

Abstract
Background & objectives:
Duchenne and Becker muscular dystrophies are X-linked allelic disorders which are caused by mutations in the DMD gene. Carrier analysis in DMD is complicated due to the heterozygous nature of the X chromosome. Several techniques have been tried for carrier analysis in families where the mutation is identified including quantitative multiplex PCR (qmPCR), Southern blot, and now multiplex ligation-dependent probe amplification (MLPA). Linkage analysis is used in cases without identifiable mutations. The present study was undertaken to determine the status of probable carriers in families where the DMD deletion/duplication has been identified for the affected index cases.
Methods:
Carrier status was present in 150 probable carriers from 110 apparently unrelated families where the patients’ mutations were known. Of these 110 families, 100 were deletions, 9 duplications and 1 point mutation. Multiplex ligation-dependent probe amplification (MLPA) was used to assess the copy number changes and direct sequencing was used for the case with the point mutation.
Results:
Of the 150 cases, 49 were found to be carriers. Among the sporadic cases, it was observed that the rate of de novo mutations was very high (71%) as compared to the hereditary cases (29%), which was higher than the calculated rate (30%). It was observed that this difference was more apparent in deletion mutations than in duplications.
Interpretation & conclusions:
Identifying the DMD carrier rates in the families with unidentified deletions and duplications and where the causative mutation could be small insertions/deletions or point mutations could throw more light into this observation. MLPA was found to be useful in detecting copy number changes in DMD carriers and this could be the method of choice for DMD carrier analysis, when the mutation is detected in the affected child.
Keywords: Carrier diagnosis, de novo mutations, Duchenne muscular dystrophy, multiplex ligation-dependent probe amplification (MLPA)
Majority of genetic disorders place a considerable burden on the families perpetuating the condition for the lack of effective treatment. Duchenne/Becker muscular dystrophies (D/BMD) are such lethal disorders caused by mutations in the dystrophin gene. DMD is a common paediatric neuromuscular disorder affecting 1/3500 live male births, while BMD is milder and less frequent1. The diseases are manifested with muscular weakness, hypertrophy of the calf muscles, positive Gower's sign and Pradhan's valley sign2. Due to the X-linked nature of the disorder, males carrying the mutated gene are affected, while females become carriers of the disease. Diagnosis of patients with D/BMD is usually definitive based on clinical, pathological and biochemical findings, although it is increasingly being confirmed by molecular and genetic analysis.
The first essential step in genetic counselling is to verify the diagnosis in the index case. Next, a detailed family tree is to be constructed before investigation of the possible carrier is begun. Practically, if the mother of an affected boy (proband) has another affected relative, she must be an obligate carrier. If there is an affected brother or at least one affected son, she is a possible carrier3. But in most families there is only one affected patient. Therefore, maternal female relatives of affected males are candidates for carrier assessment.
Establishing the carrier status in X-linked recessive disorders is one of the basic dilemmas in genetic counselling because female carriers are usually asymptomatic. Several biochemical and molecular methods have been used4. Essentially, in families where the proband deletion or duplication is known, dosage testing for the deleted/duplicated exons is the recommended method. For all other cases where the causative mutations could be small changes like small deletions, insertions and point mutations, gene sequencing can be attempted, though linkage analysis can be applied with a certain amount of accuracy in these families.
In this study, the carrier status of probable carriers was examined in families where the DMD gene deletion/duplication was identified for the affected index case. In addition, the usefulness of multiplex ligation-dependent probe amplification (MLPA) in carrier analysis was also studied, while also validating and comparing MLPA results with results obtained from other commonly used methods.
Material & Methods
Patients and samples: The carrier status of female relatives including mother, maternal grandmother, maternal aunt, and sisters of affected males whose DNA analysis results were available and confirmed to have Duchenne / Becker muscular dystrophy was analysed. All cases were referred by the clinician from ICH&HC, Egmore, Chennai, a Government specialty pediatric hospital to Molecular Diagnostic Facility at Sundaram Medical Foundation, Chennai, for carrier diagnosis between May 2007 and July 2009. The study protocol was approved by the institutional ethical committee of Sundaram Medical Foundation. A total of 150 probable carriers were analysed from 110 apparently unrelated families. Molecular analyses done to confirm diagnosis in probands were multiplex PCR and MLPA. For one of the cases (B28), DMD gene mutation was confirmed by direct sequencing elsewhere and was found to have a frameshift point mutation (c.7348dupG) in exon 51. Carrier analysis was attempted for possible carriers in each of the families by using MLPA. Linkage by STR-(CA) segregation analysis and quantitative multiplex fluorescence PCR (qmfPCR) were used to validate MLPA results on 33 samples.
Blood samples (3 ml) in EDTA were collected from female relatives suspected to be probable carriers for genetic analysis after obtaining informed consent. DNA was extracted by salting out method5, quantified and stored at -20C until tested. Multiplex PCR analysis was performed for 30 exons at the central and 5’end hot spot regions as already reported6.
Quantitative mPCR: Quantitatiave multiplex PCR7 was standardized with the same conditions used for mPCR, except that the starting DNA quantity used was 250 μg/ml for all samples. Samples from male and female controls were amplified with the test samples and 12 μl of the PCR product was resolved in 2 per cent agarose gel. The resulting band was viewed in the Gel documentation system (Biorad, USA). The band intensities were analysed using the Quantity One software (Biorad, USA) and the adjusted band volumes of the test samples were compared with that of male and female controls. The results were interpreted only if the male control band volumes were half of that of the female controls.
Quantitative multiplex fluorescence PCR (qmfPCR): QmfPCR was performed for 51 exons as described by Yau et al8. Two fluorescently labelled multiplex PCR assays were done to amplify 21 exons (1, 2, 3, 4, 5, 6, 7, 8, 9, 11, 12, 13, 17, 19, 21, 24, 25, 29, 30, 32 and 37) from the proximal deletion hotspot of the dystrophin gene (5’ assay) and 22 exons (42, 43, 44, 45, 46, 47, 48, 49, 51, 52, 53, 54, 55, 56, 58, 59, 60, 62, 63, 68, 71 and 75) from the central deletion hotspot (3’ assay). All forward primers in the assays were labelled with either fluorescent phosphoramidite fluorescein amidite 6-FAM (5’ assay) or HEX hexachlorofluorescein (3’ assay) (Applied Biosystems, USA). Primer sequences were obtained from Leiden Muscular Dystrophy, http://www.dmd.nl, accessed January 20089.
The peak areas were then entered into the excel sheets and the dosage quotient was calculated and interpreted for deletions and duplications8.
Multiplex ligation-dependent probe amplification (MLPA): MLPA analysis was carried out using PO34 and PO35 probes purchased commercially from MRC, Holland (Amsterdam, Netherlands). The procedures and analysis was carried out as published earlier6.
STR-(CA) segregation analysis: STR-(CA) segregation analyses were performed for 11 markers spanning the DMD gene, 19n8, 3’m, 1671, 1623, i50, i49, i45, i44, 7n4, 5n4, and 5n3, according to the method described in Carsana et al10. using previously reported primer oligonucleotides (Leiden Muscular Dystrophy, http://www.dmd.nl, accessed January 2008)9. The forward primers were labelled with 5-carboxyfluorescein (FAM), PET, NED, or VIC fluorochromes. PCR products (0.5 μl from each multiplex reaction) were mixed with 0.5 μl of Gene-Scan-500 LIZ size standard (Applied Biosystems, USA) and were separated by capillary gel electrophoresis (15 kV at 60°C for 30 min) on the ABI Prism 3130 Genetic Analyzer (Applied Biosystems) using the POP-7 polymer. The Genemapper 3.7 (Applied Biosystems) software was used for data analysis and creating a macro that allowed to labell the peaks and identify the alleles of each marker automatically.
Results
A total of 150 cases from 110 families were tested for carrier status in this study. Of the proband mutations, 100 were single or multiple exon deletions, 9 were duplications and one point mutation. Family history (more than one affected member in the family) was seen in 16 families.
Validation of MLPA results: MLPA results of 33 probable carriers from 23 families whose index cases showed DMD gene deletions or duplications were validated by STR-(CA) segregation analysis and qmfPCR (Table I). STR-(CA) segregation analysis was done for 32 of the 33 cases in the study. It was seen that it was helpful in assessing the carrier status in most of the cases when it was combined with one of the direct methods. STR-(CA) segregation analysis was non-informative in six cases (17.6%). Many of the markers were homozygous for most of the cases suggesting the need to use alternative markers designed specifically for the Indian population. A representation of STR-(CA) segregation analysis results in one of the families is shown in Fig. 1.
Table I.
Validation of MLPA results using CA analysis and qmfPCR
Fig. 1.
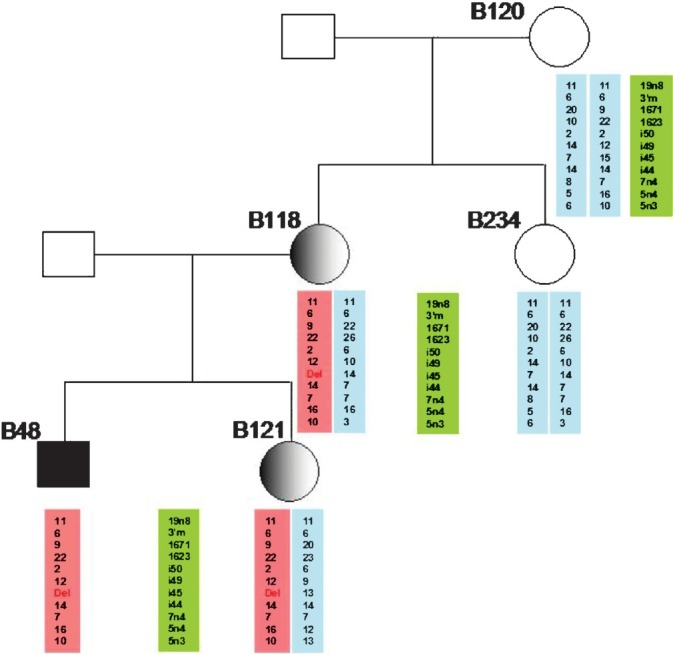
Figure showing the use of STR-(CA) segregation repeat analysis for carrier detection in a family with DMD. CA-repeat microstatellite markers flanking the DMD gene locus were used for haplotyping. The affected polymorphic allele showing the deletion of intron 45 is shown in pink. In this family the proband showed exon 45 deletion of the DMD gene. It can be observed that the affected haplotype (in pink) from the mother being transmitted to both the index case his female sibling.
QmfPCR analysis was done for all the 33 cases and the results were the same as that of MLPA.
Carrier analysis results: MLPA analysis on probable carriers revealed an average Dosage Quotients of 0.51 (range 0.34 to 0.61) for deletion and 1.46 (range 1.34 to 1.62) for duplication. Table II shows the consolidated results and carrier status of the probable carriers. Of the 150 cases, 105 were mothers, 37 sisters, six maternal aunts and two maternal grandmothers. Among the 110 families tested, 16 showed family history, 93 were sporadic cases and for one case the family history details were not available. Forty nine cases (32.7%) were tested positive for carrier status, of whom 42 were mothers (40% of all mothers) and seven were sisters of the index cases. Of the 49 cases, carrier status of 48 was confirmed by MLPA and one of the cases by direct sequencing (Fig. 2). MLPA results of one representative family each with deletion and duplication are shown in Fig. 3.
Table II.
Consolidated results of all familial and sporadic cases tested in the study

Fig. 2.
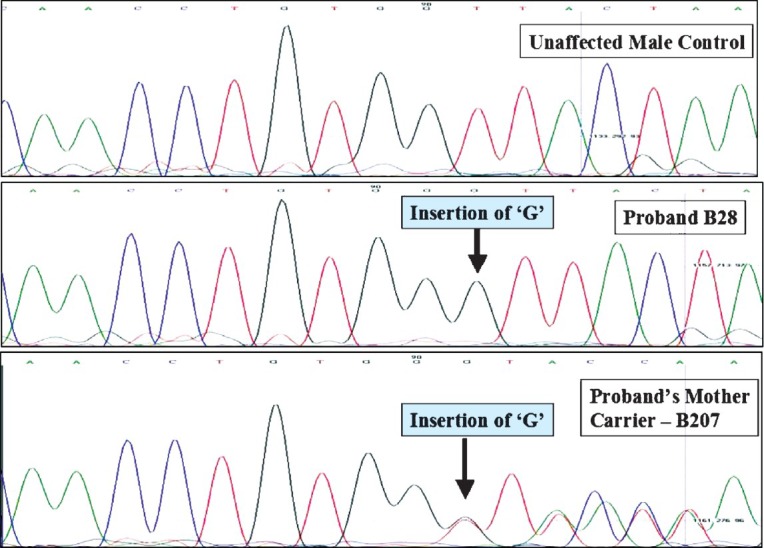
Sequence analysis of the DMD gene in a control sample, proband and his mother showing point mutation in exon 51 and the corresponding mutation in the mother causing a frameshift in one of the alleles.
Fig. 3.

A representative figure showing MLPA results in a normal control, proband and his carrier mother for probes PO34 and PO35. The x-axis displays the exons and the DQ is in the y-axis. Deletion can be seen as a reduction in DQ to around a value of 0.5 and in duplication DQ can be seen more than 1.5.
All mothers who were obligate carriers by pedigree history were tested positive for carrier status. Among the 110 families, mother's samples were not available for five and only sisters’ samples were available in these cases. Of these five families, four sisters were carriers. The family where the sisters were non-carriers was not included while calculating the number of hereditary cases in the study. Therefore, of the 93 cases with sporadic forms, 27 families (29%) were tested positive for carrier status and the remaining 66 (71%) were tested negative and hence could be a de novo mutation in the index case or germline mosaicism in the mother.
Seven sisters to index cases (of the 37 tested) were positive for carrier status. Of these seven, two belonged to families where there was a family history of the disorder.
Inheritance of single and multi exon deletions was observed and no difference was seen among them when the complete data were taken into account. However, when only the mothers were taken into account, multi exon deletions showed 12% more inheritance than the single exon deletions (Table III).
Table III.
Hereditary nature of single and multi exon deletions

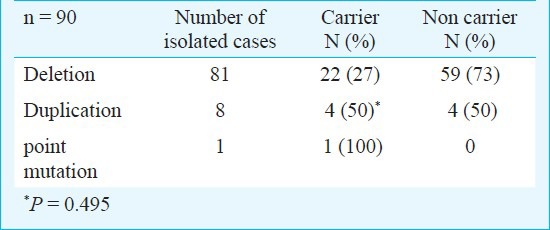
Among the isolated cases, the carrier rates of deletion mutations (27%) are not significantly different from that of duplications (50%). This may be due to the low numbers of duplication cases observed in the study and the results may show significant difference with more cases with duplication mutations added to the study (Table IV).
Table IV.
Difference in carrier rates between deletion and duplication mutations among isolated cases of DMD
Discussion
The identification of female carriers of deletions/duplications of the DMD gene is a crucial point to prevent the birth of children affected by DMD or BMD. Since, theoretically, about 30 per cent of the DMD mutations are de novo, the risk of recurrence of the disease in families with a single affected male is related to the carrier or non-carrier status of the mother of the patient3. Although several approaches for the identification of female carriers are available, many of these cannot be readily used for routine diagnostics. In diagnostic laboratories, DMD carrier testing has been principally based on linkage studies, initially using restriction fragment length polymorphisms detected by Southern blot analysis11, and subsequently by PCR amplification of short tandem repeat (STR) loci12. Linkage studies can be used to identify deletion mutation carriers on the basis of the presence of an informative STR locus within the deletion interval, and the demonstration of either heterozygosity or apparent non-Mendelian inheritance, but are frequently compromised by the nonavailability of DNA samples of other members of the family, the distribution and informativeness of STRs, and the possibility of gonadal mosaicism. Hence, to resolve such cases, adjunct techniques such as fluorescence in situ hybridisation and pulsed field gel electrophoresis have been used13,14,15.
The use of MLPA for DMD carrier detection in families where the proband mutation is a deletion/duplication has been well documented16. There are a few studies in India assessing the carrier status in DMD families using STR-(CA) segregation repeat analysis and quantitative multiplex PCR17,18,19,20. In this study, we investigated the usefulness of the MLPA approach for the detection of female carriers of deletions/duplications of the DMD gene.
The results showed MLPA analysis as a powerful tool for the detection of female carriers in families with a DMD- or BMD-affected male, with known DMD gene deletion/duplication. This technique was able to pick up carrier status in cases that could not be confirmed by STR-(CA) segregation repeat analysis. Compared to qmfPCR, this technique is easier to perform with only two reactions per sample and studying all the 79 exons of the DMD gene, as opposed to the 51 exons studies by qmfPCR. It was also observed that MLPA cannot be used to pick up point mutations and direct sequencing or other point mutation detection methods are required to detect carriers in families where index case mutation is a point mutation1.
Current genetic counselling practice is to cite a maternal carrier risk of two-thirds for the mother of an isolated case of DMD2. This risk for an X linked disorder with early lethality assumes that there is equilibrium between mutation and selection, the mutation rates for all mutational classes observed to cause DMD are the same in the ova and the sperm, and carrier women have the same reproductive fitness as non-carrier women21. Several studies have shown that the observed carrier frequency among the mothers of isolated cases is much lower than the expected theoretical value19,20,22,23. In our study, it was observed that carrier frequency among the isolated cases was only 29 per cent and de novo mutations accounted for the rest of the 71 per cent. Therefore it can be seen that the occurrence of de novo mutations among sporadic cases of DMD was high compared to the theoretical data (30%). Our results were consistent with other studies from India which also showed a high percentage of de novo mutations among DMD cases19,20. The studies from India and other earlier studies showed a low carrier frequency mainly for deletion mutations13,15,16,17,18,19,20,21,22. It has been seen that the decreased carrier rates are restricted to deletion mutations and the likelihood of being a carrier for the other classes of mutation being in the theoretical range of 55-63 per cent21. A similar trend was observed in our study that in deletion mutations the carrier rates were 27 per cent as compared to the 50 per cent among the mothers of isolated cases with duplication mutations, which could be due to the less number of duplication cases. The mother of the isolated case with point mutation was also a carrier. Hence, the carrier risk calculations for the mother of an isolated case of DMD are not valid for deletion mutations, but appear to be valid for other mutational classes and suggest a basic biological difference in the effect of a deletion mutation compared with a non-deletion mutation21. Or it can be related to the viability of the gametes with deletion mutations as compared to the other classes of mutations21. It can also be predicted that the carrier rate in isolated cases in our study would have increased had we included the other cases who did not show a deletion or duplication mutation.
Mutation detection protocols using mPCR followed by MLPA were able to pick up mutations in 75 per cent of DMD probands. These demonstrate the importance of mutation detection protocols. Recently, MLPA assay has been suggested as the first screening test for clinically suspected DMD/BMD patients as well as for women who have a DMD/BMD family history. However, according to the best-practice guidelines for medical genetics laboratories, it is recommended that at least two independent alternative methods are available for confirmation of each genotype24.
In conclusion, the MLPA approach seems to be a simple, rapid and reliable tool for the screening of the carrier status in cases where index case mutations are deletions or duplications of the DMD gene. This avoids unnecessary invasive prenatal tests with the inherent risk of miscarriage. Prenatal testing is still recommended for non-carrier mothers of an isolated male proband due to the residual chance of gonadal mosaicism. MLPA analysis could represent a method of first choice for the detection of disease-causing deletions/duplications in female relatives of affected males, especially in those cases who cannot be investigated by other approaches.
Acknowledgment
Authors acknowledge Stitching Porticus Grant, (Country?) for supporting the study, and also M.S. Swaminathan Research Foundation, Chennai, India for allowing to use the Genetic Analyser at the Centre. Authorse thank Dr Chandramohan, ICH&HC, Egmore, Chennai, and all the other clinicians who referred patients to us for testing.26
References
- 1.Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA) Hum Genet. 2005;117:92–8. doi: 10.1007/s00439-005-1270-7. [DOI] [PubMed] [Google Scholar]
- 2.Emery AE. Population frequencies of inherited neuromuscular diseases - a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 3.Dubowitz V. The female carrier of Duchenne muscular dystrophy. BMJ. 1982;284:1423–4. doi: 10.1136/bmj.284.6327.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panigrahi I, Mittal B. Carrier detection and prenatal diagnosis in Duchenne/Becker muscular dystrophy. Indian Pediatr. 2001;38:631–9. [PubMed] [Google Scholar]
- 5.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murugan S, Chandramohan A, Lakshmi BR. Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis. Indian J Med Res. 2010;132:303–11. [PubMed] [Google Scholar]
- 7.Kumar D, Mittal A, Gupta M, Goyle S. Deletion analysis of the dystrophin gene in Duchenne and Becker muscular dystrophy patients: use in carrier diagnosis. Neurol India. 2003;51:223–6. [PubMed] [Google Scholar]
- 8.Yau SC, Bobrow M, Mathew CG, Abbs SJ. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J Med Genet. 1996;33:550–8. doi: 10.1136/jmg.33.7.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. [accessed on January 23, 2008]. Available from: http://www.dmd.nl .
- 10.Carsana A, Frisso G, Tremolaterra MR, Ricci E, De Rasmo D, Salvatore F. A larger spectrum of intragenic short tandem repeats improves linkage analysis and localization of intragenic recombination detection in the dystrophin gene: an analysis of 93 families from southern Italy. J Mol Diagn. 2007;9:64–9. doi: 10.2353/jmoldx.2007.060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakker E, Bonten EJ, De Lange LF, Veenema H, Majoor-Krakauer D, Hofker MH, et al. DNA probe analysis for carrier detection and prenatal diagnosis of Duchenne muscular dystrophy: a standard diagnostic procedure. J Med Genet. 1986;23:573–80. doi: 10.1136/jmg.23.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemens PR, Fenwick RG, Chamberlain JS, Gibbs RA, de Andrade M, Chakraborty R, et al. Carrier detection and prenatal diagnosis in Duchenne and Becker muscular dystrophy families, using dinucleotide repeat polymorphisms. Am J Hum Genet. 1991;49:951–60. [PMC free article] [PubMed] [Google Scholar]
- 13.Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. 1989;45:835–47. [PMC free article] [PubMed] [Google Scholar]
- 14.Ligon AH, Kashork CD, Richards CS, Shaffer LG. Identification of female carriers for Duchenne and Becker muscular dystrophies using a FISH-based approach. Eur J Hum Genet. 2000;8:293–8. doi: 10.1038/sj.ejhg.5200450. [DOI] [PubMed] [Google Scholar]
- 15.Voskova-Goldman A, Peier A, Caskey CT, Richards CS, Shaffer LG. DMD-specific FISH probes are diagnostically useful in the detection of female carriers of DMD gene deletions. Neurology. 1997;48:1633–8. doi: 10.1212/wnl.48.6.1633. [DOI] [PubMed] [Google Scholar]
- 16.Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics. 2005;6:29–35. doi: 10.1007/s10048-004-0204-1. [DOI] [PubMed] [Google Scholar]
- 17.Basak J, Dasgupta UB, Mukherjee SC, Das SK, Senapati AK, Banerjee TK. Deletional mutations of dystrophin gene and carrier detection in eastern India. Indian J Pediatr. 2009;76:1007–12. doi: 10.1007/s12098-009-0214-y. [DOI] [PubMed] [Google Scholar]
- 18.Kumari D, Mital A, Gupta M, Goyle S. Deletion analysis of the dystrophin gene in Duchenne and Becker muscular dystrophy patients: use in carrier diagnosis. Neurol India. 2003;51:223–6. [PubMed] [Google Scholar]
- 19.Mukherjee M, Chaturvedi LS, Srivastava S, Mittal RD, Mittal B. De novo mutations in sporadic deletional Duchenne muscular dystrophy (DMD) cases. Exp Mol Med. 2003;35:113–7. doi: 10.1038/emm.2003.16. [DOI] [PubMed] [Google Scholar]
- 20.Sinha S, Mishra S, Singh V, Mittal RD, Mittal B. High frequency of new mutations in North Indian Duchenne/Becker muscular dystrophy patients. Clin Genet. 1996;50:327–31. doi: 10.1111/j.1399-0004.1996.tb02383.x. [DOI] [PubMed] [Google Scholar]
- 21.Taylor PJ, Maroulis S, Mullan GL, Pedersen RL, Baumli A, Elakis G, et al. Measurement of the clinical utility of a combined mutation detection protocol in carriers of Duchenne and Becker muscular dystrophy. J Med Genet. 2007;44:368–72. doi: 10.1136/jmg.2006.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alcantara MA, Villarreal MT, Del Castillo V, Gutierrez G, Saldana Y, Maulen I, et al. High frequency of de novo deletions in Mexican Duchenne and Becker muscular dystrophy patients. Implications for genetic counseling. Clin Genet. 1999;55:376–80. doi: 10.1034/j.1399-0004.1999.550514.x. [DOI] [PubMed] [Google Scholar]
- 23.Bakker E, Veenema H, Den Dunnen JT, van Broeckhoven C, Grootscholten PM, Bonten EJ, et al. Germinal mosaicism increases the recurrence risk for ‘new’ Duchenne muscular dystrophy mutations. J Med Genet. 1989;26:553–9. doi: 10.1136/jmg.26.9.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR. Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul Disord. 2010;20:422–7. doi: 10.1016/j.nmd.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Chaturvedi LS, Mittal RD, Srivastava S, Mukherjee M, Mittal B. Analysis of dinucleotide repeat loci of dystrophin gene for carrier detection, germline mosaicism and de novo mutations in Duchenne muscular dystrophy. Clin Genet. 2000;58:234–6. doi: 10.1034/j.1399-0004.2000.580312.x. [DOI] [PubMed] [Google Scholar]
- 26.Piko H, Vancso V, Nagy B, Ban Z, Herczegfalvi A, Karcagi V. Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families - detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord. 2009;19:108–12. doi: 10.1016/j.nmd.2008.10.011. [DOI] [PubMed] [Google Scholar]