

Abstract
Dengue virus is responsible for ≈50–100 million infections, resulting in nearly 24,000 deaths annually. The capsid (C) protein of dengue virus is essential for specific encapsidation of the RNA genome, but little structural information on the C protein is available. We report the solution structure of the 200-residue homodimer of dengue 2 C protein. The structure provides, to our knowledge, the first 3D picture of a flavivirus C protein and identifies a fold that includes a large dimerization surface contributed by two pairs of helices, one of which has characteristics of a coiled-coil. NMR structure determination involved a secondary structure sorting approach to facilitate assignment of the intersubunit nuclear Overhauser effect interactions. The dimer of dengue C protein has an unusually high net charge, and the structure reveals an asymmetric distribution of basic residues over the surface of the protein. Nearly half of the basic residues lie along one face of the dimer. In contrast, the conserved hydrophobic region forms an extensive apolar surface at a dimer interface on the opposite side of the molecule. We propose a model for the interaction of dengue C protein with RNA and the viral membrane that is based on the asymmetric charge distribution of the protein and is consistent with previously reported results.
Dengue virus, a member of the flavivirus genus of enveloped RNA viruses, is one of the most significant mosquito-borne viral pathogens, given the impact of the recent resurgence of dengue fever and dengue hemorrhagic fever (1). Other members of the flavivirus genus are also important human pathogens and include such viruses as yellow fever, West Nile virus, and Japanese encephalitis (2). The structure of dengue virus was recently described by using cryo-electron microscopy, which permitted visualization of certain viral protein components (3). The organization of the major envelope glycoprotein (E) in the virion was obtained by fitting the atomic structure of the ectodomain from the related tick-borne encephalitis (TBE) E protein into the outermost layer of density (4). Density internal to the E ectodomain demonstrated a host-derived lipid bilayer with transmembrane helices from E and the small structural protein M (5). Interior to the bilayer is the nucleocapsid core that comprises multiple copies of the capsid, or core, protein (C) and a single 10.7-kb genome RNA. Surprisingly, the organization of the C protein within the nucleocapsid core layer is not discernable. The lack of strong density for C may suggest a rather unique architecture of the flavivirus nucleocapsid core that is distinct from the structural organization of morphologically related viruses such as alphaviruses, a genus in the Togaviridae family of mosquito-borne viruses.
The dengue C protein is essential in virus assembly to ensure specific encapsidation of the viral genome. The critical role of C is evident from the existence of subviral particles that are released from infected cells but lack C protein and genome RNA (6). The mechanism by which encapsidation occurs is not understood but may require participation of nonstructural viral proteins as well as C protein (7). The mature form of dengue 2 C protein (DEN2C) is a highly basic protein of 12 kDa after removal of the C-terminal hydrophobic signal sequence by the virally encoded NS2B-NS3 protease before virus assembly (8, 9). Although the structural organization of DEN2C in the virus particle is unknown, DEN2C likely interacts with RNA and the lipid bilayer. We recently reported the secondary structure assignment of DEN2C determined by NMR techniques and that DEN2C is a dimer in solution (10). Here, we report the solution structure of the DEN2C dimer, the first, to our knowledge, 3D structure of a flavivirus C protein. DEN2C is found to have an alternate protein fold. Other flavivirus C proteins are likely to have a similar fold given the sequence conservation among C proteins (Fig. 1). A model for interaction of DEN2C in the virion is suggested based on the unusual arrangement of charge revealed by the structure analysis.
Fig. 1.

A multiple sequence alignment of flavivirus C proteins reproduced from multalign (30). Residues with high similarity (>50%) are red, and the conserved residues are highlighted. The secondary structure is indicated at the top. Helices are V26-L35, K45-T58, A63-W69, and K74-N96. The conserved hydrophobic region of flaviviruses is shaded gray. DEN2, dengue type 2; DEN1, dengue type 1; DEN3, dengue type 3, DEN4, dengue type 4; KUN, Kunjin; WNV, West Nile virus; MVE, Murray Valley encephalitis; JEV, Japanese encephalitis; SLE, St. Louis encephalitis; YFV, yellow fever; TBE; LIV, louping ill; LAN, Langat; POW, Powassan virus.
Materials and Methods
Sample Preparation. The cDNA encoding the mature form of DEN2C (amino acids 1–100) from the PR-159S1 strain of DEN2 (11) was cloned into the pET30a (Novagen) expression plasmid. Expression of uniformly 15N/13C and 15N-labeled proteins were accomplished by growth in M9 minimal media containing 15NH4Cl (1 g/liter) with or without 13C6-glucose (2 g/liter, Spectra Stable Isotopes, Columbia MD), respectively. Purification of DEN2C was performed as described (10). The protein was dissolved in 90% H2O/10% D2O with 50 mM sodium phosphate and 200 mM NaCl, pH 6.0. NMR samples were prepared in Shigemi (Shigemi, Tokyo) tubes with DEN2C protein concentrations ≈0.8–2.0 mM in ≈300-μl solutions.
NMR Spectroscopy. All NMR spectra were acquired at 300 K (unless stated otherwise) on a Varian Inova 600 spectrometer equipped with 5-mm [1H, 15N, 13C] triple-resonance z axis pulsed-field gradient probes. Backbone assignments were made by using standard triple-resonance experiments 3D HNCACB, CBCA(CO)NH, C(CO)NH, HC(CO)NH, and 2D 15N heteronuclear single quantum coherence, 2D constant time 13C heteronuclear single quantum coherence recorded on uniformly 15N/13C-labeled DEN2C. Side-chain assignments were obtained from 3D HCCH-TOCSY spectrum. Aromatic resonances were assigned by using 2D hbCBcgcdHD or hbCBcgcdHDHE experiments (12). Proton–proton distance constraints were derived from 3D 13C-edited [1H,1H] NOESY for aliphatic regions (tmix = 150 ms) or aromatic regions (tmix = 100 ms), 3D 15N-edited [1H,1H] NOESY (tmix = 150 ms, 305 K), and 3D simultaneously 15N/13C-edited [1H,1H] NOESY (tmix = 100 ms) (13, 14). Restraints for φ torsion angles were derived from 3JHNHα measured from a 3D HNHA-J spectrum (15). NMR data were processed with nmrpipe (16) and were analyzed in sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Indirect dimensions were normally extended by linear prediction and were zero filled before Fourier transformation, and only spectral regions containing signals were retained.
Interpretation of NOESY Crosspeaks. Assignment of the NOESY crosspeaks with respect to either intramonomeric (monomer) or intermonomeric (dimer) interactions used a “secondary structure sorting” strategy, and an iterative procedure that involved defining the structure of the isolated monomer, followed by the structure of the dimer. To begin, the residue positions of the four α-helices were identified by both chemical shift indexing (17) and the nuclear Overhauser effect (NOE) patterns for 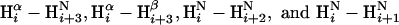 . The remaining parts were defined as loop regions. All observed NOE interactions were sorted according to the secondary structure element in which the NOE proton pair is located. For example, an NOE between residue 26 and residue 51 is classified as an α1-to-α2 interaction. Secondary structure sorting resulted in the groups α1 to α2, α1 to α3, α2 to α2, α2 to α3, α2 to α4, α3 to α4, α4 to α4, αi to loop, and loop to loop. A small number of crosspeaks with unique chemical shifts were immediately and unambiguously assigned as a dimer interaction because the two protons involved were positioned within secondary structure elements in a manner inconsistent with a monomer NOE interaction. Such dimer interactions, called (αα)D, were observed between protons near the C terminus of α4 and either the N terminus of α4 or the C terminus of α3, as well as between protons at the N- and the C termini of α2. The initial stage of the structure calculation was the definition of a crude monomer structure by using selected NOE restraints between two α-helices and excluding the known dimer interactions (αα)D. Our strategy was to identify a minimal set of α-to-α restraints to avoid potential erroneous inclusion of dimer interactions, yet sufficient to define the monomer core structure. The monomer structure calculations were reiterated with additions and editing of the restraint list of secondary structure-sorted restraints to convergence. A reasonable monomer structure was achieved with ≈1,200 restraints. The next phase was to dock two monomers by using the (αα)D interactions and rigid-body translation/rotation of the monomers. The docked structure was then conformationally relaxed by performing restrained simulated annealing on a fully flexible dimer with the minimal set of monomer restraints and the restraints defined by the (αα)D NOE interactions. Multiple docked orientations were used. Additional NOE crosspeaks were subsequently assigned in an iterative fashion as either monomer, dimer or both (ambiguous) interactions based on structural convergence. Chemical shift degeneracy was accounted for by using a restraint energy term that included degenerate NOE proton pairs as a summation of r-6 terms, i.e., the SUM option of x-plor.
. The remaining parts were defined as loop regions. All observed NOE interactions were sorted according to the secondary structure element in which the NOE proton pair is located. For example, an NOE between residue 26 and residue 51 is classified as an α1-to-α2 interaction. Secondary structure sorting resulted in the groups α1 to α2, α1 to α3, α2 to α2, α2 to α3, α2 to α4, α3 to α4, α4 to α4, αi to loop, and loop to loop. A small number of crosspeaks with unique chemical shifts were immediately and unambiguously assigned as a dimer interaction because the two protons involved were positioned within secondary structure elements in a manner inconsistent with a monomer NOE interaction. Such dimer interactions, called (αα)D, were observed between protons near the C terminus of α4 and either the N terminus of α4 or the C terminus of α3, as well as between protons at the N- and the C termini of α2. The initial stage of the structure calculation was the definition of a crude monomer structure by using selected NOE restraints between two α-helices and excluding the known dimer interactions (αα)D. Our strategy was to identify a minimal set of α-to-α restraints to avoid potential erroneous inclusion of dimer interactions, yet sufficient to define the monomer core structure. The monomer structure calculations were reiterated with additions and editing of the restraint list of secondary structure-sorted restraints to convergence. A reasonable monomer structure was achieved with ≈1,200 restraints. The next phase was to dock two monomers by using the (αα)D interactions and rigid-body translation/rotation of the monomers. The docked structure was then conformationally relaxed by performing restrained simulated annealing on a fully flexible dimer with the minimal set of monomer restraints and the restraints defined by the (αα)D NOE interactions. Multiple docked orientations were used. Additional NOE crosspeaks were subsequently assigned in an iterative fashion as either monomer, dimer or both (ambiguous) interactions based on structural convergence. Chemical shift degeneracy was accounted for by using a restraint energy term that included degenerate NOE proton pairs as a summation of r-6 terms, i.e., the SUM option of x-plor.
NMR Restraints and Structure Calculations. Interpretation of the NOE crosspeak intensities in terms of distances was by the ratio of peak intensities and calibration based on the known distance for αiNi+3 NOE interactions found in helical conformations or for δ–β interactions of phenylalanine and tryptophan residues. Distance restraints involving ambiguous NOEs, nonstereospecifically assigned methylene protons, methyl groups and Hδ and Hε protons of Phe, were represented as a (Σr-6)-1/6 summation over the set of distances (18). For distances involving methyl protons, 0.5 Å was added to the upper distance bound to account for larger intensity values due to internal mobility associated of methyl protons (19). Hydrogen bond restraints were included for the helix regions identified as described above. For each hydrogen bond, distance restraints were used between the amide hydrogen and carbonyl oxygen atom (RNH-O = 1.7–2.2 Å) and between the amide nitrogen and carbonyl oxygen atom (RN-O = 2.7–3.2 Å).
The structure was calculated by using standard restrained simulated annealing protocols and x-plor. Nonbonded contacts were represented by the “repel” energy term in the target function used for simulated annealing. The final force constants in the target function were as follows: 1,000 kcal·mol-1·Å-2 for bond energy; 500 kcal·mol-1·rad-2 for angle and improper torsion energies; 4 kcal·mol-1·Å-4 for the repel nonbonded contact energy; 50 kcal·mol-1·Å-2 for the experimental distance restraints (NOE distances and hydrogen bonds); and 200 kcal·mol-1·rad-2 for dihedral angle restraints. The van der Waals radii were set to 0.8 times their actual value in the charmm param19/20 protein parameters. The NOE distance restraints were represented by a soft-square-well potential, and a harmonic potential was used for the dihedral angle restraints.
Results and Discussion
Solution Structure Determination. Earlier, we reported that Escherichia coli-expressed DEN2C (residues 1–100) is a dimer with four helices (10). The complete chemical shift assignment of 1H, 15N, and 13C nuclei lead to a single set of NMR resonances for residues 21–100 to demonstrate that DEN2C is a symmetric dimer. The solution structure of DEN2C (Fig. 2A) was determined from 21 dihedral angle restraints and 2,042 NOE distance restraints, including 73 intersubunit restraints assigned by using a secondary structure sorting approach (see Materials and Methods and Table 1). All NOE crosspeaks were included in the final structure calculation, excluding extensively overlapped crosspeaks in the aliphatic region. No distance restraint was violated by >0.3 Å. The good agreement between the structure and all NOE data validates the assignment of intersubunit interactions from the secondary structure sorting procedure. The structure for residues 21–100 in the dimer of DEN2C is well defined (Fig. 2 A); the main-chain atom rms deviation = 0.85 Å. Residues in α2 to α4, and the loops connecting these helices, are defined to high precision, whereas larger deviations among the NMR structures are observed for the α1/α2 loop and to a lesser extent for α1 and the C-terminal residues 98–100. Residues 1–20 are conformationally labile, as indicated by small values of the heteronuclear 15N-NOE (range of 0.3 to -1.5), the paucity of medium-range, and absence of long-range 1H-1H NOE interactions. These residues were not included in the structure calculations.
Fig. 2.

Structure of DEN2C, residues 21–100. Labels for one of the two dimer subunits are designated with prime (′). Helix α1 is blue, α2 is green, α3 is yellow, and α4 is magenta. (A) Stereoview of backbone tracing for set of 20 refined solution structures prepared by using insight ii (Molecular Simulation). (B) Ribbon diagram of DEN2C dimer and separated monomer. (C) A 90° rotation of B about a horizontal axis. (D) As in C, rotated 90° about a vertical axis. Figs. 2, 3, 4 were generated by using molscript (31) and raster3d (32).
Table 1. Structural statistics for the DEN2C dimer.
| No. of restraints* | |
| Total distance restraints | 2,042 |
| Intrasubunit NOE | |
| Intraresidue | 574 |
| Sequential |I - j| = 1 | 500 |
| Medium |i - j| = 2, 3, 4 | 367 |
| Long-range |i - j| >4 | 56 |
| Ambiguous | 337 |
| Intersubunit NOE | 73 |
| Intra- or intersubunit NOE | 115 |
| α-Helices hydrogen bond | 70 |
| Dihedral ϕ angle restraints | 21 |
| rms deviation in NOE restraints and idealized geometry† | |
| NOE, Å | 0.02 |
| Bond length, Å | 0.003 |
| Bond angles, ° | 0.62 |
| Improper dihedral angles, ° | 0.45 |
| Coordinate rms deviation from average structure† | |
| All heavy atoms, Å | 1.12 |
| Backbone atoms, Ň | 0.85 |
| α-Helix backbone atoms, ҧ | 0.52 |
| procheck Ramachandran analysis† | |
| Most favored regions, % | 75.7 |
| Additional allowed regions, % | 17.9 |
| Generously allowed regions, % | 2.9 |
| Disallowed regions, % | 3.5 |
Indicated number is per subunit residues 21-100, i.e., half the number in the structure calculation
Analysis of 53 accepted structures of 54 total calculated structures
rms deviation for N, Cα, and C′ backbone atoms
α-Helix residues are V26 to F33, L45 to L58, A63 to W69, and K74 to N96
Dengue C Structure, An Alternate Protein Fold. This first structure of a flavivirus C structure reveals an alternate protein fold. A search of the Protein Data Bank (PDB) structures by using the dali method of structure alignment (20) found no acceptable match for the monomeric domain. In comparison with other viral C proteins, DEN2C is most similar to the C-terminal C domain of retroviruses. Both protein domains belong to the α-fold class with four helices, but the spatial orientation of the helices differs considerably between the two C proteins. In each DEN2C monomer, α1-α3 form a 3-helix core arranged in a right-handed bundle with the axes of α2 and α3 close to antiparallel, and at an ≈45° angle to the α1 helix axis (Fig. 2B). The longer helix, α4 (residues 74–96), extends out from the 3-helix core, and has little intramolecular contact, but has extensive intersubunit interaction with α2′, α3′, and α4′. The hydrophobic interior of the 3-helix core is packed by side chains from V26, L29, F47, V51, I65, R68, and W69 (Fig. 3). Residue R68, which is conserved among certain flaviviruses (Fig. 1), terminates α3, and its side chain is directed inwardly toward the protein interior, yet with the guanidinium group partially solvent-exposed (Fig. 3, indigo side chain). The position of R68 is supported by unusual chemical shifts for CHδ and NHε, by a large
Fig. 3.

Close-up view of the environment of W69 and hydrophobic interactions (orange) of the monomer core. R68 is shown in indigo. (Inset) The context of the full dimer.
1H-15Nε NOE value that is similar to those of the rigid backbone-amide groups, and by numerous long-range NOE interactions to other side chains in the monomer core. W69, another absolutely conserved residue (Fig. 1), lies at the dimer interface, on the exterior of the 3-helix core and with the outer edge of the indole ring packed against the C terminus of α4′ from the second molecule (Fig. 3). The critical role for this conserved residue appears to be structural stabilization of the dimer interface.
Dimerization of Dengue C Protein. A large fraction of the solvent-accessible surface of dengue C protein is buried after dimerization. The contact surface is >1,650 Å2, or approximately one-fourth of the solvent accessible surface area of each subunit. This large contact surface area explains the energetic preference for dimerization, and is consistent with the estimate of Kd < 10 nM (data not shown).
Two pairs of antiparallel helices, α2–α2′ and α4–α4′, form the majority of the dimer contact surface (Fig. 2), with both interfaces being stabilized by extensive hydrophobic interaction (Fig. 4). The only charge interaction at the interface involves E87 interacting with K45 and R55′ (Fig. 4B). The α2–α2′ interface is formed by part of the conserved internal hydrophobic region of flavivirus C proteins. In DEN2C, this conserved region corresponds to residues 46–66 and includes α2, α3, and the intervening loop. Dimerization therefore joins two internal hydrophobic sequences to create a large contiguous apolar patch comprising L46, F53, and L57 from both subunits (Fig. 4B), and is located in the center of the protein when viewed down the twofold symmetry axis, as shown in Fig. 2B. Membrane association of the dengue C protein may involve the conserved internal hydrophobic region (21). The accessibility of this hydrophobic patch formed at the exposed surface of the α2–α2′ interface lends credence to the premise that the conserved region interacts with the membrane.
Fig. 4.

(A) Cross-eye stereoview of the side-chain interactions that form the hydrophobic α4–α4′ interface along the buried edge of the helix pair. (B) Cross-eye stereoview of the dimer interface formed by the α2–α2′ helix pair (green), and interactions between α2 and α4′ (magenta), which are equivalent to α2′ and α4. The primed subunit is in front (α2′) and on the left (α4′). Ionic interactions involving the single acidic residue, E87 (yellow), and K45 (dark blue) and R55′ (cyan) are indicated with dotted lines. Residues L46, F53, and L57 (purple) from both subunits (top of view) are exposed to solvent and form a large continuous hydrophobic surface. Buried residues F56 and F84 (purple) form dimer contacts.
The α4–α4′ interface is stabilized by packing along the full length of the two helices. The conformation of α4–α4′ has characteristics of a coiled-coil structure, but differs from known coiled-coils, as judged by failure to find a match from either the sequence-based prediction program multicoil (22) or a structure-based search by using dali. It is worth noting that the terminal helix of the C protein from TBE, another flavivirus, is predicted to form a coiled-coil structure (23). Characteristics of α4-α4′ in DEN2C that are similar to coiled-coils include the slight superhelical twist of the helices, a pitch of ≈3.5 residues per turn, and the packing of leucine and isoleucine side chains across the interface at positions a and d of a heptad repeat. Side chains of I78, L81, I88, L92, and L95 on one monomer form hydrophobic interactions with their counterparts on the adjacent monomer (Fig. 4A), as occurs in coiled coils. The amino acid corresponding to the a position missing from this list is R85, an unlikely amino acid type for coiled-coil structures.
The DEN2C structure provides insight into recent studies on TBE C protein (23). Kofler et al. (23) demonstrated that TBE virus containing large deletions in the conserved internal hydrophobic sequence was viable. The largest viable deletion corresponds to a removal of DEN2C residues 32–50 (from the sequence alignment in Fig. 1), spanning the C-terminal half of α1 and the N-terminal half of α2 in DEN2C. Larger deletions that extended to the N terminus of α3, residues 32–64, failed to produce viable virus, but second-site revertants to these lethal deletions restored virus viability (24). Many of these revertants involved single amino acid substitutions to an apolar residue. The authors suggested that this increased hydrophobicity might restore membrane-binding activity that is deficient in the lethal TBE C deletion mutants. Mapping of the single-site reversions on the structure of DEN2C suggests an alternative explanation. The reversions occur in the terminal helix, which corresponds to α4 in DEN2C, and have the potential to increase the stability of the TBE C dimer. The ability of single amino acid substitutions within this region of the C protein to compensate for such large internal deletions indicates that the terminal helix plays a critical role in the function of the flavivirus C protein. Nevertheless, internal deletions of the size introduced in TBE C are expected to have a profound effect on the C protein so that the structure resulting from the mutations is uncertain.
Asymmetric Charge Distribution. A striking feature of DEN2C is its charge: at neutral pH and assuming solution-state pKa values, the dimer net charge would be +46, an unusually large value. There are 26 basic amino acids and only three acidic residues per 100-residue subunit. Whereas 29/100 is not unusual for the number of charged residues in a protein (25), it is common that the charge is more evenly distributed between acidic and basic residues so that the net charge is considerably smaller in magnitude (26). Moreover, the spatial charge distribution of the DEN2C dimer is remarkably nonuniform. The region with the highest density of positive charge is the solvent-exposed edge of α4-α4′ at the “bottom” of DEN2C as viewed in Fig. 2 C and D. Twenty-two of the 52 basic residues, or nearly half, lie on this face of the protein. In contrast, the concave portion contributed by α2-α2′ at the “top” in Fig. 2D is apolar. The solvent-accessible surface colored by residue type (Fig. 5) illustrates the distinctly asymmetric charge distribution. Surfaces due to basic residues are cyan, acidic residues are red (none visible), and all others are gray. The α4-α4′ surface (Fig. 5A) shows a large fraction of basic residues, while on the opposite face (Fig. 5B) the surface contributed by α2-α2′ and α1-α1′ is largely devoid of charge. Helices α2–α2′ form the bottom of a deep cleft (shown in stereo) from all apolar residues (L46, L50, F53, L54, and L57 of both subunits). The two charged residues, located on both upper edges of the cleft, are K31 and R32. Interestingly, this surface has a highly concave topography, which may have functional significance, as discussed below.
Fig. 5.

Solvent-accessible surfaces colored by residue type for the DEN2C dimer. Lysine and arginine are blue, and glutamic acid is red (not visible in these views). A main-chain tracing in the same orientation is shown with the surface representation. (A) Charged surface of the α4–α4′ helix pair. (B) Cross-eyed stereoview of the apolar surface formed by the α2–α2′ helix pair. Generated by using grasp (33).
Functional Implications from the DEN2C Solution Structure. The solution structure of the dengue C protein reveals an alternate fold for which dimerization emphasizes features likely to be functionally important. Oligomerization of DEN2C into a protein-only core is unlikely because of the large positive (+46) charge of the dimer. Alternatively, the structure suggests regions of interaction with other viral components in the virus particle. Based on the nonuniform charge distribution of DEN2C and the concave shape of the hydrophobic cleft, we propose that the α4-α4′ region, which is rich in basic residues, interacts with RNA, whereas the apolar α2-α2′ region interacts with the viral membrane (Fig. 6). In addition to the electrostatic potential, the concave groove at the α2-α2′ region provides an energetic argument for membrane association based on protein solvation. That is, surface topography has been implicated in the free energy of hydrophobic hydration (27). Examination of the physical properties of water molecules at a hydrophobic patches with alternative topographies, such as flat and convex shapes, finds that hydration of concave surfaces destabilizes the average specific water interaction energy relative to that at flat surfaces or bulk water molecules. Thus, hydrophobic association between the α2-α2′ surface and either membrane or some other protein surface would include both entropic and enthalpic components, favoring the release of water molecules from the hydrophobic cleft.
Fig. 6.
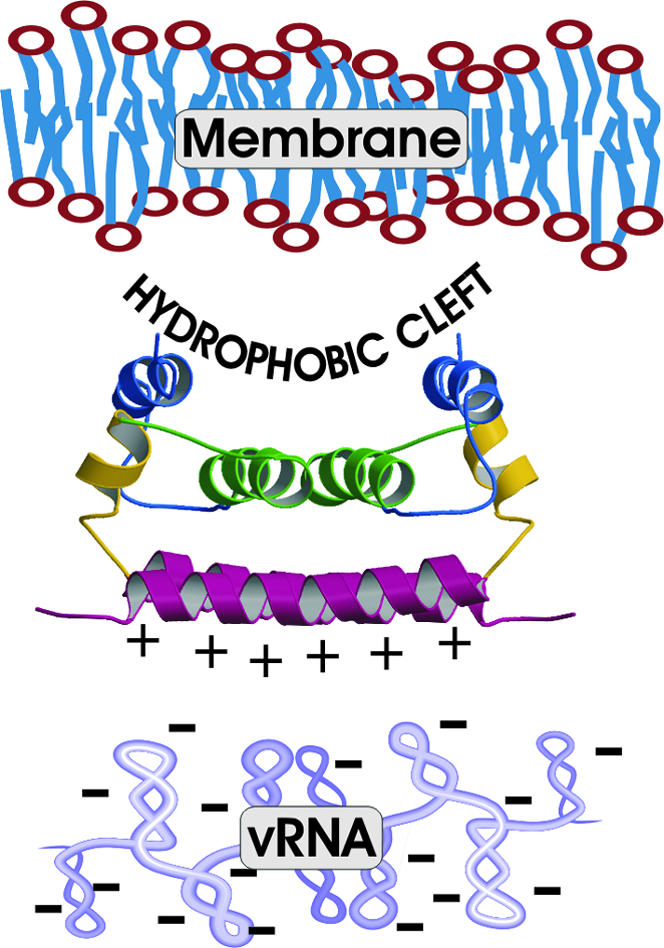
Model for molecular interactions between structural components of flavivirus. The viral membrane is shown on top near the hydrophobic cleft, and the viral RNA is shown on the bottom near the positively charged surface of DEN2C.
The interactions suggested from the DEN2C structure are consistent with the conclusion that the internal hydrophobic sequence including α2 mediates membrane association reported by others (21). In addition, the model agrees with interpretations of genetic studies that the C-terminal region of DEN2C is involved with RNA association (28). A similar model was proposed for the M1 protein of influenza virus whereby membrane and RNA interact with opposite faces of this C protein (29), but includes a substantial conformational rearrangement to insert a hydrophobic helix in the membrane. In contrast, the equilibrium solution structure observed for DEN2C includes an exposed hydrophobic region that is readily accessible without analogous structural changes. Nevertheless, given the negative surface potential of the impinging viral membrane, it is likely that some structural changes are necessary to facilitate the process of virus assembly. The positive charge on exposed regions of α1 may play a role in initial binding, followed by rearrangements to further expose the hydrophobic surface in the cleft.
Acknowledgments
We thank Dr. Klaas Hallenga for assistance with NMR spectroscopy, the staff of the Purdue University Interdepartmental NMR Facility, and Jeff Bolin for many helpful comments. This work was supported by National Institutes of Health grants (to C.B.P. and R.J.K.), a Purdue University reinvestment grant, and a grant from the Purdue Cancer Center. C.T.J. and T.D.G. were trainees of the National Institutes of Health Training Grant in Biophysics and received Purdue Research Foundation Graduate Fellowships.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NOE, nuclear Overhauser effect; C, capsid; DEN2C, dengue 2 C protein; E, envelope glycoprotein; TBE, tick-borne encephalitis.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1R6R).
References
- 1.Gubler, D. J. (2002) Trends Microbiol. 10, 100-103. [DOI] [PubMed] [Google Scholar]
- 2.Lindenbach, B. D. & Rice, C. M. (2001) in Fields Virology, eds. Knipe, D. M. & Howley, P. M. (Lippincott, Williams & Wilkins, Philadelphia), pp. 991-1041.
- 3.Kuhn, R. J., Zhang, W., Rossmann, M. G., Pletnev, S. V., Corver, J., Lenches, E., Jones, C. T., Mukhopadhyay, S., Chipman, P. R., Strauss, E. G., et al. (2002) Cell 108, 717-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rey, F. A., Heinz, F. X., Mandl, C., Kunz, C. & Harrison, S. C. (1995) Nature 375, 291-298. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, W., Chipman, P. R., Corver, J., Johnson, P. R., Zhang, Y., Mukhopadhyay, S., Baker, T. S., Strauss, J. H., Rossmann, M. G. & Kuhn, R. J. (2003) Nat. Struct. Biol., 10, 907-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferlenghi, I., Clarke, M., Ruttan, T., Allison, S. L., Schalich, J., Heinz, F. X., Harrison, S. C., Rey, F. A. & Fuller, S. D. (2001) Mol. Cell 7, 593-602. [DOI] [PubMed] [Google Scholar]
- 7.Kummerer, B. M. & Rice, C. M. (2002) J. Virol. 76, 4773-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamshchikov, V. F. & Compans, R. W. (1995) J. Virol. 69, 1995-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amberg, S. M., Nestorowicz, A., McCourt, D. W. & Rice, C. M. (1994) J. Virol. 68, 3794-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones, C. T., Ma, L. X., Burgner, J. W., Groesch, T. D., Post, C. B. & Kuhn, R. J. (2003) J. Virol. 77, 7143-7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn, Y. S., Galler, R., Hunkapiller, T., Dalrymple, J. M., Strauss, J. H. & Strauss, E. G. (1988) Virology 162, 167-180. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki, T., Forman-Kay, J. D. & Kay, L. E. (1993) J. Am. Chem. Soc. 115, 11054-11055. [Google Scholar]
- 13.Pascal, S. M., Muhandiram, D. R., Yamazaki, T., Forman-Kay, J. D. & Kay, L. E. (1994) J. Magn. Reson. B 103, 197-201. [Google Scholar]
- 14.Kay, L. E., Keifer, P. & Saarinen, T. (1992) J. Am. Chem. Soc. 114, 10663-10665. [Google Scholar]
- 15.Bax, A., Vuister, G. W., Grzesiek, S., Delaglio, F., Wang, A. C., Tschudin, R. & Zhu, G. (1994) Methods Enzymol. 239, 79-105. [DOI] [PubMed] [Google Scholar]
- 16.Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277-293. [DOI] [PubMed] [Google Scholar]
- 17.Wishart, D. S. & Sykes, B. D. (1994) J. Biomol. NMR 4, 171-180. [DOI] [PubMed] [Google Scholar]
- 18.Nilges, M. (1993) Proteins Struct. Funct. Genet. 17, 297-309.8272427 [Google Scholar]
- 19.Wagner, G., Braun, W., Havel, T. F., Schaumann, T., Go, N. & Wuthrich, K. (1987) J. Mol. Biol. 196, 611-639. [DOI] [PubMed] [Google Scholar]
- 20.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123-138. [DOI] [PubMed] [Google Scholar]
- 21.Markoff, L., Falgout, B. & Chang, A. (1997) Virology 233, 105-117. [DOI] [PubMed] [Google Scholar]
- 22.Wolf, E., Kim, P. S. & Berger, B. (1997) Protein Sci. 6, 1179-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kofler, R. M., Heinz, F. X. & Mandl, C. W. (2002) J. Virol. 76, 3534-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kofler, R. M., Leitner, A., O'Riordain, G., Heinz, F. X. & Mandl, C. W. (2003) J. Virol. 77, 443-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creighton, T. (1993) Proteins: Structures and Molecular Properties (Freeman, New York).
- 26.Barlow, D. J. & Thornton, J. M. (1986) Biopolymers 25, 1717-1733. [DOI] [PubMed] [Google Scholar]
- 27.Cheng, Y. K. & Rossky, P. J. (1998) Nature 392, 696-699. [DOI] [PubMed] [Google Scholar]
- 28.Wang, S. H., Syu, W. J., Huang, K. J., Lei, H. Y., Yao, C. W., King, C. C. & Hu, S. T. (2002) J. Gen. Virol. 83, 3093-3102. [DOI] [PubMed] [Google Scholar]
- 29.Sha, B. D. & Luo, M. (1997) Nat. Struct. Biol. 4, 239-244. [DOI] [PubMed] [Google Scholar]
- 30.Corpet, F. (1988) Nucleic Acids Res. 16, 10881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 32.Merritt, E. A. & Bacon, D. J. (1997) Methods Enzymol. 277, 505-524. [DOI] [PubMed] [Google Scholar]
- 33.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins Struct. Funct. Genet. 11, 281-296. [DOI] [PubMed] [Google Scholar]