitor Aβ oligomerization in the 3xTg-AD mouse model.35 Brain homogenates from individual 12-month-old AD or AD/Acat1−/− mice that had received brain injections with 1.0 µl of PBS, AAV-NC, or AAV-Acat1 in the hippocampi at 10 months of age were analyzed by dot blotting (Figure 8a). The A11 immunoreactivity was quantified for each mouse (Figure 8b). Analysis of variance analysis showed that there was a statistically significant difference between the group means (P = 0.03). Post-hoc t-tests showed that treatment of AD mice with AAV-Acat1 led to a statistically significant decrease in oligomeric Aβ, compared to treatment with either PBS or AAV-NC (P ≤ 0.05). There was no significant difference in oligomeric Aβ levels when comparing AD mice treated with PBS versus AAV-NC. Compared to AD mice treated with PBS, AD/Acat1−/− mice treated with PBS had significantly decreased oligomeric Aβ levels (P < 0.05).
Figure 8.

Analysis of oligomeric amyloid-β in brain injected mice. (a) Brain homogenates from individual 12-month-old mice that received brain injections at 10 months of age were prepared in sucrose buffer, spotted onto a nitrocellulose membrane, and subjected to dot blot with the A11 antibody. (b) Quantification of the intensity of A11 antibody immunoreactivity in each group of mice. The intensity of each spot in the dot blot was measured using ImageJ, and the intensity of the background (an adjacent area of membrane with no protein spotted) was subtracted from the measurement. The mean of each group is shown; the error bars represent mean ± SEM. The number of mice in each group is as follows: AD, PBS: n = 6; AD, AAV-NC: n = 8; AD, AAV-Acat1: n = 13; AD/Acat1−/−, PBS: n = 9; AD/Acat1−/−, AAV-NC: n = 4; AD/Acat1−/−, AAV-Acat1: n = 4. Analysis of variance analysis showed that there was a statistically significant difference between the group means (P = 0.03). This was followed by post-hoc t-test which showed that there was a statistically significant difference between AD, PBS and AD, AAV-Acat1 groups (P = 0.02) and between AD, AAV-NC and AD, AAV-Acat1 groups (P = 0.05), but not between AD, PBS and AD, AAV-NC groups (P = 0.79). There was also a statistically significant difference between AD, PBS and AD/Acat1−/−, PBS groups (P = 0.04). *P ≤ 0.05. AAV, adeno-associated virus; ACAT, acyl-CoA:cholesterol acyltransferase; AD, Alzheimer's disease; PBS, phosphate-buffered saline.
Discussion
The present study shows that AAV targeting Acat1 for gene knockdown delivered to the brains of AD mice decreased the levels of total brain amyloid-β, oligomeric amyloid-β, and full-length hAPP to levels similar to complete genetic ablation of Acat1, thus providing support for further testing of Acat1 knockdown gene therapy for AD. This study offers several novel findings. The first is that Acat1 can be specifically and partially knocked down, and still ameliorate biochemical markers of AD in AD mice. Previous studies10,11 have utilized ACAT inhibitors in AD mice, which provide partial (not 100%) inhibition of ACAT activity, however, the ACAT inhibitors CP-113,818 and CI-1011 used in10 and11 respectively, inhibit both ACAT1 and ACAT2,36,37 and may even convey other off-target effects by inhibiting other members of the membrane-bound O-acyltransferase enzyme family38 or alteration of cellular membrane properties. Another study utilized specific genetic knockout of Acat1 in AD mice,12 but did not address whether complete loss of Acat1 function was necessary to ameliorate the AD phenotype, or if partial loss would be sufficient. Yet another study relied on small interfering RNA knockdown of Acat1 and showed that Acat1 knockdown decreased Aβ secretion, but the study was done in cultured cells overexpressing hAPP.39 In the current work, evidence is presented that Acat1 can be targeted specifically, but need not be completely ablated, to confer benefit in this AD mouse model. The second novel finding of the current study is that Acat1 can be knocked down beginning at 10 months of age, instead of throughout the lifetime of the mouse, and still ameliorate biochemical markers of AD. Previous studies10,11 administered ACAT inhibitors to AD mice later in life, and observed beneficial effects on the AD phenotype. However, as described above, it was not conclusively demonstrated that the inhibitors' effect was mediated through specific targeting of Acat1. The third novel finding from this study is that Acat1 can be targeted for knockdown in brain tissue only, and still ameliorate biochemical markers of AD in AD mice. Previously, it was hypothesized, but not demonstrated, that the brain was the important location for ACAT1 inhibition or genetic loss ameliorating the AD phenotype. Previous studies in mice either utilized ACAT inhibitors,10,11 which potentially could distribute throughout the entire body (including the brain), or whole-body genetic ablation of Acat1.12 Interestingly, in our study a single injection of 1.0 µl of AAV in the hippocampus produced a ~50% decrease in ACAT activity in the whole mouse hemibrain. We chose to target the hippocampus because the hippocampus houses the highest levels of Acat1 mRNA among mouse brain regions based on in situ hybridization data;12 also, it is a brain region with high amyloid pathology in the 3xTg-AD mouse model13 as well as in humans with AD. The current study provides evidence that a relatively small amount of AAV delivered to a key brain area can have appreciable effects on both brain ACAT activity and the AD-like phenotype.
An earlier study using cultured cells which overexpressed hAPP relied on small interfering RNA knockdown of Acat1, and showed that Acat1 knockdown decreased Aβ secretion without affecting full-length hAPP protein levels.39 In contrast, we observed in the present study that full-length hAPP was decreased in AD mice with AAV-Acat1 treatment or Acat1 genetic knockout. The discrepancy in results between the study by Huttunen and colleagues and the current work cannot be explained at present, but may be due to the difference in model systems employed.
A previous study linked decreased levels of HMGR protein and brain cholesterol content with amelioration of the AD phenotype in male AD/Acat1−/− mice.12 We were able to replicate these findings in young mice (4 months old), but we also observed that increased interindividual variation in HMGR occurred with age, and we were unable to observe the same large statistically significant decrease in HMGR levels in the current study in older AD/Acat1−/− mice that had received brain injections. Therefore, we concluded that decreased HMGR in young male AD/Acat1−/− mice may be an early life event, and may not be necessary to mediate the decrease in Aβ42 and full-length hAPP in 12-month-old AD mice treated with AAV-Acat1. Another possibility is that our neurosurgical procedure alters HMGR levels in the brains in some way and that this was responsible for the lack of HMGR effect in the AAV-treated mice, or that we did not have a large enough sample size to observe a relatively subtle difference between groups. Consistent with our previous study,12 our current results show that total or partial ablation of ACAT1 enzymatic activity leads to reductions in full-length hAPP and in its key metabolite Aβ42. However, the mechanism(s) by which Acat1 knockout or knockdown leads to these events are not completely understood at the molecular level and will require further studies.
An unexpected result of the current study was that compared to treatment with PBS, the control treatment with AAV-NC led to a mild decrease in Aβ42 in AD mice, although its effect was less robust than that of AAV-Acat1 treatment. AAV-NC also showed a tendency toward reducing full-length hAPP; however, unlike the effect of AAV-Acat1, its effect was smaller and did not reach statistical significance. In measurements of oligomeric Aβ, there was no detectable decrease with AAV-NC compared to PBS treatment. We also showed that in AD/Acat1−/− mice, there were no significant differences in hAPP, total Aβ42, or oligomeric Aβ levels between any of the groups (treated with PBS, AAV-NC, or AAV-Acat1). This result strongly suggests that the effect of AAV-Acat1 on reducing hAPP, total Aβ42, and oligomeric Aβ levels in AD mice is mediated specifically through Acat1. Why the control virus would affect biochemical markers of AD in a beneficial manner is unknown but warrants further investigation. The observation that oligomeric Aβ was not decreased by AAV-NC treatment, but total Aβ42 was, leads us to speculate that treatment with any AAV may nonspecifically increase clearance of Aβ via a low-level activation of microglia and astrocytes that was not detected in our assays for brain inflammation.
Our current work provides evidence that further investigation of Acat1 knockdown gene therapy as an approach to AD treatment is warranted. Future preclinical studies on AD mice will need to include cognitive testing to rigorously demonstrate the viability of AAV-Acat1 for AD. There may be functional roles of APP and/or its cleavage products in neurons, however AAV-Acat1 treatment does not completely diminish human APP, it only lowers its levels, so we believe it is unlikely that there would be any adverse effects from this lowering effect. Multiple mouse studies utilizing different approaches have provided proof-of-concept that AAV gene therapy benefits AD in mouse models,24,25,26,27 and at least one therapeutic AAV is in phase II clinical testing in humans with AD28 (ClinicalTrials.gov identifier: NCT00876863). However, AAV brain injection has not currently been demonstrated as a viable treatment strategy for human AD. The ACAT inhibitors pactimibe and avasimibe have already demonstrated safety in human clinical trials, even though they failed to show efficacy for treatment of atherosclerosis.40,41 Avasimibe (CI-1011) specifically has been tested in an AD mouse model and shown to improve brain amyloid pathology11 and lower the maturation of APP through the secretory pathway.42 These drugs could potentially be investigated for the treatment of human AD if preclinical studies provide enough evidence in favor of ACAT inhibition for AD, with the caveat that they would likely need to be able to reach the brain and cause significant inhibition of ACAT1 there to mediate treatment of AD.
Materials and Methods
Mice. All mouse husbandry and procedures were performed at Dartmouth, in accordance with Dartmouth IACUC approved protocols. Mice were given ad libitum access to food and water and housed in rooms with a 12-hour light/dark cycle.
For measurement of brain ACAT activity, wild-type, Acat1−/−,33 and Acat2−/−34 mice were on a C57BL/6 genetic background. For the rest of the experiments in this study, the mouse lines used were the same as first described.12 Briefly, the 3xTg-AD mouse13 was generously provided to the Chang Lab by Dr Frank LaFerla, UC Irvine, in 2006. The 3xTg-AD mouse was crossed with the Acat1−/− mouse33 to create 3xTg-AD mice with or without Acat1, referred to in the current work as AD or AD/Acat1−/−. The mice were on a mixed 129;C57BL/6 genetic background. The mouse lines were maintained as follows: 3xTg-AD mice heterozygous for Acat1 (AD/Acat1+/− mice) were crossed to one another to produce AD/Acat1+/+ and AD/Acat1−/− offspring, which were siblings of each other. The AD/Acat1+/+ or AD/Acat1−/− mice were crossed to a nonsibling mouse of the same genotype to produce litters of either AD/Acat1+/+ or AD/Acat1−/− mice, which were then used for experiments. The AD/Acat1+/+ and AD/Acat1−/− mice used for experiments were cousins of each other.
All mice were housed in barrier housing until undergoing surgery, then moved to nonbarrier housing until being euthanized. All mice for experiments were euthanized between the hours of 11 am and 2 pm by CO2 asphyxiation and their brains were rapidly extracted, hemisected, and frozen until use in downstream analysis.
Surgical procedures. Neurosurgical AAV delivery was performed as described43 with some modifications. Mice were anesthetized with Avertin (250 mg/kg), positioned in a stereotaxic frame (Kopf, Tujunga, CA) over a heated pad, and monitored for breathing, maintenance of deep anesthesia, and body temperature. They were administered ketoprofen (5 mg/kg) at this time for postoperative analgesia. Using a stereomicroscope (Leica S6E; Leica, Buffalo Grove, IL), the head was shaved and disinfected. An incision was made in the scalp, exposing the skull. (For mice who received “sham surgery,” the following step involving opening craniotomies and injecting solution into the brain was skipped). Craniotomies were opened bilaterally with a dental drill (Fine Science Tools, Foster City, CA) at (−2.1 mm, 2.0 mm) from the bregma line and midline resectively, and a stainless steel syringe (Hamilton, Reno, NV) was inserted to 1.5 mm depth, corresponding to the dorsal hippocampus. Approximately 1.0 µl of liquid—either sterile PBS or AAV solution—was injected manually into the brain, at a rate of 0.5 µl/min. Each mouse received injections of ~1.0 µl of virus, bilaterally in the hippocampi at the stereotaxic coordinates above (−2.1 mm, 2.0 mm, 1.5 mm). The needle was left in place for an additional 2 minutes to prevent backflow of the liquid. The syringe was then withdrawn and the craniotomy closed with bone wax; the scalp incision was closed with Vetbond (3M) and treated with triple antibiotic ointment. Mice were allowed to recover in individual cages with wet food and careful monitoring. They were administered ketoprofen (5 mg/kg) again at 24 and 48 hours after surgery, and observed daily for 2 weeks post-surgery for any signs of discomfort or difficulty in healing of the surgical wound. For experiments, adult mice of various ages were housed postoperatively for 1 or 2 months and then euthanized.
Statistical methods and calculations. All results are expressed as a mean, with error bars representing the SEM. Statistical significance was determined by unpaired t-test, and a P value of <0.05 was considered statistically significant. When there were multiple groups, analysis of variance analysis was used followed by post-hoc t-testing.
Design of artificial miRNA sequences to target Acat1. We utilized the block-it RNAi designer (Invitrogen, Carlsbad, CA; http://rnaidesigner.invitrogen.com/rnaiexpress) to design artificial miRNAs targeting four different regions within the mouse Soat1 mRNA (Genbank accession #NM_009230). We also selected a scrambled control sequence that did not match the sequence of any mouse gene. Artificial miRNA sequences are available in Supplementary Table S1. Artificial miRNA sequences were synthesized by Invitrogen.
Cloning into pcDNA 6.2-GW/miR. pcDNA 6.2-GW/miR was part of the BLOCK-iT Pol II miR RNAi Expression Vector Kit (Invitrogen). miRNA sequences were ligated into the mammalian expression vector pcDNA 6.2-GW/miR according to the manufacturer's instructions.
Transfection of mouse 3T3 fibroblasts. Mouse 3T3 fibroblast cells (generously provided by Dr Henry Higgs, Dartmouth) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin (Medium A). Cells were cultured to 80% confluency and then transfected with Lipofectamine LTX (Invitrogen) according to the manufacturer's instructions.
Cloning into pAM/CAG-pL-WPRE-bGHpA AAV vector. The pAM/CAG-pL-WPRE-bGHpA AAV1/2 vector was graciously provided by M.T.H. (Max Planck Institute, Heidelberg, Germany). pAM/CAG-pL-WPRE-bGHpA was amplified in SURE (Stop Unwanted Recombination Events) Escherichia coli, which were generously provided by Dr Ronald Taylor at Dartmouth, and made competent in-house.44 After amplification, SmaI (New England Biolabs, Ipswich, MA) digest was performed to verify the integrity of the viral inverted terminal repeats, which are prone to recombination events. The vector was purified by miniprep (Qiagen, Valencia, CA), and a 1,058 bp fragment containing emerald GFP plus either the Soat1 #54, Soat1 #55, or negative control miRNA sequence, obtained from the mammalian expression vector pcDNA 6.2-GW/miR was subcloned into pAM/CAG-pL-WPRE-bGHpA between the SacI and EcoRV sites to create three AAV constructs. After subcloning and amplification, the integrity of the viral inverted terminal repeats sites was again verified by SmaI digest (Figure 1c).
Recombinant AAV production. Replication incompetent, recombinant AAVs of hybrid capsid type 1/2 were produced as described.20 Briefly, viral DNA constructs plus helper plasmids were transfected into HEK293 cells, packaged viruses were released by repeated freeze-thawing, and purified by heparin column. The purity of the virus and presence of three major viral proteins was verified by protein gel. AAV stocks were shipped in individual aliquots of 10 µl to the Chang Lab and stored at −80 °C until use. Any remainder of an aliquot was stored at 4 °C after thawing and used within 1 week. The viral titer was tested in cell culture, was similar between AAV aliquots and was >1011 viral particles/µl.
Primary cell culture of mouse neurons and glia. Neurons were isolated as described45 with modifications, including saving the glial layers from the density gradient and plating the mixed glia in Medium A. Neurons were plated and grown at a density of 350 cells/mm2. Glia were plated at a similar density and grown to confluency.
Oleate pulse. Cultured mouse neurons were treated with AAVs during plating. After 14 days in culture, the neurons were observed by fluorescence microscopy for GFP expression. After confirmation of GFP expression, the neurons were assayed for ACAT activity by oleate pulse.30 Neurons were pulsed with 3H-oleate-BSA for 3 hours at 37 °C. A well with no cells was included in the experiment as a blank control.
Brain ACAT assays. Brain ACAT activity assays were performed in vitro in mixed micelles as described.12,32
Brain western blots. For hAPP analysis, brains were processed according to Schmidt et al.46 with modifications described.12 Membrane proteins were collected by 100,000 g centrifugation and used for western blot. For HMGR analysis, frozen hemibrains were homogenized in 10% sodium dodecyl sulfate with 1× protease inhibitor cocktail (Sigma, St Louis, MO). Protein determination was done by Lowry assay. Standard tris-glycine or tricine sodium dodecyl sulfate polyacrylamide gel electrophoresis protocols were used. Primary antibodies used were: 6E10 (Covance, Princeton, NJ) for hAPP; IgG-A9 hybridoma cell line supernatant for HMGR (ATCC CRL-1811).47
Dot blots. Two microgram of protein in a 2 µl volume from mouse brain homogenates in sucrose buffer were spotted on a nitrocellulose membrane divided into 1 cm2 squares. The primary antibody was 1 µg/ml A11 (Invitrogen) antibody for oligomeric Aβ in 1% skim milk/TBST.
Amyloid β (1-42) ELISA assay. Sample pools in homogenization buffer (250 mmol/l sucrose, 20 mmol/l Tris-HCl (pH 7.4), 1 mmol/l EDTA, 1 mmol/l EGTA, 1 mmol/l PMSF, Leupeptin, Antipain, Pepstatin A, and phosphatase inhibitor cocktail II & III (Invitrogen)) were subjected to formic acid extraction.46 Neutralized formic acid extracts were used with the Human β Amyloid (1-42) ELISA kit (Wako Chemicals, Richmond, VA). Formic acid extracts were made in triplicate for each sample, and ELISA assays were run in duplicate or triplicate for each sample.
Real-time PCR. RNA from mouse brains was extracted with Trizol (Invitrogen) reagent. One microgram of RNA per sample was used to synthesize cDNA, on which real-time PCR was performed. Real-time PCR primers are available in Supplementary Table S2.
Image quantification. Western blot densitometry was performed using Image J software. Band intensity (background value subtracted) was quantified relative to loading control or Ponceau S staining in the lane.48
SUPPLEMENTARY MATERIAL Figure S1. Macroscopic GFP expression in adult mouse brain, 1 month after brain injection with 1.0 µl of AAV solution in the dorsal hippocampus. Figure S2. Temporal characterization of Aβ pathology in 3xTg-AD mice used in this study. Figure S3. Amyloid-β (1-40) (Aβ40) ELISA assay in brain homogenates from brain injected mice. Table S1. Sequences of artificial miRNAs. Table S2. Primers used for real-time PCR.
Acknowledgments
We thank Charles Barlowe (Dartmouth) and Maximillian Rogers (Dartmouth) for helpful discussion about this project; Henry Higgs (Dartmouth) for helpful discussion and provision of mouse 3T3 fibroblasts; Ronald Taylor (Dartmouth) for providing us with SURE Escherichia coli; and Alireza Kheirolla (Dartmouth) for assistance with the mouse surgery. This work was supported by National Institutes of Health grant AG37609 to T.-Y.C. and by grants from Max Planck Society and the Fritz Thyssen Stiftung to M.T.H. S.R.M. received additional support from the National Institute of General Medical Sciences Award #T32GM008704. The authors declared no conflict of interest.
Supplementary Material
References
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy S, DeKosky ST. Toward the treatment and prevention of Alzheimer's disease: rational strategies and recent progress. Annu Rev Med. 2013;64:367–383. doi: 10.1146/annurev-med-092611-084441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006262. doi: 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab. 2009;297:E1–E9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T, Kuchenbecker J, Grimm MO. Alzheimer's disease: the lipid connection. J Neurochem. 2007;103 suppl. 1:159–170. doi: 10.1111/j.1471-4159.2007.04715.x. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer's disease beta-amyloid production. Biochim Biophys Acta. 2010;1801:860–867. doi: 10.1016/j.bbalip.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R, Kovacs DM. ACAT inhibition and amyloid beta reduction. Biochim Biophys Acta. 2010;1801:960–965. doi: 10.1016/j.bbalip.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron. 2004;44:227–238. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Havas D, Peach C, Barren C, Duller S, Xia W, et al. The acyl-coenzyme A: cholesterol acyltransferase inhibitor CI-1011 reverses diffuse brain amyloid pathology in aged amyloid precursor protein transgenic mice. J Neuropathol Exp Neurol. 2010;69:777–788. doi: 10.1097/NEN.0b013e3181e77ed9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci USA. 2010;107:3081–3086. doi: 10.1073/pnas.0913828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, et al. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsova Z, Malik JM, Michel U, Bähr M, Kügler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- Royo NC, Vandenberghe LH, Ma JY, Hauspurg A, Yu L, Maronski M, et al. Specific AAV serotypes stably transduce primary hippocampal and cortical cultures with high efficiency and low toxicity. Brain Res. 2008;1190:15–22. doi: 10.1016/j.brainres.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova E, Hartelt N, Mironova LA, Hasan MT, Kügler S. Remodelling of the respiratory network in a mouse model of Rett syndrome depends on brain-derived neurotrophic factor regulated slow calcium buffering. J Physiol (Lond) 2009;587 Pt 11:2473–2485. doi: 10.1113/jphysiol.2009.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Aller MI, Baron U, Cambridge S, Bausen M, Herb J, et al. Silencing and un-silencing of tetracycline-controlled genes in neurons. PLoS ONE. 2007;2:e533. doi: 10.1371/journal.pone.0000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hasan MT, Seung HS. Laser-evoked synaptic transmission in cultured hippocampal neurons expressing channel rhodopsin-2 delivered by adeno-associated virus. J Neurosci Methods. 2009;183:165–175. doi: 10.1016/j.jneumeth.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassim SH, Li H, Bell P, Somanathan S, Lagor W, Jacobs F, et al. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther. 2013;24:19–26. doi: 10.1089/hum.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikel T, Marx V, Freudenberg F, Zivkovic A, Resnik E, Hasan MT, et al. Select overexpression of homer1a in dorsal hippocampus impairs spatial working memory. Front Neurosci. 2007;1:97–110. doi: 10.3389/neuro.01.1.1.007.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Lebrón E, Gouvion CM, Moore SA, Davidson BL, Paulson HL. Allele-specific RNAi mitigates phenotypic progression in a transgenic model of Alzheimer's disease. Mol Ther. 2009;17:1563–1573. doi: 10.1038/mt.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DA, Mastrangelo MA, Narrow WC, Sullivan MA, Federoff HJ, Bowers WJ. Abeta-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in Alzheimer's disease mice. Mol Ther. 2010;18:1471–1481. doi: 10.1038/mt.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty NC, Nash K, Lee D, Mercer M, Gottschall PE, Meyers C, et al. Adeno-associated viral (AAV) serotype 5 vector mediated gene delivery of endothelin-converting enzyme reduces Abeta deposits in APP + PS1 transgenic mice. Mol Ther. 2008;16:1580–1586. doi: 10.1038/mt.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer's mice. J Neurosci. 2010;30:13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel RJ. CERE-110, an adeno-associated virus-based gene delivery vector expressing human nerve growth factor for the treatment of Alzheimer's disease. Curr Opin Mol Ther. 2010;12:240–247. [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Doolittle GM, Chang TY. Cycloheximide sensitivity in regulation of acyl coenzyme A:cholesterol acyltransferase activity in Chinese hamster ovary cells. 1. Effect of exogenous sterols. Biochemistry. 1986;25:1693–1699. doi: 10.1021/bi00355a038. [DOI] [PubMed] [Google Scholar]
- Sakashita N, Miyazaki A, Chang CC, Chang TY, Kiyota E, Satoh M, et al. Acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) is induced in monocyte-derived macrophages: in vivo and in vitro studies. Lab Invest. 2003;83:1569–1581. doi: 10.1097/01.lab.0000095687.17383.39. [DOI] [PubMed] [Google Scholar]
- Chang CC, Lee CY, Chang ET, Cruz JC, Levesque MC, Chang TY. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J Biol Chem. 1998;273:35132–35141. doi: 10.1074/jbc.273.52.35132. [DOI] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, et al. Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc Natl Acad Sci USA. 1996;93:14041–14046. doi: 10.1073/pnas.93.24.14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, et al. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med. 2000;6:1341–1347. doi: 10.1038/82153. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, et al. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Chang CC, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem. 2000;275:28083–28092. doi: 10.1074/jbc.M003927200. [DOI] [PubMed] [Google Scholar]
- Cases S, Novak S, Zheng YW, Myers HM, Lear SR, Sande E, et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J Biol Chem. 1998;273:26755–26764. doi: 10.1074/jbc.273.41.26755. [DOI] [PubMed] [Google Scholar]
- Chang CCY, Sun J, Chang T-Y. Membrane-bound O-acyltransferases (MBOATs). Front Biol. 2011;6:177–182. [Google Scholar]
- Huttunen HJ, Greco C, Kovacs DM. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007;581:1688–1692. doi: 10.1016/j.febslet.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE) Investigators et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354:1253–1263. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Grégoire J, L'Allier PL, Anderson TJ, Bertrand O, Reeves F, Avasimibe and Progression of Lesions on UltraSound (A-PLUS) Investigators et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–3377. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Peach C, Bhattacharyya R, Barren C, Pettingell W, Hutter-Paier B, et al. Inhibition of acyl-coenzyme A: cholesterol acyl transferase modulates amyloid precursor protein trafficking in the early secretory pathway. FASEB J. 2009;23:3819–3828. doi: 10.1096/fj.09-134999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin A, Komai S, Eliava M, Seeburg PH, Osten P. Stereotaxic gene delivery in the rodent brain. Nat Protoc. 2006;1:3166–3173. doi: 10.1038/nprot.2006.450. [DOI] [PubMed] [Google Scholar]
- Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Nucleic Acids Res. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-β quantitation. Amyloid Proteins : Methods and Protocols Series: Methods in Molecular Biology. 2005;vol. 299:pp 267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- Liscum L, Luskey KL, Chin DJ, Ho YK, Goldstein JL, Brown MS. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J Biol Chem. 1983;258:8450–8455. [PubMed] [Google Scholar]
- Romero-Calvo I, Ocón B, Martínez-Moya P, Suárez MD, Zarzuelo A, Martínez-Augustin O, et al. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal Biochem. 2010;401:318–320. doi: 10.1016/j.ab.2010.02.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.