

Abstract
A rapidly growing understanding of the complex circuitry of microRNA (miRNA)-mediated gene regulation is attracting attention to miRNAs as new drug targets. Targeted miRNA suppression is achieved in a sequence-specific manner by antisense RNA “decoy” molecules. Such synthetic miRNA inhibitors have reached the clinic with remarkable pace and may soon appear as new therapeutic modalities in several diseases. Shortcomings, however, include high production costs, the requirement for repeated administration, and difficulty achieving tissue-specific delivery. With the many recent landmark achievements in clinical gene therapy, new and refined vector-encoded miRNA suppression technologies are attractive for many applications, not least as tools in innumerable daily studies of miRNA biology in laboratories worldwide. Here, we provide an overview of the strategies that have been used to adapt vector-encoded inhibitors for miRNA suppression and discuss advantages related to spatiotemporal and long-term miRNA attenuation. With the remarkable new discovery of miRNA management by naturally occurring circular RNAs, RNA circles generated by trans-splicing mechanisms may prove to be well-suited carriers of decoy-type miRNA inhibitors. The community will aspire to combine circles with high-affinity miRNA decoy methodologies, and such “vectorized” RNA circles may represent new solid ways to deliver miRNA inhibitors, perhaps even with therapeutic applications.
Introduction
With the discovery of abundant expression of microRNAs (miRNAs) in several organisms, these small noncoding RNAs catapulted onto the stage of posttranscriptional gene regulation a bit more than 10 years ago.1 Originating from longer primary miRNA transcripts, approximately 22 nucleotides long double-stranded miRNAs are formed by successive processing steps, after which one strand is incorporated into the RNA-induced silencing complex (RISC), which exerts posttranscriptional gene silencing. The miRNA guides RISC to complementary mRNA target sequences mainly located in 3' untranslated regions (3' UTRs). In humans, the sequence complementarity between mRNA and miRNA is usually imperfect, but base pairing involving the seed region, nucleotides 2-7 of the miRNA as counted from the 5'-end, is particularly important for target recognition and in many cases sufficient to facilitate miRNA-directed gene silencing.2 Such partial mRNA:miRNA complementarity promotes mRNA deadenylation or translational repression, whereas near-perfect complementarity promotes mRNA cleavage at a position opposite to nucleotides 10-11 of the miRNA.3 More than 60% of all human genes are predicted to be regulated by a total of over 2,000 mature miRNAs found in humans so far.4 Some miRNAs are expressed in virtually all cell types, whereas others are highly tissue-specific with a distinct function in a particular cell type or organ.
Given their comprehensive involvement in gene regulation, it has become widely accepted that miRNAs play a key role in almost any biological process. Not surprisingly, perturbed miRNA expression has been functionally linked to numerous diseases, such as diabetes, rheumatoid arthritis, schizophrenia, coronary artery disease, and cancer—just to list a few. In several cancer types, oncogenic miRNAs as well as tumor suppressor miRNAs have been identified. These may serve as powerful diagnostic and prognostic biomarkers, or as potential therapeutic targets, further stressing the urge for crafting effective molecular tools for manipulating miRNA activity. Hence, the appearance of miRNAs on the scene was soon followed by methods of manipulating their function to experimentally validate miRNA target genes and to study gain- and loss-of-function phenotypes. Overexpression of natural miRNAs is readily achieved by expression of the genomic region encoding the primary miRNA transcript, or custom-designed miRNAs may alternatively serve as RNA interference effectors, allowing targeting of for example viral RNA genomes.5,6 The miRNA inhibitors (previously referred to as anti-miRs, antagomiRs, AMOs [Anti-miRNA antisense inhibitors], sponges, or decoys) are commonly based on antisense molecules that act to bind and sequester miRNAs from their natural targets. Two main approaches for delivery of miRNA inhibitors have been utilized, namely (i) direct cellular delivery of chemically synthesized inhibitors and (ii) delivery of a vector from which intracellular transcription of RNA inhibitors occurs. Synthetic miRNA inhibitors have been thoroughly reviewed elsewhere.7,8 Here, we focus on vector-encoded inhibitors, and give an overview of current suppression and miRNA targeting strategies, including some of the newcomers on the market, and their use in studying miRNA biology and as novel therapeutics.
Express Your miRNA Inhibitor—Why Bother?
Synthetic miRNA inhibitors are suitable for many experimental applications, allowing easy accessible in vitro studies of the immediate effect of suppressing miRNAs. In vivo miRNA inhibition has been obtained as well using synthetic miRNA inhibitors, and such inhibitors are slowly reaching drug status.9 So, why should we bother about “vectorizing” miRNA inhibitors after all? Though powerful, the effect of synthetic RNA is transient due to degradation and loss of the inhibitors over time, and repeated administration is required to obtain a sustained effect.10 Moreover, issues concerning high production costs, reduced delivery to some cell types, and lack of tissue-specific delivery further reduce the applicability of synthetic inhibitors for some uses. Vector-encoded inhibitors possess several advantageous features conferred by the great repertoire of different vectors available to date. Nonviral vectors, such as naked plasmid DNA and DNA minicircles,11 can be engineered with tissue-specific or drug-inducible promoters, thus providing spatiotemporal expression of the miRNA inhibitor. However, such carriers still share some of the disadvantages of synthetic inhibitors including poor uptake in certain cell types and tissues as well as clearance over time. Viral vectors, in contrast, are very efficient gene vectors capable of transducing a wide array of cell types, and the tropism of the virus may be modulated by pseudotyping the virus for directed delivery of a miRNA inhibitor cassette to a specific tissue. Different serotypes of adeno-associated virus-derived vectors allow potent transduction of many tissues, and this high-titer vector type promises to be an optimal carrier of inhibitor-encoding gene cassettes.12 By the use of retro- or lentiviral vectors, sustained inhibitor expression is readily achieved,13 but nonviral integration technologies such as integrases, transposons, and recombinases also offer chromosomal insertion and thus stable inhibitor expression. With the development of tools for site-directed genetic engineering, based on the DNA-cleaving activity of zinc-finger nucleases, TALE nucleases, or CRISPR-Cas systems, methods of stably introducing miRNA inhibitors into specified “safe harbors” of the genome may be employed. For both viral and nonviral vectors, it is straightforward to include a reporter gene to monitor inhibitor expression. For example, a fluorescent or luminescent marker can be included for tracking cells expressing the inhibitor by fluorescence-activated cell sorting or in vivo bioimaging of tissues and animals.12,13,14,15,16,17,18
Numerous miRNA knockout mouse models have been created and are available from libraries of miRNA knockout mice or mouse embryonic stem cells.19,20,21 Park et al. reported the generation of 162 miRNA targeting vectors, 64 targeted embryonic stem cell lines, and 46 germline-transmitted miRNA knockout mice, which were compatible with germline- or tissue-specific Cre recombinase-transgenic mice to generate conditional knockouts. For some miRNAs, the creation of knockout mice is not straightforward because some miRNAs originate from two distinct loci in the genome. Furthermore, some miRNAs have seed family members that share mRNA targets,22 so complete abrogation of miRNA activity in a particular cellular pathway would be a laborious process. Additionally, many miRNAs are located in an intron of a gene in which knockout of the miRNA sequence might add confounding effects to gene expression. Vector-encoded miRNA inhibitors offer an alternative approach for knocking down a specific miRNA or families of miRNAs in transgenic animals, and spatially and temporally regulated promoters may be employed to study physiological miRNA function at distinct time points in specific tissues during development.16,23 Several studies have shown stable miRNA inhibition in transgenic organisms including plants,24 flies,16,25 and mice.17,23 Stable miRNA inhibition has been obtained in the complete human hematopoietic system by transplanting bone marrow-ablated mice with human hematopoietic stem/progenitor cells genetically engineered with a Sponge targeting miR-223.13 Importantly, the obtained phenotype matched that obtained in miR-223 knockout mice. In cases where no direct validation can be made by comparison to the knockout phenotype, miRNA inhibition phenotypes may be experimentally supported by a reciprocal phenotype when overexpressing the miRNA.25 However, since inhibition efficiency is highly dependent on the cellular concentration of the miRNA and the inhibitor, transgenic animals expressing miRNA inhibitors may not always faithfully replicate the null phenotype produced by a knockout.
Suppressing miRNA Activity by Decoy-Type Inhibitors
RNA molecules carrying target sequences for the desired miRNA constitute the most widely employed type of miRNA inhibitor. The basic strategy is to provide a surplus of miRNA recognition sites exposed in a structural RNA context that optimizes affinity and specificity for the mature miRNA. By attracting the miRNA-loaded silencing complex, inhibitors should serve to reduce miRNA binding to the natural mRNA target (see Figure 1a for schematic representation). For vector-encoded inhibitors, expressed RNAs containing miRNA recognition sites range from short and simple antisense transcripts without additional sequences to structured scaffolds encompassing multiple miRNA binding sites (Figure 1b). Depending on the specific application, both RNA Pol III- and RNA Pol II-transcribed decoy-type inhibitors (the latter possibly fused to a protein-coding sequence) would be applicable for suppressing miRNA function.
Figure 1.
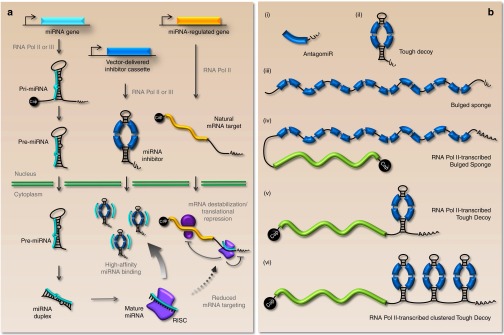
A schematic view of the anti-miR function of vector-encoded miRNA inhibitors. (a) Overview of miRNA biogenesis, miRNA regulation of protein-coding mRNAs, and miRNA inhibition by vector-encoded inhibitors. Long primary miRNA transcripts are transcribed from the genome and sequentially processed and transported to the cytoplasm, where they exert the posttranscriptional repression of target genes via the RNA-induced silencing complex (RISC). A miRNA inhibitor carrying miRNA binding sites (here represented by a Tough Decoy-type inhibitor) is intracellularly produced and interferes with natural miRNA regulation by attracting miRNAs by the exposure of high-affinity miRNA binding sites. (b) Schematic representation of commonly used inhibitor designs. (i) An antagomir consisting of a single miRNA binding site (shown in blue) followed by a short stretch of uridines originating from the RNA Pol III termination signal. (ii) An RNA Pol III-transcribed Tough Decoy consists of a ~60-bp long hairpin-shaped RNA carrying an internal loop exposing two miRNA binding sites each with a central bulge for prevention of fast endonucleolytic turnover of the transcript upon miRNA binding. The stretch of terminal uridines from the RNA Pol III termination signal provides the necessary 3' overhang for Exportin-5 recognition (iii) An RNA Pol III-transcribed Bulged Sponge design is generally expressed as a nonstructured RNA harboring 4–12 consecutive miRNA binding sites (eight are shown) with central bulges. (iv and v) RNA Pol II-transcribed variants of the Bulged Sponge and Tough Decoy resemble the Pol III-transcribed inhibitors, but are capped and polyadenylated and may be fused to a protein-coding sequence (shown in green). (vi) The Pol II-transcribed clustered Tough Decoy carrying three consecutive Tough Decoys with a total of six miRNA binding sites.
Scherr et al. developed a lentiviral vector encoding small transcripts, which they designated “antagomirs” and which contained only a single antisense sequence with perfect complementarity to the miRNA followed by a small 3' extension.26 The use of an RNA Pol III promoter enabled transcription of small well-defined RNAs that were neither capped nor polyadenylated, but contained only a short stretch of uridines added to the 3' end (Figure 1b, (i)). Carè and co-workers employed an adenoviral vector to express tandemly arranged perfect miRNA target sequences, which they termed “miRNA decoys.”27 The miRNA decoys were expressed as RNA Pol II transcripts, and the target sites were placed in the 3' UTR of the eGFP gene allowing monitoring of miRNA targeting. Soon after, an endogenous miRNA inhibitor was discovered in plants in which a noncoding RNA was found to suppress miR-399 activity through a single target site.24 Notably, this target contained a central mismatch opposite to position 10-11 of the miRNA. When this bulge was mutated to create perfect complementarity to the miRNA, the authors observed a decrease in miRNA inhibition potency. This signified that an internal bulge opposite to position 10-11 of the miRNA prevented fast endonucleolytic turnover of the inhibitor thereby prolonging the interaction between the miRNA and the inhibitor. This concept was further strengthened by Ebert et al. who showed superior potency of target sites harboring internal mismatches rather than perfect target sites.14 They also showed that these inhibitors, which were termed “Sponges,” were efficacious when expressed both as short RNAs from RNA Pol III promoters (Figure 1b, (iii)) or fused to the 3' UTR of a reporter gene (GFP) expressed from an RNA Pol II promoter (Figure 1b, (iv)).
As one would expect, an increase of the number of miRNA target sites has on several occasions been found to enhance miRNA suppression efficiency. However, at a certain threshold, reportedly somewhere between six and twelve target sites, there is only a marginal increase in potency, although the effect of additional target sites may be more pronounced in the event of a low inhibitor concentration.14,28 This apparent saturation could be caused by a threshold ratio between the number of bound miRNAs and the degradation rate of the inhibitor and may very well depend on the expression levels of both miRNA and inhibitor. In addition, one should bear in mind that increasing the number of target sites also elevates the risk of recombination during cloning and during transduction of lentiviral vectors. Lentiviral vectors are particularly prone to genetic rearrangements promoted by direct repeat sequences,29 and rearrangements have been observed in lentiviral vectors encoding sponge inhibitors.30 Another approach to maximize the potency of miRNA inhibitors is to render the target sites more accessible for the miRNA. Otaegi et al. envisioned that the previously employed short spacer regions of 6–8 nucleotides between the tandemly arranged target sites would restrict RISC-binding at adjacent target sites.28 This was based on previous findings by Chi and co-workers who used high-throughput sequencing of RISC-crosslinked mRNA to show that RISC spans 45–62 nucleotides of the mRNA,31 indicating that spacer lengths shorter than 10 nucleotides could potentially cause the binding of a single RISC to sterically block the adjacent target sites. However, in a comparative study using 6-, 29-, or 42-nucleotide spacers the authors showed that the 6-nucleotide spacer actually generated a more robust inhibition of the miRNA. This is in line with another report showing that two juxtaposed target sites both contribute to miRNA-mediated repression.32 In accordance, it could be speculated that the use of short spacers reduces the risk of forming alternative RNA secondary structures that do not support miRNA recognition.
Haraguchi and co-workers elaborated on the sponge design and used a rationally designed scaffold called a Tough Decoy (often referred to as a “TuD”) to expose two miRNA target sites.33 The Tough Decoy is an ~60-bp long hairpin-shaped RNA with an internal loop exposing two miRNA binding sites (Figure 1a,b, (ii)). It was originally designed to be expressed as a short RNA by an RNA Pol III promoter and designed to be actively exported from the nucleus to the cytoplasm via the Exportin-5 pathway like miRNAs. This was facilitated by including the structural requirements, i.e. an 18-bp stem region with a short 3' overhang, for Exportin-5 recognition. In the Tough Decoy hairpin, the two miRNA binding sites have a central bulge to bypass Ago2-mediated cleavage, and RISC-mediated destabilization of the inhibitor is believed to be impaired by the double-stranded structure, which should also confer resistance to cellular RNases. Studies addressing targeting of miR-21 and miR-122/let-7 found that the Tough Decoy inhibitors were more potent than Sponge inhibitors containing four and seven target sites, respectively.12,33 In our recent work, we compared the anti-miRNA capacity of Tough Decoy hairpins and Sponge inhibitors containing eight target sites. In comparisons of five unrelated miRNAs, one miRNA was found to be inhibited most efficiently by the Tough Decoy, one most efficiently by the Sponge, and the remaining three equally well by the two decoy-type inhibitors.30 Collectively, these studies indicated that Tough Decoy inhibitors carrying only two target sites are just as potent and for some miRNAs more potent than Sponge inhibitors carrying several target sites. Another important aspect of the comparison between the two types of inhibitors is whether inhibitor-encoding vector RNA—as relevant for lentiviral delivery strategies—is targeted by highly expressed endogenous miRNAs during vector production (and/or transduction) leading to reduced inhibitor transfer capacity. Interestingly, this effect was more pronounced for Sponge inhibitors than for Tough Decoy inhibitors, most probably indicating that vector RNA carrying a higher number of miRNA target sites was more vulnerable to degradation or less efficiently incorporated in virus particles. This effect could suspectedly be avoided by inserting the inhibitor cassette in the opposite direction thereby excluding target sites from virally packaged, positive-sensed vector RNA strands. Recently, Mullokandov et al. generated a library of lentiviral vectors encoding RNA Pol III-transcribed Tough Decoys targeting 291 different conserved miRNAs.15 Such vectors were utilized in a pooled manner for loss-of-function studies and have been made available to other researchers. Our studies showed that Tough Decoy inhibitors could also be embedded into the 3' UTR of an actively translated gene expressed from an RNA Pol II promoter (Figure 1b, (v)).30 When comparing Tough Decoy inhibitors expressed from the H1 promoter (RNA Pol III) and from the PGK promoter (RNA Pol II) embedded in the 3' UTR of eGFP, we generally observed comparable inhibitory potencies, but such efficiencies would obviously depend on the choice of promoter. This RNA Pol II strategy was further improved by arranging several Tough Decoys in tandem (“clustered Tough Decoy”) (Figure 1b, (vi)).30,34 The level of miRNA-mediated repression of an eGFP transcript carrying eight miRNA target sites did not differ between RNAs carrying the miRNA target sequences in the clustered Tough Decoy and Sponge configurations. However, when the same transcripts were assessed for their ability to inhibit miRNA function, inhibition by the clustered Tough Decoy was markedly higher relative to the Sponge, indicating that the miRNA inhibition potential was not directly proportional to the extent of miRNA-mediated repression of the inhibitor transcript.30 It is remarkable that the exact same target sites are targeted equally in the two sequence contexts, but that the Tough Decoy configuration supports increased miRNA inhibition. Perhaps the secondary structure of the Tough Decoy provides unique features that favor prolonged miRNA binding and enhance the overall anti-miRNA properties. One may speculate that the hairpin partially stalls miRNA-recruited exonucleases, thereby increasing the half-life of the inhibitor after miRNA binding. However, this notion is not supported by current experimental evidence, and future endeavors to optimize miRNA inhibitors should address such mechanistic differences between Tough Decoy and Sponge inhibitors. Notably, for certain miRNAs the Tough Decoy secondary structure can also pose a problem. Hence, using synthetic Tough Decoys based on two fully 2'-O-methylated RNAs, it was found that mutual affinity and binding between the two miRNA binding sites could impair miRNA binding and thus inhibition potency.35 Obviously, such intramolecular interactions may also exist in Sponge-type inhibitors, but this has not been investigated and, unlike the Tough Decoy configuration, the binding sites in sponges are not structurally aligned for annealing. Adverse effects caused by structural collapse of the inhibitor will clearly depend on the sequence of the targeted miRNA and could potentially be alleviated by introduction of mutations in the binding sites, although this will come at the expense of decreasing miRNA affinity.35
Mechanisms of miRNA Inhibition: Elucidating the Fate of Inhibitor-Captured miRNAs
The fate of miRNAs that are attracted to intracellularly expressed antisense RNA molecules is somewhat controversial. Some studies have not been able to show a decrease in miRNA levels following miRNA inhibition.13,14,18 Ebert et al. quantified the targeted miRNA by Northern Blot analyses and found that part of the miRNA pool migrated with the inhibitor, but they did not observe any noticeable difference in total miRNA abundance. Along the same lines, miRNA inhibition with Tough Decoy and Sponge inhibitors has been reported not to reduce levels of miRNAs, as quantified by miRNA qRT-PCR.13,18 In contrast, we observed a 54–83% reduction in miRNA levels following inhibition with vector-encoded Sponge or Tough Decoy inhibitors carrying bulged target sites.30 Haraguchi et al. could also detect decreased miRNA levels upon miRNA inhibition with Tough Decoy inhibitors as detected by qRT-PCR, but Northern blotting performed at elevated temperatures to avoid duplex formation between the miRNA and the Tough Decoy failed to confirm such a decrease.33 Xie et al. employed Tough Decoy inhibitors and showed lowered miRNA levels both by qRT-PCR and Northern Blotting, though qRT-PCR showed a much higher reduction than Northern Blotting, but Northern Blotting was not performed at elevated temperatures.12 Interestingly, high-throughput sequencing of miRNAs documented reduced levels of miRNAs with extensive complementarity to the binding sites in the inhibitor. This was caused by 3' to 5' exonucleolytic trimming, which is in line with previous reports showing miRNA trimming upon normal regulation of endogenous mRNA targets, but only for miRNAs with extensive complementarity to the target.36,37 Collectively these studies suggest that miRNA inhibition by decoy-type inhibitors may be caused by both sequestration and degradation and that care should be taken when evaluating miRNA inhibition by miRNA quantification. Indeed, Scherr et al. have shown that antisense transcripts render miRNAs unavailable for cDNA synthesis and Northern Blot probing,26 and, hence, quantification of the targeted miRNA may not accurately reflect the level of miRNA inhibition.
Toward Simultaneous Suppression of Multiple miRNAs by a Single Inhibitor
To maximize miRNA affinity, decoy-type inhibitors are designed to allow complete base pairing with a specific miRNA. However, miRNAs from the same miRNA family are also inhibited through seed sequence recognition, although with an efficacy that expectedly depends on the overall complementarity between the target sites and the miRNA.14,33 This could pose a problem in studies of the unique contribution of a single miRNA to a given phenotype. However, it enables studies of whole miRNA families for which a functional phenotype is only revealed by simultaneous targeting of the whole family. Xie et al. employed high throughput sequencing of small RNA libraries to look for off-target effects imposed by Tough Decoys inhibitors targeting miR-122 and let-7.12 No overall change in the abundance of miRNAs other than the targeted miRNAs was observed. However, because this approach only measures miRNA abundance, it fails to identify miRNAs that are in fact inhibited but not degraded upon targeting by the inhibitor. Since off-target effects may occur through limited complementarity and nonseed recognition of the inhibitor, the unintentionally targeted miRNAs are not subject to trimming and will not be identified by high throughput sequencing. Thus, off-target effects of miRNA inhibitors should be carefully assessed using a functional screening assay, such as the recently described “Sensor-seq” high-throughput assay that involves a miRNA sensor library to simultaneously monitor the activity of hundreds of miRNAs.15 In addition, a comparison between different inhibitor designs regarding their propensity to off-target effects has yet to be performed. Finally, it is also imperative to assess unintended miRNA perturbations caused by the vector alone and by additional sequences present in the RNA Pol II inhibitor transcripts.
For studies analyzing the synergistic effects of two unrelated miRNAs, we have developed a “dual-targeting Tough Decoy” carrying two different target sites (Figure 2, left). Using this approach, we achieved potent inhibition of the two miRNAs comparable to that obtained with Tough Decoys targeting a single miRNA.34 Since the activity is comparable for dual-targeting and standard Tough Decoy inhibitors one may wonder whether the Tough Decoy hairpin structure allows simultaneous miRNA binding at the two opposing target sites. In accordance, it is not currently known whether concomitant suppression of two unrelated miRNAs by dual-targeting Tough Decoys is achieved through binding of both miRNAs, or just one miRNA, to a single inhibitor molecule. If the Tough Decoy inhibitor binds only one miRNA, one of the two target sites of the Tough Decoy hairpin could be converted to a custom-designed sequence that minimizes base pairing with the target site to ensure optimal exposure. By combining the dual-targeting Tough Decoy with the clustered Tough Decoy approach, simultaneous inhibition of six unrelated miRNAs was achieved by three clustered dual-targeting Tough Decoys placed in the 3' UTR of eGFP (schematic representation shown in Figure 2, right). Expression of a multiplexed inhibitor from a single expression cassette ensures synchronized inhibition of several miRNAs, which may not easily be accomplished with a split design based on expression of six different inhibitors. A multiplexed design could have experimental and therapeutic potential for coordinated and parallel suppression of distinct miRNAs, as well as an entire miRNA family.
Figure 2.

Simultaneous managing of unrelated miRNAs by multitargeting inhibitors. The figure illustrates suppression of two out of a total of six endogenously expressed miRNAs by dual-targeting Tough Decoy inhibitors (left) and synchronized suppression of all six miRNAs by clustered dual-targeting Tough Decoy hairpins (right). As indicated by the Tough Decoy molecules bound by “yellow” and “green” miRNAs (left), it is yet unclear whether single Tough Decoy molecules are bound by two miRNAs or whether miRNAs interact with Tough Decoy inhibitors in a 1:1 ratio.
Suppression of miRNA By Nondecoy-Type Inhibitors
In addition to decoy-type inhibitors that bind and sequester the miRNA from their natural targets, several other approaches have been utilized for miRNA attenuation. Two studies have successfully employed an RNA interference approach by which short hairpin RNAs were expressed to target either mature miRNA or pre-miRNA for degradation.38,39 In our hands, however, short hairpin RNAs designed to target mature miR-16 and miR-203 were not functional.30 Concerns have previously been raised about the applicability of an RNA interference approach to target miRNAs due to the anticipated limited accessibility of the miRNA imposed by the hairpin structure or protection by RISC,7 and it has been shown that short hairpin RNA-loaded RISC is unable to unfold and target structured RNA.40
One perhaps undesired feature of the decoy-type inhibitors is that they repress the broad function of a miRNA; this means that the miRNA-suppressive effect is relieved on all mRNA targets. An approach for abolishing the interaction between a miRNA and a specific mRNA, without interfering with the action of the miRNA on its remaining pool of mRNA targets, has been explored by the use of a target mask.41,42 However, successful masking of the miRNA target site has only been achieved using chemically modified ~25-nucleotide long oligonucleotides, whereas vector-encoded RNA masks do not seem functional.30 This may be due to differences in RNA binding affinities between synthetic and transcribed RNA masks. It remains to be tested, however, if target site masking may be achieved using longer intracellularly transcribed RNAs.
Natural RNA Sponges Shape Future miRNA Inhibition Strategies
The basis of miRNA biology is founded on miRNAs being the active regulators of protein-coding mRNAs. However, a hypothesis has also been provided for the reciprocal notion, that a miRNA-targeted RNA is capable of titrating miRNAs, thereby reducing the number of free miRNA molecules that are available to repress other mRNA targets.43 Hence, a 3' UTR subject to miRNA regulation may not only regulate protein synthesis in cis, but may also work in a complex trans-regulatory network to regulate protein synthesis of mRNAs sharing the same miRNA target sites (reviewed in ref. 44). This type of regulation may involve pseudogenes that are able to regulate their parent protein-coding gene by competing for miRNAs. Similar mechanisms involve noncoding RNAs in plants and humans as well as in viruses. Such competing endogenous RNAs (ceRNAs) may sequester miRNAs and potentially derepress expression from the authentic miRNA target mRNAs. The potency of ceRNAs is likely to depend on the concentration of both the particular miRNA and the targeted mRNA, but may be affected also by the affinity of miRNA-loaded RISC for the miRNA binding site. Based on studies of the functional properties of miRNA binding to and dissociation from its target sequence, Wee and coworkers suggested that ceRNAs, even with highly complementary miRNA binding sites, may have limited effect on abundant miRNAs, whereas the function of intermediate and low abundance miRNAs is more likely to be affected by high-affinity sponges.45 It is proposed that such sponges may therefore only be functionally relevant to a smaller subset of miRNAs that meet certain criteria related to abundance and target affinity. Furthermore, quantifications of the number of miRNA and mRNA target molecules indicate that a miRNA is not irreversibly sequestered with its target, lending support to the notion that a substantial number of sponge molecules may be required to suppress miRNA activity.37 Hence, considerations related to each specific miRNA and the abundance of miRNA as well as target mRNA may have importance in relation to the design and use of decoy-inhibitors. It should be noted that a considerable number of potentially ceRNA-targeted cellular miRNAs may in fact be too scarcely expressed to have a biologically meaningful effect.15,45 In addition, some ceRNA-targeted miRNAs may have insignificant regulatory potential due to weak seed-pairing stability and/or high target site abundance.46 While still a matter of debate, further investigation of endogenous miRNA sponges is needed to characterize their functional importance in regulating miRNA activity.
Only recently, circular RNAs (circRNAs) have emerged as a new, highly prevalent, and conserved class of RNAs, which are derived from head-to-tail splicing of exons.47,48 Many of these circRNAs contain putative miRNA target sites and may therefore function as ceRNAs.47,49 One such human circRNA, circular RNA sponge for miR-7 (ciRS-7; also referred to as CDR1as), was found to harbor approximately 70 seed matches to miR-7 and to serve as a potent miR-7 decoy.47,48 CiRS-7 is highly expressed in specific regions of the brain and is suggested to play a key role in regulating miR-7 activity in neurons. Similarly, sex-determing region Y (Sry) circRNA that is produced mainly in the testes serves as a sponge for miR-138.47 None of the miR-7 target sites contained in ciRS-7 displayed sufficient complementarity to miR-7 to promote cleavage, and ciRS-7 was therefore insensitive to miRNA-mediated activation of exonucleolytic degradation. Hence, due to its high stability conferred by the circular structure combined with imperfect miRNA target sites, ciRS-7 appears as an evolutionarily optimized inhibitor of miRNA activity.
With the discovery of naturally occurring circular RNA sponges and their involvement in miRNA management, the focus on circular RNAs and their role as miRNA inhibitors will intensify. Circles may prove to be well-suited carriers of decoy-type miRNA-inhibitors, and the community will aspire to combine circles with high-affinity miRNA inhibitors. Hansen and co-workers established an expression cassette for production of circRNA and demonstrated potent circRNA production in cells transfected with ciRS-7-encoding plasmid DNA.47 As an alternative to linear sponges (Figure 3a), such “vectorized” RNA circles may represent new solid ways to deliver stable and persistent miRNA inhibitors (Figure 3b), perhaps even with therapeutic applications. It seems likely that an adaptation to a circularized design would render vector-encoded Sponge or Tough Decoy inhibitors more potent, but this remains to be tested. At this stage, we can only speculate that a new generation of intracellularly expressed inhibitors that combine the stability of circRNA with the high affinity of clustered Tough Decoy hairpins (Figure 3c) may suppress miRNA activity to an unprecedented level.
Figure 3.
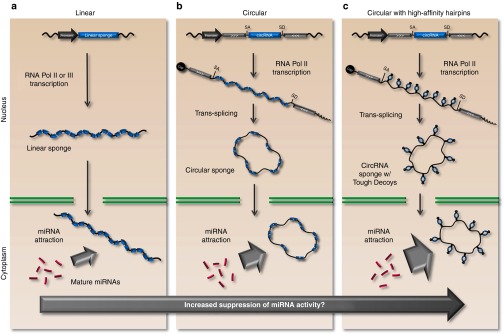
Natural circular RNA sponges inspire next-generation miRNA inhibitors. (a) Current vector-encoded miRNA inhibition technologies rely on linear RNA decoys, such as the Bulged Sponge or Tough Decoy designs. (b) Naturally occurring circular RNA sponges contain numerous miRNA seed matches thereby avoiding endonucleolytic cleavage upon miRNA recognition. Furthermore, high stability is conferred by the circular structure which evades miRNA-promoted decay by exonucleolytic degradation. (c) Circular RNA sponges may be optimized with high-affinity hairpins (such as those in the Tough Decoy) to further increase suppression of miRNA activity.
A Future Perspective
Inhibition of miRNAs by synthetic RNA molecules is already widely used to study miRNA biology in vitro, but like other small RNA-based drugs (e.g. siRNAs) such molecules face multiple delivery challenges such as lack of targeted delivery. In Hepatitis C virus (HCV) infection, miR-122 serves as an important host factor for virus replication and therefore represents a potential therapeutic target.50 miR-122 binds two adjacent target sites in the 5' UTR of the HCV genome where it acts in a noncanonical manner to facilitate positive regulation of viral replication through a so far unknown mechanism.50 The first clinical trials treating HCV with synthetic miRNA inhibitors based on Locked Nucleic Acid chemistry with a phosphorothioate backbone are already in phase 2a.9 Two key features favor a therapeutic strategy targeting miR-122 in liver using these inhibitors. First, small oligonucleotides with a phosphorothioate backbone have been shown to accumulate in the liver eliminating the need for a tissue-specific delivery technology. Secondly, miR-122 is almost exclusively expressed in the liver, which minimizes the risk of side effects in irrelevant tissues. Preliminary efficacy and safety data from the clinical trials suggest that synthetic miR-122 inhibitors for the treatment of HCV may become the first-in-line anti-miRNA therapeutic and pave the way for increased focus and investments in miRNA-directed therapy.9 Since adeno-associated virus vectors can readily be targeted to the liver, an analogous approach employing vector-encoded miR-122 inhibitors may serve as a long-term and more cost-effective treatment for HCV. Potent inhibition of HCV replication has already been achieved in vitro by transducing cells with adenoviral vectors encoding Tough Decoy inhibitors targeting miR-122.18 As methods of miRNA suppression are optimized—for example by inspiration from naturally occurring miRNA sponges—and the complexity of miRNA biology (not least the endogenous managing of miRNA activity) is further unraveled, vector-encoded miRNA inhibitors should soon move much closer to clinical applicability.
Acknowledgments
Studies of miRNA management in the laboratory of JGM has been carried out through support from the Lundbeck Foundation, the Novo Nordisk Foundation, Aase og Ejnar Danielsens Fond, Agnes og Poul Friis Fond, Kgl. Hofbuntmager Aage Bangs Fond, Grosserer A. V. Lykfeldt og Hustrus Legat, Else og Mogens Wedell-Wedellsborgs Fond, Fonden af 17-12-1981, Kong Christian den Tiendes Fond, Civilingeniør Frode V. Nyegaard og hustrus Fond, Clara Hansens Mindelegat, Kirsten Anthonius Mindelegat, Snedkermester Sophus Jacobsen & Hustru Astrid Jacobsens Fond og Frits, Georg og Marie Cecilie Gluds Legat. JGM is a member of the Aarhus Research Center for Innate Immunology (ARCII) established through funding by the AU-Ideas program at Aarhus University. ROB is the recipient of a PhD fellowship from the Faculty of Health Sciences at Aarhus University funded by the Lundbeck Foundation and Aarhus University. The authors declared no conflict of interest.
References
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Ellwanger DC, Büttner FA, Mewes HW, Stümpflen V. The sufficient minimal set of miRNA seed types. Bioinformatics. 2011;27:1346–1350. doi: 10.1093/bioinformatics/btr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YP, Haasnoot J, ter Brake O, Berkhout B, Konstantinova P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008;36:2811–2824. doi: 10.1093/nar/gkn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs'. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Kay MA, He CY, Chen ZY. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28:1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner B, Schira G, Giustacchini A, Amendola M, Brown BD, Ponzoni M, et al. Stable knockdown of microRNA in vivo by lentiviral vectors. Nat Methods. 2009;6:63–66. doi: 10.1038/nmeth.1277. [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, et al. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loya CM, Lu CS, Van Vactor D, Fulga TA. Transgenic microRNA inhibition with spatiotemporal specificity in intact organisms. Nat Methods. 2009;6:897–903. doi: 10.1038/nmeth.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon- Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- Sakurai F, Furukawa N, Higuchi M, Okamoto S, Ono K, Yoshida T, et al. Suppression of hepatitis C virus replicon by adenovirus vector-mediated expression of tough decoy RNA against miR-122a. Virus Res. 2012;165:214–218. doi: 10.1016/j.virusres.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Park CY, Choi YS, McManus MT. Analysis of microRNA knockouts in mice. Hum Mol Genet. 2010;19 R2:R169–R175. doi: 10.1093/hmg/ddq367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser HM, Koike-Yusa H, Cooper JD, Law FC, Bradley A. A resource of vectors and ES cells for targeted deletion of microRNAs in mice. Nat Biotechnol. 2011;29:840–845. doi: 10.1038/nbt.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Jeker LT, Carver-Moore K, Oh A, Liu HJ, Cameron R, et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep. 2012;1:385–391. doi: 10.1016/j.celrep.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, et al. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell. 2005;9:403–414. doi: 10.1016/j.devcel.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Sun W, Okano K, Chen Y, Zhang N, Maeda T, et al. Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J Biol Chem. 2011;286:31749–31760. doi: 10.1074/jbc.M111.259028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39:1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- Tan H, Poidevin M, Li H, Chen D, Jin P. MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2012;8:e1002681. doi: 10.1371/journal.pgen.1002681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, et al. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Otaegi G, Pollock A, Sun T. An Optimized Sponge for microRNA miR-9 Affects Spinal Motor Neuron Development in vivo. Front Neurosci. 2011;5:146. doi: 10.3389/fnins.2011.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Telesnitsky A. Frequency of direct repeat deletion in a human immunodeficiency virus type 1 vector during reverse transcription in human cells. Virology. 2001;286:475–482. doi: 10.1006/viro.2001.1025. [DOI] [PubMed] [Google Scholar]
- Bak RO, Hollensen AK, Primo MN, Sørensen CD, Mikkelsen JG. Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy' inhibitors. RNA. 2013;19:280–293. doi: 10.1261/rna.034850.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37:e43. doi: 10.1093/nar/gkp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollensen AK, Bak RO, Haslund D, Mikkelsen JG. Suppression of microRNAs by dual-targeting and clustered Tough Decoy inhibitors. RNA Biol. 2013;10:406–414. doi: 10.4161/rna.23543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Nakano H, Tagawa T, Ohki T, Ueno Y, Yoshida T, et al. A potent 2'-O-methylated RNA-based microRNA inhibitor with unique secondary structures. Nucleic Acids Res. 2012;40:e58. doi: 10.1093/nar/gkr1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, et al. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, Brown BD. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr Biol. 2011;21:369–376. doi: 10.1016/j.cub.2011.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Han L, Zhang A, Wang G, Jia Z, Yang Y, et al. Adenovirus-mediated shRNAs for co-repression of miR-221 and miR-222 expression and function in glioblastoma cells. Oncol Rep. 2011;25:97–105. [PubMed] [Google Scholar]
- Druz A, Son YJ, Betenbaugh M, Shiloach J. Stable inhibition of mmu-miR-466h-5p improves apoptosis resistance and protein production in CHO cells. Metab Eng. 2013;16:87–94. doi: 10.1016/j.ymben.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130:101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Wang Z. The principles of MiRNA-masking antisense oligonucleotides technology. Methods Mol Biol. 2011;676:43–49. doi: 10.1007/978-1-60761-863-8_3. [DOI] [PubMed] [Google Scholar]
- Seitz H. Redefining microRNA targets. Curr Biol. 2009;19:870–873. doi: 10.1016/j.cub.2009.03.059. [DOI] [PubMed] [Google Scholar]
- Almeida MI, Reis RM, Calin GA. Decoy activity through microRNAs: the therapeutic implications. Expert Opin Biol Ther. 2012;12:1153–1159. doi: 10.1517/14712598.2012.693470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, Zamore PD. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012;151:1055–1067. doi: 10.1016/j.cell.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]