

Abstract
Aggressive regrowth of recurrent tumors following treatment-induced dormancy represents a major clinical challenge for treatment of malignant disease. We reported previously that recurrent prostate tumors, which underwent complete macroscopic regression followed by aggressive regrowth, could be cured with a vesicular stomatitis virus (VSV)–expressed cDNA library derived from recurrent tumor cells. By screening the protective, recurrence-derived VSV-cDNA library, here we identify topoisomerase-IIα (TOPO-IIα) as a recurrence-specific tumor antigen against which tolerance can be broken. Tumor recurrences, in two different types of tumor (prostate and melanoma), which had evaded two different frontline treatments (immunotherapy or chemotherapy), significantly overexpressed TOPO-IIα compared with their primary tumor counterparts, which conferred a novel sensitivity to doxorubicin (DOX) chemotherapy upon the recurrent tumors. This was exploited in vivo using combination therapies to cure mice, which would otherwise have relapsed, after suboptimal primary therapy in both models. Our data show that recurrent tumors—across histologies and primary treatments—express distinct antigens compared with the primary tumor which can be identified using the VSV-cDNA library technology. These results suggest that it may be possible to design a few common second-line therapies against a variety of tumor recurrences, in some cases using agents with no obvious activity against the primary tumor.
Introduction
Potentially fatal, aggressive tumor recurrence, following a period of tumor dormancy induced by apparently effective frontline therapies, represents a major clinical challenge for successful treatment of malignant disease.1,2,3,4 Escape from frontline therapy is common, in part because of the heterogeneity of tumor populations, which include treatment-resistant subpopulations of tumor cells.5,6 In this respect, effective immunotherapies for cancer must target a broad repertoire of tumor-associated antigens to minimize the chances that highly plastic tumor cells can evolve novel phenotypes which escape the frontline immune pressure.7,8,9 As self-antigens on normal tissues can serve as rejection antigens on associated tumors,10,11,12 we hypothesized that expression of a cDNA library of a normal tissue, from a vesicular stomatitis virus (VSV) platform,13,14,15 would allow the immune system to sample a wide range of self-antigens, some of which may serve as tumor-associated antigens against a tumor of the same histological type16,17,18,19,20,21 in a highly immune stimulatory environment provided by the viral infection.22,23 This would, in theory, prime T-cell responses to those antigens against which tolerance could be broken and, although T-cell responses against each self-antigen may be individually relatively weak, cumulatively they would impose a strong selective pressure against immune escape.22,23,24,25
Using human prostate cDNA for vaccination against murine prostate tumors to exploit the immunogenicity of altered-self epitopes,26,27,28 we generated an Altered-Self Epitope Library (ASEL) expressed in VSV.22 The Th17 response to prostate antigens induced by nine intravenous injections of the ASEL-cured–established TC2 tumors. However, six injections induced complete macroscopic tumor regressions, a period in which no palpable tumor could be detected, followed by rapid regrowth of aggressive recurrent tumors (TC2R). These recurrent tumors expressed a new profile of gene expression,22,29,30,31 suggesting that they had evolved away from the broad antigenic profile targeted by the ASEL-induced Th17 response. Therefore, we constructed a second VSV-cDNA library (Immune Escape Epitope Library, IEEL) using cDNA from TC2R tumors.22 ASEL-treated mice, vaccinated with the IEEL rejected TC2R tumors, dependent upon early treatment with the IEEL during recurrence. Significantly, the ASEL-primed response was Th17 associated, CD4 dependent, TC2/prostate specific, and blind to TC2R tumors/cells. In contrast, the IEEL-primed response was IFN-γ associated, CD8 dependent, TC2R specific, and blind to TC2 cells/tumors.
In the current report, by screening the IEEL for VSV-cDNA viruses encoding antigens targeted by the IFN-γ CD8+ T-cell response that clears TC2R tumors, we identified topoisomerase-IIα (TOPO-IIα) as a tumor-associated antigens associated with escape variants of both prostate cancer and melanoma. In addition, we show that a subset of TOPO-IIαHi cells within tumor cell cultures can respond to frontline selective pressure and repopulate the tumor with escape variants which allowed recovery of cultures subjected to an otherwise sub-lethal immune pressure. Recurrence-specific overexpression of TOPO-IIα also conferred a novel sensitivity to doxorubicin (DOX),32,33 which we exploited in vivo to generate combination therapies to cure up to 100% of mice which would otherwise have relapsed.
Results
The TC2–TC2R transition is induced by immune selective pressure
Nine i.v. ASEL injections cured 80–100% of mice of TC2 tumors (Figure 1a–c). Six injections typically induced complete macroscopic regressions early after treatment followed, in many mice, by aggressive tumor recurrences (TC2R) (Figure 1b), which had significantly different histologies compared with parental TC2 tumors.22 Freshly explanted TC2R cells also displayed markedly different profiles of gene expression compared with TC2 tumors with increased expression of N-cadherin, a gene often associated with an epithelial-mesenchymal transition,22,29,30,31 as well as different growth properties and morphology with higher frequency of large multi-nucleated syncytia and spindle-shaped cells (Figure 1d,g). The TC2–TC2R transition was also induced in vitro by high E:T ratio coculture of TC2 cells with splenocytes and lymph node (LN) cells from mice cured of TC2 tumors by multiple i.v. ASEL injections (Figure 1d–f).
Figure 1.

Selective pressure drives TC2R recurrent phenotype evolution. (a–c) Mice bearing 7-day–established TC2 tumors were injected intravenously with either (a) VSV-GFP (5 × 108 pfu, days 7, 9, 11, 14, and 16) or (b) with ASEL (107 pfu) six times (days 7, 9, 11, 14, 16, and 18) or (c) nine times (days 7, 9, 11, 14, 16, 18, 21, 23, and 25). Three representative animals for each group, from which tumor cell lines were derived and used in following experiments, are shown. (d) Parental TC2 cells were cocultured (E:T ratio of 10:1) with pooled splenocytes/LN from (e) untreated C57BL/6 mice or from (f) 6×ASEL-treated mice which had been restimulated as described in Materials and Methods. Five days after the last addition of splenocytes/LN, cultures were examined by microscopy and cDNA was screened by PCR for expression of E-cadherin and N-cadherin. (g) Early passage culture (5 days post-explant) of a tumor from a 6×ASEL-treated mouse. Scale bar = 50 μm. (h) Functional cloning of viruses encoding tumor rejection antigens for recurrent prostate tumors. LN/splenocyte cultures (104/well) from mice cured of TC2 and TC2R tumors by vaccination with a combination of ASEL and IEEL23 were screened for secretion of IFN-γ induced by infection with aliquots of ~104 pfu of the parental IEEL virus stock in the presence of recombinant hsp70.23 Aliquots which contained virus competent for inducing the IFN-γ recall response were pooled and expanded in BHK cells (24–36 hours). New LN/splenocyte cultures from IEEL-vaccinated mice were infected with serial dilutions of this expanded stock in the presence of recombinant hsp70, and assayed for IFN-γ production. The highest dilution of the virus stock which induced IFN-γ at levels significantly above background was amplified by passaging through BHK cells for 24–36 hours. Serial dilutions of this expanded stock were screened for their ability to induce IFN-γ. A 10 µl aliquots of the highest dilution of the virus which induced IFN-γ were used as the starting point for limiting dilution cloning on BHK cells to identify the dilution at which a single virus particle generated cytopathic effect (+). ASEL, Altered-Self Epitope Library; IEEL, Immune Escape Epitope Library; LN, lymph node; S, syncytia; VSV, vesicular stomatitis virus.
Rejection antigens from recurrent TC2R tumors
TC2R recurrences were effectively treated by a second VSV-cDNA library (IEEL) derived from the pooled cDNA of three different TC2R tumors. Splenocytes/LN from mice cured of TC2R recurrences by the IEEL secreted IFN-γ (but not IL-17) following restimulation with the IEEL and TC2R lysates, but not IEEL and TC2 lysates.22 In contrast, TC2 tumor-bearing mice vaccinated with the IEEL, as opposed to the ASEL, were not protected against continued growth of TC2 primary tumors (data not shown). Therefore, the antigenic repertoire which protected against TC2R recurrences (in the IEEL) differed significantly from that which protected against TC2 tumors (in the ASEL). We used an in vitro assay23 to screen for individual viruses within the IEEL which induced TC2R-specific IFN-γ recall responses (Figure 1h). Of multiple viruses recovered from this screen, nine encoded 5′ sequences of the murine TOPO-IIα gene (#NM011623) and five encoded the 5′ end of the murine CD44 gene (#NM009851). VSV-GFP was unable to induce IFN-γ following restimulation of splenocyte/LN cultures from IEEL-vaccinated mice, even at high MOI (Figure 2a). Restimulation of splenocytes/LN from IEEL-treated mice with either VSV-TOPO-IIα clone#2 or VSV-CD44 clone#17 alone induced very low, but reproducibly above background (20 pg/ml), levels of IFN-γ. However, a combination of VSV-TOPO-IIα#2 and VSV-CD44#17 induced levels of IFN-γ up to 1 log higher (>1,000 pg/ml) than either virus alone (but always lower than levels induced by restimulation with TC2R cell lysates) (Figure 2a) confirming the combination of TOPO-IIα and CD44 as potential tumor antigens in TC2R tumors. The ability of VSV-cDNA viruses to induce IFN-γ in combination depended on the nature of the cDNA insert because combining either alone with VSV-GFP did not induce IFN-γ.
Figure 2.

Topoisomerase-IIα is a tumor antigen for recurrent prostate tumors. (a) Splenocyte/LN cultures from IEEL-vaccinated mice were screened for IFN-γ secretion following no infection, stimulation with lysates (TC2, TC2R1, and TC2R2 cells), infection with VSV-GFP (107 pfu), or infection/restimulation with VSV-cDNA#2 (CD44), VSV-cDNA#17 (TOPO-IIα) or a combination (5 × 106 pfu/each) of VSV-cDNA#2 and #17, VSV-cDNA#2 and VSV-GFP, or VSV-cDNA#17 and VSV-GFP. (b) cDNA from freshly explanted TC2.1–3, TC2R1-3 (Figure 1b), or TC2R4 tumor from a separate experiment were screened by PCR (15 cycles), or (c,d) quantitative real-time PCR for expression of murine TOPO-IIα. *Samples positive for TOPO-IIα after 30 cycles. Relative quantities of TOPO-IIα mRNA were determined using GAPDH as calibrator gene (*P < 0.05). (d) Amplification curves from a representative quantitative real-time PCR experiment using cDNA (1:100) from TC2.1 and TC2R1 are shown. (e) Nuclear extracts (107 cells, 1:100) from two freshly explanted TC2 or TC2R tumors, or from TC2-cultured cells were incubated with kDNA to test TOPO-IIα activity. (f,g) TC2 cells (105/well) were either left untreated or cocultured with pooled LN/splenocytes (106/well) from untreated mice, or from 6×ASEL-treated mice, all of which previously restimulated as described in Materials and Methods. cDNA were screened by (f) PCR (15 cycles) or (g) three independent quantitative real-time PCR experiments for expression of TOPO-IIα at different time points following the coculture initiation. (h) cDNA from a freshly explanted TC2 tumor, TC2 cells cocultured with ASEL-restimulated LN/splenocytes from a C57BL/6 mouse, a TC2R explanted tumor at different time points post-explant, or TC2 cells cocultured for 1 week or 1 month with ASEL-restimulated LN/splenocytes were screened for expression of TOPO-IIα. (i) cDNA were prepared from freshly explanted TC2 or TC2R tumors at different time points following explant and screened by quantitative real-time PCR for expression of TOPO-IIα. ASEL, Altered-Self Epitope Library; IEEL, Immune Escape Epitope Library; LN, lymph node; VSV, vesicular stomatitis virus.
TC2R tumors overexpress TOPO-IIα
Although TOPO-IIα was expressed in TC2 tumors freshly explanted from untreated mice, an approximately three log higher level of TOPO-IIα mRNA was expressed in freshly explanted TC2R tumors (Figure 2b–d). Similarly, freshly explanted TC2R tumors displayed higher levels of TOPO-IIα enzyme activity compared with TC2 tumors (Figure 2e). Consistent with Figure 1d–g, the TOPO-IIαLo to TOPO-IIαHi TC2–TC2R transition could be induced in vitro by high E:T ratio coculture of TC2 cells with splenocytes/LN from ASEL-cured mice (cycle threshold (CT) for detection of GAPDH minus CT for TOPO-IIα increased from −13 to 0) (Figure 2f,g). However, after extended culture, levels of both mRNA (Figure 2f–i) and enzyme activity (data not shown) returned to those expressed in TC2 cells whether the TC2R transition had been induced in vitro (Figure 2f,g) or in vivo (Figure 2h,i) (no difference in ΔCT GAPDH-TOPO-IIα).
ASEL-induced novel chemosensitivity
High levels of TOPO-IIα are associated with sensitivity to DOX.32,33 Consistent with this, although TOPO-IIαLo TC2 cells were poorly sensitive to DOX, about 80% of freshly explanted TOPO-IIαHi TC2R cells were killed within 48 hours by DOX (Figure 3a). In contrast, and consistent with use of taxanes against prostate cancer, paclitaxel (PAC) was significantly more toxic to TC2 than TC2R cells.
Figure 3.

Immune escape variants acquire a novel chemosensitivity. (a) TC2 parental cells or freshly explanted TC2R tumor cells were treated in vitro with different concentrations of PAC or DOX as indicated. After 48 hours, cell viability was assessed using an MTT assay (mean ± SD). *P < 0.05; ***P < 0.001. (b,c) TC2 cells were left untreated or were treated for 48 hours with DOX (0.1 mg/ml) or PAC (10 nmol/l). Cells were then cocultured with ASEL-restimulated LN/splenocytes as described in Materials and Methods or with LN/splenocytes from an untreated C57BL/6 mouse. Twenty-four hours following washing, cDNA were screened for expression of TOPO-IIα by (b) PCR (15 cycles) or (c) quantitative real-time PCR. mRNA relative quantities were determined using GAPDH as calibrator gene (*P < 0.05). ASEL, Altered-Self Epitope Library; LN, lymph node; PAC, paclitaxel.
Survival of DOX-treated TC2 cells was not significantly different from that of untreated TC2 (Figure 3a). However, pretreatment with DOX completely prevented the TOPO-IIαLo–TOPO-IIαHi transition following coculture with splenocytes/LN from ASEL-treated mice (Figure 3b,c). In contrast, pretreatment with PAC did not prevent induction of the TOPO-IIαHi TC2R phenotype.
A DOX-sensitive subpopulation responds to immune pressure
We tested the hypothesis that a DOX-sensitive, pre-existing subset of TOPO-IIαHi TC2 cells responds to immune pressure. TC2 cells cultures, separately transduced with lentiviral vectors encoding different fluorescent marker genes, were combined to generate a mixed culture (TC2 Rainbow) (Figure 4a–i). Following in vitro coculture with splenocytes/LN from C57BL/6 mice, the relative proportions of each colored population was consistently unchanged across several such experiments (Figure 4b,i). In contrast, coculture with splenocytes/LN from ASEL-treated mice invariably resulted in the emergence of a population in which a single color predominated, although the same single color did not always emerge across experiments (Figure 4c,d,i). Pretreatment with PAC did not alter the ability of TC2 Rainbow cultures to become repopulated by a predominant color population in response to immune pressure (Figure 4e,i)—although PAC did not alter the proportions if no immune pressure was applied (Figure 4f,i). In contrast, and consistent with Figure 3b,c, DOX pretreatment completely inhibited the ability of TC2 Rainbow cells to respond to the immune pressure by selecting a predominant color population of TOPO-IIαHi cells (Figure 4g–i).
Figure 4.
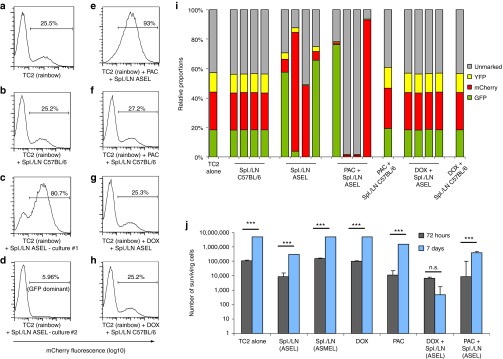
A doxorubicin (DOX)-sensitive subpopulation senses immune selective pressure. (a–d) TC2 cells were separately infected with three lentiviral vectors encoding fluorescent marker genes (GFP/mCherry/YFP) and mixed with unmarked TC2 cells at approximately 1:1:1:1. The resulting TC2 Rainbow population contained a stable level of cells of each lineage over long-term (>2 weeks) culture. TC2 Rainbow cells (105/well) were either (a) left alone or (b–d) cocultured with (b) pooled LN/splenocytes (106/well) from untreated or (c,d) 6×ASEL-treated mice, which had been restimulated as described above. Populations were analyzed by flow cytometry for fluorescent markers expression. Percentages of mCherry+ cells are shown. (e–f) The experiment was repeated as above and TC2 Rainbow cultures were treated (48 hours) with either (e,f) PAC (10 nmol/l) or (g,h) DOX (0.1 mg/ml) before being washed and cocultured with ASEL-restimulated LN/splenocytes from (e,g) ASEL-treated or (f,h) untreated mice. (i) The percentages of cells expressing the fluorescent markers following treatment (described in a–h) are shown. (j) TC2 cells were left untreated, or were cocultured with LN/splenocytes from prostate-specific ASEL-, or melanoma-specific ASMEL-treated mice. Alternatively, TC2 cells were treated with DOX or PAC with or without subsequent coculture with LN/splenocytes from ASEL-treated mice as described in e–h. MTT assays were carried out on surviving cell populations 72 hours or 1 week after coculture. ASEL, Altered-Self Epitope Library; LN, lymph node; PAC, paclitaxel.
Within 72 hours of coculture of TC2 cells with prostate-specific splenocytes/LN from ASEL-treated mice, significant numbers of cells were killed (Figure 4j), although the remaining population could recover, and was expanding, 1 week later. Coculture with splenocytes/LN from ASMEL-treated mice (melanoma-derived VSV-cDNA library)23 was not cytotoxic to TC2 cells. Treatment with DOX alone had no significant effects either 72 hours or 1 week after exposure. TC2 cell survival following a 48 hours pretreatment with DOX, followed by prostate-specific splenocyte/LN coculture, was equivalent to the coculture alone (no DOX) at 72 hours but, significantly, prevented long-term recovery of the cultures by nearly three logs. Treatment of TC2 cells with PAC led to significant tumor cell killing in the short term, but the cultures were able to recover over 7 days either with, or without, subsequent splenocyte/LN coculture.
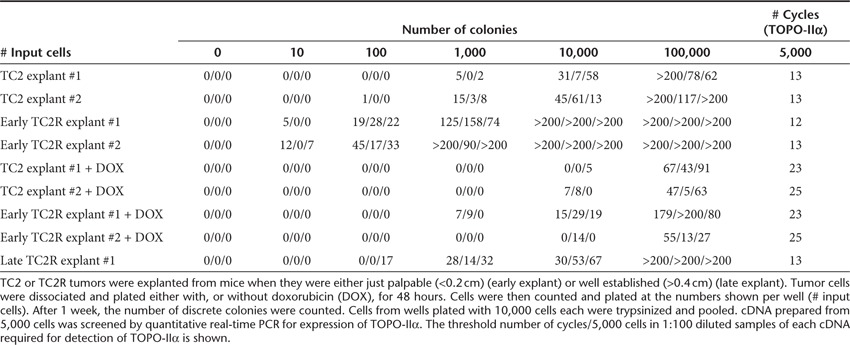
Consistent with TOPO-IIαHi cells having stem cell–like properties, early explanted TC2 tumors contained colony-forming cells at a significantly lower frequency (~1/100 cells) than TC2R tumors (~1/5 cells) (Table 1). Those cells which developed into colonies, from both TC2 and TC2R tumors, expressed high levels of TOPO-IIα mRNA, as shown by a CT at about 12–13 cycles/5,000 cells. Forty-eight hours in vitro purging with DOX before plating significantly reduced the frequency of these colony-forming cells in both TC2 (to ~5/10,000 cells) and TC2R (to ~1/500 cells) explants as seen by both the absolute number of viable colony-forming units and by an increased CT (quantitative real-time PCR). Finally, explants from more well-established TC2R tumors had a significantly lower frequency of colony-forming cells (~1/50 cells) than from early explants (~1/5 cells), although those cells had similarly high levels of TOPO-IIα expression as from the early explants.
Table 1. TOPO-IIαHi cells have high colony-forming capacity in vitro.
Immune escape variants are highly sensitive to DOX in vivo
In vitro, TC2 cells alone, or cocultured with splenocytes/LN from either control mice or mice vaccinated with the melanoma-specific, ASMEL VSV-cDNA library,23 were all sensitive to PAC but insensitive to DOX (Figure 5a). However, TC2 cells pretreated with splenocytes from ASEL-cured mice, or freshly explanted TC2R tumor cells, were significantly less sensitive to PAC but were highly sensitive to DOX. This sensitivity to DOX was lost if TC2R cells were kept for more than 3 weeks in culture (data not shown). Consistent with these results, TC2 tumors were sensitive to PAC (P < 0.001 compared with phosphate-buffered saline (PBS)) in vivo but completely insensitive to DOX (no significant difference with PBS) (Figure 5b). As before (Figure 1), six i.v. ASEL injections delayed tumor growth significantly compared with PBS or even PAC (P = 0.01) but generated no cures. However, when chemotherapy was administered as a second-line treatment for recurrent disease at the time of tumor recurrence, DOX cured 100% of TC2R bearing mice (tumor-free >60 days) (Figure 5c). In contrast, all mice with recurring TC2R tumors died of tumor with second-line PAC (P < 0.0001). If DOX or PAC therapy was started when the recurrences were greater than 0.5 cm in diameter, no significant therapy was achieved with either drug—consistent with the TOPO-IIαHi phenotype being lost, or diluted out, with time. Characteristic of treatment with the ASEL, all ASEL/DOX survivors developed an IL-17 response to TC2 cells (IL-17 secretion above background levels of 50 pg/ml), which was not observed in response to TC2R cells (IL-17 <50 pg/ml from all mice tested) (Figure 5d). However, in contrast to survivors of the ASEL/IEEL combination immunotherapy (Figure 2a), splenocytes of survivors of ASEL/DOX did not secrete IFN-γ in response to either TC2 or TC2R cells (Figure 5e).
Figure 5.

TC2R recurrences are cured by early treatment with doxorubicin (DOX). (a) TC2 and TC2R cells were either left alone or cocultured with pooled LN/splenocytes from 6×ASEL-treated, untreated C57BL/6, or 6×ASMEL-treated mice. All LN/splenocyte cultures had been restimulated as described in Materials and Methods. Two days after the last addition of LN/splenocytes, cocultures were washed three times and then cultured in medium containing no drug (Dulbecco's Modified Eagle's Medium, DMEM), PAC (10 nmol/l), or DOX (0.1 mg/ml) for 5 days. Surviving cells were visualized by crystal violet staining. (b) C57BL/6 mice (n = 7/group) bearing 4-day–established TC2 tumors were treated with i.v. PBS, PAC (10 mg/kg), DOX (10 mg/kg), or ASEL (107 pfu) on days 4, 6, 8, 11, 13, and 15. Survival with time is shown. (c) C57BL/6 mice (n = 6/group) bearing 4-day–established TC2 tumors were treated with i.v. ASEL (107 pfu) on days −14, −12, −10, −7, −5, and −3. Mice were then treated with either PAC or DOX on days 0, 2, 4, 7, 9, and 11 (six mice/group). (d,e) Splenocytes/LN from four of the mice which survived the sequential treatment of ASEL followed by DOX in c above were cocultured with either nothing (control) or with lysates of B16 murine melanoma cells, TC2 murine prostate cells or TC2R cells every 24 hours for 3 days. After 48 hours, supernatants were assayed for (d) IL-17 or (e) IFN-γ secretion. *P < 0.05; ***P < 0.001. ASEL, Altered-Self Epitope Library; LN, lymph node; PBS, phosphate-buffered saline; PAC, paclitaxel.
We also investigated the relevance of these findings in a different tumor type and with a different frontline selective pressure. When B16 murine melanoma cells expressing the Herpes Simplex Virus thymidine-kinase (HSVtk) gene were treated with ganciclovir (GCV) for 5 days in vitro, most of the cells were killed (Figure 6a). However, with time, the surviving cells repopulated the cultures (Figure 6b). Pretreatment with DOX before GCV did not increase overall cell killing over the 5 days of GCV (Figure 6a) but significantly inhibited the ability of the population to recover 1 week later (Figure 6b). B16tk cells which survived GCV chemotherapy expressed higher levels of TOPO-IIα enzyme activity (Figure 6c) and mRNA (data not shown), but pretreatment with DOX before GCV prevented selection of these TOPO-IIαHi cells. Finally, although treatment of B16tk tumors in C57BL/6 mice with DOX had no significant therapeutic effect compared with PBS alone (Figure 6d), it largely prevented the emergence of GCV-resistant recurrent tumors following GCV therapy in vivo compared with mice treated with GCV alone (P < 0.01).
Figure 6.

Doxorubicin (DOX)-sensitive, TOPO-IIαHi melanoma cells drive escape from chemotherapy. (a,b) B16tk cells were treated with PBS, DOX (0.1 mg/ml), or PAC (10 nmol/l) for 48 hours before being treated with ganciclovir (GCV, 5 µg/ml) for 5 days. Surviving cells were (a) counted (mean ± SD) at the end of GCV treatment or were (b) harvested and replated at 500 cells/well and grown in normal medium for 7 days before being counted. Statistical significance is shown for comparisons of the same treatments between a and b. (c) B16tk cells were treated with PBS or DOX for 48 hours before being treated with GCV for 3 days. After 24 hours, cells were harvested and TOPO-IIα activity was assessed. (d) C57Bl/6 mice (n = 8/group) bearing 5-day–established B16tk tumors were treated daily with PBS or DOX (10 mg/kg) on days 6–10 and then daily with PBS or GCV (50 mg/kg) on days 13–17 and 20–24. Tumor survival with time is shown. GCV, ganciclovir; NS, not significant; PBS, phosphate-buffered saline; PAC, paclitaxel.
Discussion
We show here that, when exposed to suboptimal frontline immunotherapy, prostate tumors evolved a highly reproducible escape phenotype radically different from the primary tumors, suggesting that escape was only possible by acquisition of one, or a few, different antigenic profiles.22,29,30,31 Relapsing mice could be treated with a VSV-cDNA library prepared from the TC2R tumors (IEEL) to attack the antigenic profile that TC2R tumors acquired to evade the ASEL-mediated immune pressure. By cloning individual viruses from the IEEL, we identified TOPO-IIα and CD44 as potential antigens, which, in combination, were important for vaccination against recurrent prostate tumors which escaped initial immunotherapy. Consistent with this, TOPO-IIα was expressed at significantly higher levels in freshly explanted recurrent TC2R, compared with TC2, tumors, a phenotype which could be recapitulated in vitro, at least transiently, by coculture of TC2 cells with ASEL-restimulated splenocytes/LN.
To investigate the immune-induced TC2(TOPO-IIαLo)–TC2R(TOPO-IIαHi) transition, we exploited the ability of DOX to target TOPO-IIα.32,33 DOX had no significant cytotoxicity against bulk populations of TC2 cells (Figure 3). However, DOX pretreatment prevented induction of the TOPO-IIαHi phenotype. Moreover, the ability of the TC2 population to react to immune pressure with the evolution of a TOPO-IIαHi, predominantly clonally-derived, population was completely inhibited by pretreatment with DOX, which also prevented the recovery of the TC2 population as a whole from initial splenocyte/LN killing. Therefore, the data of Figure 4, combined with our ongoing studies, suggest a model in which an immune pressure applied to the overall tumor population (by T cells or natural killer cells) leads to high levels of killing of the majority of the tumor cells. However, a pre-existing, minority population of cells, by virtue of their high level expression of TOPO-IIα and other proteins associated with control of DNA replication and cell cycle progression, have the ability to generate rapidly a pool of potential escape variants, one, or a few of which, are successful in evading the primary immune pressure. Once selected, these cells rapidly proliferate and expand as the nascent recurrent tumor.
These data suggest that a minority population of DOX-sensitive, TOPO-IIαHi TC2 cells represent a stem cell–like population which can directly respond to, and deviate phenotypically away from, applied immune pressure.22,29,30,31 However, DOX purging did not affect the long-term viability of TC2 cultures as a whole, suggesting that these TOPO-IIαHi plastic cells are not responsible for repopulating/maintaining the entire tumor. Nonetheless, DOX-sensitive, TOPO-IIαHi cells were present at significantly increased frequency in TC2R, as opposed to TC2, cultures, associated with a greater colony-forming capacity in vitro, similar to the conventional properties of stem cells. It is possible, therefore, that these cells represent a plastic/stem cell–like cell type, which responds to strong selective pressures but is not necessarily, or exclusively, the cancer stem cell per se.34,35
Consistent with a model in which these plastic/stem cell–like, DOX-sensitive, TOPO-IIαHi cells drove the evolution of recurrent tumors capable of escaping ASEL-mediated immune pressure, TC2R tumors showed a de novo vulnerability to DOX chemotherapy (Figures 3 and 5). Significantly, we used this recurrence-specific property of the escape tumors to design a rational combination therapy in which 100% of mice were cured of tumors if they were administered DOX at the time of tumor recurrence. Studies are underway to investigate whether combined initial vaccination with both the ASEL (protective against TC2 primary tumors) and the IEEL (protective against recurrent TC2R tumors following ASEL immunotherapy) can eliminate the TOPO-IIαHi cells in the primary tumor population to prevent aggressive tumor recurrence. Despite the possible immunogenic effects of chemotherapy,36,37 DOX-mediated clearance of TC2R tumors did not prime the same IFN-γ–based immune responses as IEEL vaccination. In addition, we showed that this strategy has relevance across different tumor models (prostate and melanoma) and can be independent of the nature of the initial selective therapeutic pressure (immunotherapy or chemotherapy). Thus, we were also able to cure mice of B16 melanoma recurrences with a combination therapy of GCV and DOX chemotherapies. These data with the B16 melanoma are, therefore, also consistent with a model in which a subpopulation of TOPO-IIαHi cells exists within a tumor population, which can respond to frontline selective pressure to drive tumor escape and which can be purged by pretreatment with DOX. Most importantly, DOX had no detectable cytotoxic activity against either TC2 or B16 parental cells in vitro or in vivo and so would not be selected as a viable candidate drug for the treatment of these types of tumors using classical screens. Nonetheless, its efficacy to target a recurrence-specific antigen across tumor, and frontline treatment, types shows that antigenic profiling of recurrent tumors using the VSV-cDNA technology can identify common therapy-induced phenotypic changes necessary for tumors to escape from a variety of different initial selective pressures. These findings suggest that it may be possible to design one, or a few, rational, carefully timed, second-line therapies against a variety of types of tumor recurrences using immunological-based screens of the type described here. Moreover, the agents identified as being effective against recurrences in this way may well have no obvious activity against the primary tumor.
In summary, we show here that VSV-cDNA libraries can be used to identify recurrence-specific tumor antigens, shared across tumor types and overexpressed in recurrent tumors escaping from different frontline treatments. Moreover, our data show that, even if a potent frontline immuno- or chemotherapy is not completely effective, it can still enforce the evolution of a reproducible recurrent tumor phenotype, which is highly sensitive to a conventional chemotherapy. Importantly, this effective second-line chemotherapy may be one which is not normally associated with the treatment of the parental tumor type. These results raise the intriguing possibility of pretreating patients with chemo- or immunotherapies which have minimal obvious activity against the primary tumor population, but which purge those tumors of critical, treatment-evading, cells which will eventually drive emergence of aggressive tumor recurrences.
Materials and Methods
Cells and viruses. TRAMP-C2 (TC2) cells are derived from a prostate tumor that arose in a TRAMP mouse (H-2kb) and were characterized by Esteban Celis. TC2 cells grow in an androgen-independent manner and are routinely grown as tumors in C57BL/6 male mice.19 B16tk cells are B16 murine melanoma cells (H-2kb) stably transfected with the HSVtk gene which confers sensitivity to the prodrug GCV.38,39
VSV-cDNA libraries (ASEL/IEEL) were constructed as described in ref. 22, containing cDNA from normal human prostate (Biochain, Hayward, CA) or from three-pooled TC2R recurrent tumors respectively. VSV-cDNA libraries were generated from baby hamster kidney (BHK) cells by cotransfection of pVSV-XN2-cDNA library DNA along with plasmids encoding viral genes.40 Virus was expanded by a single round of infection of BHK cells and purified by sucrose gradient centrifugation. VSV-GFP was generated by cloning GFP cDNA into the plasmid pVSV-XN2.40 Titers were measured by standard plaque assays on BHK-21 cells.
Animal studies. All procedures were approved by the Mayo Foundation Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) at 6–8 weeks of age. To establish subcutaneous tumors, 2 × 106 TC2 cells in 100 µl of PBS were injected into the flank of mice. Intravenous injections of virus were administered in 100 µl volumes. Freshly explanted tumors were kept in culture for a single passage from explant before in vitro experiments. For survival studies, tumor diameter in two dimensions was measured three times weekly using calipers and mice were killed when tumor size was approximately 1.0 × 1.0 cm in two perpendicular directions.
Induction of the TC2–TC2R transition. LN/splenocytes from either control C57BL/6 mice, or C57BL/6 mice previously treated with at least six injections of the ASEL, were restimulated in vitro by infection with ASEL (MOI of ~10) or at a lower MOI (1.0) with added recombinant hsp70 (10 µg/ml). After 48 hours, TC2 cells (105/well) were cocultured with restimulated LN/splenocytes at an E:T ratio of 10:1. After 24 hours, cultures were replenished with an additional 106 ASEL-restimulated LN/splenocytes. After 2 days, cocultures were washed three times. Thereafter, tumor cells were cultured for 48 hours in serum-free medium before cDNA preparation.
Quantitative real-time PCR. Cells were cultured for 48 hours in serum-free medium. RNA was then prepared with the QIAGEN-RNeasy-MiniKit (QIAGEN, Valencia, CA). A 1 µg total RNA was reverse-transcribed in a 20 µl volume using oligo-(dT) primers. A cDNA equivalent of 1 ng RNA was amplified by PCR with gene-specific primers using GAPDH as loading control. Expression of the murine TOPO-IIα gene was detected using the forward 5′-GAGCCAAAAATGTCTTGTATTAG-3′ and reverse 5′-GAGATGTCTGCCCTTAGAAG-3′ primers.
Quantitative real-time PCR was carried out using a LightCycler480 SYBRGreenI Master kit and a LightCycler480 instrument (Roche, Basel, Switzerland) according to the manufacturer's instructions. Typically, RNA was prepared from equal numbers of cells from each sample (usually 5,000 cells) and reverse-transcribed as described above. PCR (primers at 0.5 µmol/l, annealing = 58 °C) was run with diluted cDNA samples (neat, 1:10, 1:100, 1:1,000). GAPDH amplification was used as a control for equal loading of target cDNAs. The threshold cycle (Ct) at which amplification of the target sequence was detected was used to compare the relative levels of mRNA between samples. Relative quantities of TOPO-IIα mRNA were normalized with Ct of GAPDH amplification.
TOPO-IIα activity. The enzymatic activity of DNA TOPO-IIα was assayed from nuclei of cells using the TOPO-II Assay kit (Topogen, Port Orange, FL) according to the manufacturer's instructions. Briefly, 107 cells grown for 48 hours in serum-free medium were used to prepare nuclear extracts, were diluted (neat, 1:10, 1:100) and incubated (37 °C, 60 minutes) with kinetoplast DNA (kDNA). TOPO-IIα activity in the nuclear extracts corresponds to increasing kDNA decatenation/relaxation which can be detected by gel electrophoresis as a clearly defined band migrating out of the well.
Cytokine secretion. Supernatants were harvested from 106 LN/splenocytes previously stimulated with virus stocks as described in the text and/or with freeze thaw lysates from tumor cells in triplicate, every 24 hours for 3 days. After 48 hours, cell-free supernatants were collected and tested by ELISA for IL-17 (R&D Systems, Minneapolis, MN) or IFN-γ (BD Biosciences, San Jose, CA).
Phase contrast microscopy. Pictures were acquired using an Olympus-IX70 microscope (UplanF1 4x/0.13PhL), a SPOT Insight-1810 digital camera and SPOT Software v4.6 (Sterling Heights, MI).
Statistics. Survival data from the animal studies were analyzed using the log-rank test, and the Mann–Whitney U-test, one-way analysis of variance and two-way analysis of variance were applied for in vitro assays as appropriate. Statistical significance was determined at the level of P < 0.05.
Acknowledgments
We thank Toni Higgins and Nicole Goldman for expert secretarial assistance. This work is supported by the Richard M Schulze Family Foundation, the Mayo Foundation, Cancer Research UK, the National Institute of Health (R01CA107082, R01CA130878, and R01CA132734), and a grant from Terry and Judith Paul. The authors declare no conflict of interest.
References
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target. Nat Rev Cancer. 2010;10:871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol. 2013;10:41–51. doi: 10.1038/nrclinonc.2012.207. [DOI] [PubMed] [Google Scholar]
- McGowan PM, Kirstein JM, Chambers AF. Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol. 2009;5:1083–1098. doi: 10.2217/fon.09.73. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer. Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131:844–854. doi: 10.1002/ijc.26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–1171. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med. 2006;354:709–718. doi: 10.1056/NEJMoa053007. [DOI] [PubMed] [Google Scholar]
- Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol. 2002;2:227–238. doi: 10.1038/nri774. [DOI] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14:37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, et al. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum Gene Ther. 2011;22:1343–1353. doi: 10.1089/hum.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, Sanchez-Perez L, Diaz RM, Kottke T, Thompson J, Lai M, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol. 2004;22:1125–1132. doi: 10.1038/nbt1007. [DOI] [PubMed] [Google Scholar]
- Ferrone S. Immunotherapy dispenses with tumor antigens. Nat Biotechnol. 2004;22:1096–1098. doi: 10.1038/nbt0904-1096. [DOI] [PubMed] [Google Scholar]
- Kottke T, Pulido J, Thompson J, Sanchez-Perez L, Chong H, Calderwood SK, et al. Antitumor immunity can be uncoupled from autoimmunity following heat shock protein 70-mediated inflammatory killing of normal pancreas. Cancer Res. 2009;69:7767–7774. doi: 10.1158/0008-5472.CAN-09-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Sanchez-Perez L, Diaz RM, Thompson J, Chong H, Harrington K, et al. Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 2007;67:11970–11979. doi: 10.1158/0008-5472.CAN-07-2259. [DOI] [PubMed] [Google Scholar]
- Sanchez-Perez L, Kottke T, Daniels GA, Diaz RM, Thompson J, Pulido J, et al. Killing of normal melanocytes, combined with heat shock protein 70 and CD40L expression, cures large established melanomas. J Immunol. 2006;177:4168–4177. doi: 10.4049/jimmunol.177.6.4168. [DOI] [PubMed] [Google Scholar]
- Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, et al. Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res. 2005;65:2009–2017. doi: 10.1158/0008-5472.CAN-04-3216. [DOI] [PubMed] [Google Scholar]
- Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30:337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Breckenridge C, Chiocca EA. A viral strategy to ambush tumors. Nat Med. 2011;17:784–785. doi: 10.1038/nm0711-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogadri F, Wolchok JD. Selecting antigens for cancer vaccines. Nat Biotechnol. 2012;30:328–329. doi: 10.1038/nbt.2174. [DOI] [PubMed] [Google Scholar]
- Gold JS, Ferrone CR, Guevara-Patiño JA, Hawkins WG, Dyall R, Engelhorn ME, et al. A single heteroclitic epitope determines cancer immunity after xenogeneic DNA immunization against a tumor differentiation antigen. J Immunol. 2003;170:5188–5194. doi: 10.4049/jimmunol.170.10.5188. [DOI] [PubMed] [Google Scholar]
- Guevara-Patiño JA, Turk MJ, Wolchok JD, Houghton AN. Immunity to cancer through immune recognition of altered self: studies with melanoma. Adv Cancer Res. 2003;90:157–177. doi: 10.1016/s0065-230x(03)90005-4. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle. Nat Rev Urol. 2011;8:428–439. doi: 10.1038/nrurol.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, et al. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22:113–124. doi: 10.1016/j.smim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success. J Clin Invest. 2008;118:1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Perez L, Gough M, Qiao J, Thanarajasingam U, Kottke T, Ahmed A, et al. Synergy of adoptive T-cell therapy and intratumoral suicide gene therapy is mediated by host NK cells. Gene Ther. 2007;14:998–1009. doi: 10.1038/sj.gt.3302935. [DOI] [PubMed] [Google Scholar]
- Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H. Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th1-like profile of intratumoural cytokine expression. Int J Cancer. 1997;71:267–274. doi: 10.1002/(sici)1097-0215(19970410)71:2<267::aid-ijc23>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]