

Abstract
Pathogenicity of Yersinia pestis (Y. pestis) relies on several effector proteins, including YopH, a protein-tyrosine phosphatase. Previously, we screened a library of analogues based on the ubiquitous PTP substrate, p-nitrophenylphosphate (pNPP) and found that incorporation of a 3-phenyl substituent (6-nitro-[1,1'-biphenyl]-3-yl dihydrogen phosphate (1)) enhanced affinity. The current study reports the conversion of 1 from a substrate to inhibitor by replacing the hydrolysable phosphoryl group with a 3-isoxazolecarboxylic acid moiety and by introduction of an aminooxy group and subsequent diversification using oxime-based click chemistry. As reported herein, this approach led to the identification of non-promiscuous low micromolar affinity bidentate YopH inhibitors.
Keywords: Substrate screening, Aminooxy platform, Oxime-based click chemistry, YopH inhibitors, In silico docking studies
Introduction
Yersinia pestis (Y. pestis) has played important roles in human history by serving as the causative agent for pneumonic and bubonic plague.[1, 2] Recently, Y. pestis has gained attention as a potential biological warfare or bioterrorism agent and this has engendered renewed interest in the development of anti-plague therapeutics. For pathogenicity Y. pestis employs a Type III secretion system (T3SS) to inject into host cells a variety of “Yop” proteins that include YopH, a highly active protein-tyrosine phosphatase (PTP).[3] Inappropriate dephosphorylation by YopH can interfere with normal cellular function and lead to pathogenesis, and accordingly, YopH inhibitors could potentially provide a basis for new anti-plague therapeutics.
PTPs share a common mechanism of action, which involves substrate recognition by a conserved (H/V)CX5R(S/T) signature motif that forms the heart of the catalytic cleft. Catalysis occurs in two steps by initial transfer of the phosphoryl group to the active-site Cys residue and subsequent release of dephosphorylated substrate and hydrolysis of the phosphoprotein thioester intermediate to liberate inorganic phosphate and regenerate the free enzyme. The phosphotyrosyl (pTyr) phenylphosphate functionality plays a defining role in substrate recognition. One approach to inhibitor development is to identify high affinity substrates, which can subsequently be converted to inhibitors by replacement of the hydrolysable phosphoryl group with non-hydrolysable mimetics. Identification of substrates as platforms for inhibitor development (a known approach[4–7] that has recently been termed, “substrate activity screening” (SAS)[8]) has the potential advantage of overcoming “false positives” that can arise from inhibition by promiscuous mechanisms.[9, 10]
As an application of SAS we recently screened YopH against a library of analogues based on the ubiquitous PTP substrate, p-nitrophenylphosphate (pNPP, KM = of 600 µM) and found that incorporation of a 3-phenyl substituent resulted in enhanced affinity (6-nitro-[1,1'-biphenyl]-3-yl dihydrogen phosphate (1, Figure 1), KM = 80 µM).[11] The current study reports the conversion of 1 from a substrate to a series of inhibitors (3) by replacing the hydrolysable phosphoryl group with a 3-isoxazolecarboxylic acid moiety and by introduction of an aminooxy handle and subsequent diversification of this platform (2) using oxime-based click chemistry (Figure 1). As reported herein, this approach led to the identification of non-promiscuous low micromolar affinity bidentate YopH inhibitors.
Figure 1.

Development of oxime-containing inhibitors 3 based on starting substrate 1.
Results and Discussion
Conversion of substrate 1 to inhibitor platform 2
Substrate 1 was modified by replacement of its phosphoryl group with the non-hydrolysable phosphate mimetic, 3-isoxazolecarboxylic acid.[12, 13] Further modification by removal of the nitro group and introduction of an aminooxy handle yielded platform 2, which served as the starting point for preparing a library of oxime-based bidentate inhibitors 3. The synthesis of 3 was accomplished in five steps starting from ((3-bromophenyl)ethynyl)trimethylsilane (4, Scheme 1). Removal of the TMS group (potassium carbonate in MeOH) and treatment of the resulting alkyne with 2-chloro-2-(hydroxyimino)-acetate gave the 5-bromophenyl-3-isoxazole derivative 5 in good yield.[14] Suzuki coupling to provide the corresponding benzylic alcohol 6 and Mitsunobu reaction with N-hydroxyphthalimide gave 7. Treatment with hydrazine hydrate followed by ester hydrolysis provided the aminooxy-containing platform 2. The synthesis of oxime 3 was accomplished by reacting 2 with a library of aldehydes using AcOH as a catalyst.
Scheme 1.

Synthetic procedure of platform 3. Reagents and conditions: a) K2CO3, MeOH, RT; b) EtO2CC(Cl)=NOH, Et3N, THF, 50° C (84% yield); c) 4-hydroxymethylboronic acid, satd. K2CO3, Pd(PPh3)4, EtOH, PhMe, 70° C (69% yield); d) N-hydroxyphtalimide, PPh3, DIAD, THF, RT (91% yield); e) NH2NH2˙H2O, EtOH, RT f)1 N NaOH, THF (52% yield); g) RCHO, AcOH, DMSO, RT (>90% yield).
Oxime-based library diversification
The syntheses of oxime-containing inhibitors (3) were performed in DMSO by reacting the aminoxy-containing platform 2 (24 mM) with appropriate aldehydes and AcOH in the ratio (1:1:2). The resulting crude oxime reaction mixtures were of sufficient purity (≥ 90%) for direct biological evaluation. Inhibitory IC50 values were determined spectrophotometrically in an in vitro YopH assay using pNPP as substrate.[15] A library of 65 commercially available aldehydes was employed with structural diversity ranging from simple linear and branched alkyls to phenyl and phenylmethyls and heterocycles.[16] Inhibitory potencies (IC50 values) for the resulting compounds ranged from 3µM to 115 µM, with aromatic-containing analogues exhibiting greater inhibitory potency than the linear and branched alkyl-containing analogues. The most potent inhibitors (3a – 3d) showed IC50 values of approximately 10 µM (Table 1). Addition of a 5-carboxyl group to the indolyl portion of oxime 3d resulted in an approximate 3 - fold enhancement of inhibitory potency (3e; IC50 = 3.1 µM, Table 1). When assays were conducted in the presence and absence of 0.01% TX-100[11, 17, 18] it was found that the inhibitory potency of 3e was independent of detergent at the concentration tested. This provided strong evidence that YopH inhibition by 3e does not arise through promiscuous mechanisms. Kinetic analysis of 3e indicated inhibition by a cometitive mechanism with a Ki value of 3.9 ± 0.3 µM (Figure 4).
Table 1.
In vitro YopH IC50 values for selected inhibitors.
| No. | R | IC50 ± S.E. (µM)[a] |
|---|---|---|
| 3a | 14.2 ± 0.2 | |
| 3b |  |
9.8 ± 0.2 |
| 3c | 8.7 ± 0.3 | |
| 3d | 8.6 ± 0.3 | |
| 3e | 3.1 ± 0.2 |
IC50 values were determined as indicated in the Experimental procedures.
Figure 4.

Lineweaver-Burk plot for 3e and inhibits YopH through a competitive mechanism. The inhibitor concentrations used were 2, 4, and 8 µM.
Potential role of nitro functionality
The conversion of substrate 1 to the lead platform 2 was done with removal of a nitro group, since nitro functionality can impart undesirable biological properties,[19] and the original function of this moiety as a chromophoric agent was no longer necessary. However, it was unclear what effects deletion of the nitro group had on the binding affinities of the final ligands (3a – 3d). In order to clarify this issue, 4-nitro functionality was introduced into the inhibitors. This involved the synthesis of the aminooxy-containing platform 15 (Scheme 2). The synthesis of 15 started by the Suzuki coupling of 3-bromo-4-nitrophenol (8) with (4-(hydroxymethyl)phenyl)boronic acid to give the biphenyl product 9. Reaction of 9 with a mild triflating reagent (N-phenyl-bis(trifluoromethanesulfonimide)[20] provided 10 in quantitative yield. Sonogashira coupling of 10 with TMS-acetylene afforded 11 followed by removal of the TMS-group to give the alkyne 12. Treatment of 12 with 2-chloro-2-(hydroxyimino)-acetate gave the 3-isoxazolecarboxylic acid ester 13, which provided phthalimide 14 under Mitsunobu conditions. Deprotection by treatment with hydrazine hydrate followed by ethyl ester hydrolysis gave the nitro-containing aminooxy platform 15. Synthesis of the nitro-containing oxime-containing inhibitors (16a – 16e) was then performed in DMSO as before and YopH inhibitory potencies were determined for the resulting oximes without purification. The similarity of IC50 values for compounds with (Table 2) and without (Table 1) nitro groups indicated that this functionality apparently had little effect on binding affinity.
Scheme 2.

Synthetic procedure of oxime 16. Reagents and conditions: a) 4-hydroxymethylboronic acid, satd. K2CO3, Pd(PPh3)4, EtOH, PhMe, 70° C (76% yield); b) PhN(SO2CF3)2, DIPEA, CH3CN, RT (97% yield); c) Pd(PPh3)2Cl2, CuI, DIPEA, TMS-acetylene, CH3CN, RT (98% yield); d) K2CO3, MeOH, RT (69% yield); e) EtO2CC(Cl)=NOH, Et3N, THF, RT (59% yield); f) N-hydroxyphtalimide, PPh3, DIAD, THF, RT (60% yield); g) NH2NH2˙H2O, EtOH, RT h)1 N NaOH, THF (49% yield); i) RCHO, AcOH, DMSO, RT (>90% yield).
Table 2.
In vitro YopH IC50 values for inhibitors 16a–d.
| No. | R | IC50 ± S.E. (µM)[a] |
|---|---|---|
| 16a | 15.4 ± 0.8 | |
| 16b | 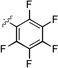 |
14.9 ± 0.5 |
| 16c | 10.0 ± 0.4 | |
| 16d | 9.2 ± 0.2 | |
| 16e | 3.9 ± 1.4 |
IC50 values were determined as indicated in the Experimental procedures.
Potential YopH binding modes of synthetic ligands
In order to identify potential YopH binding modes, in silico docking studies were performed[21, 22] starting from our earlier X-ray crystal structure of YopH in complex with the peptide Ac-Asp-Ala-Asp-Glu-F2Pmp-Leu-amide ((PDB 1QZ0),[23, 24] where “F2Pmp” represents the non-hydrolyzable pTyr mimetic, phosphonodiflouoromethylphenylalanine.[25, 26] The portion of the peptide bound within the catalytic pocket was isolated and the phosphonodiflouoromethyl group was replaced with a 3-isoxazolecarboxylic acid moiety, The resulting 5-phenyl-3-isoxazolecarboxylic acid structure was re-docked alternatively in the presence and absence of a catalytically-conserved H2O molecule.[27] Inclusion of the conserved H2O resulted in additional bridging interactions with Q357 and Q450 (Figure 2) that were not possible in the absence of the H2O. These additional interactions were reflected in more favourable calculated binding scores in subsequent docking studies of fully elaborated oxime-containing inhibitors.
Figure 2.

Docking of 5-phenyl-3-isoxazolecarboxylic acid in the YopH catalytic pocket (a) Docking performed in the presence a catalytically-conserved H2O molecule. (b) Overlay onto the docking pose of Panel A of the phopshonodifuoromethylphenyl group (shown in yellow) derived from the crystal structure of an F2Pmp-containing peptide bound to YopH (PDB 1QZ0).
Potential YopH interactions with 3d (Figure 3a) and 3e (Figure 3b) were examined. The phenyl ring originating from the meta-position of the 5-phenyl-3-isoxazolecarboxylic acid group was found to bind within a hydrophobic channel lying between residues I232 and Q446, which is identical to the region occupied by the Leu side chain in the original Ac-Asp-Ala-Asp-Glu-F2Pmp-Leu-amide peptide (PDB 1QZ0). The most favourable binding poses for both inhibitors involved insertion of their tethered indolyl rings into the previously identified pockets P1 and P2 (Figure 3).[28–30] The higher binding affinity of 3e relative to 3d could be explained by bonding interactions of its indolyl 5-carboxy group with residues in the base of each pocket (the guanidinium group of R473 in Pocket 1 and the amide nitrogen of G442 in the base of pocket 2).
Figure 3.

Predicted favoured YopH biding interactions for synthetic oxime-containing inhibitors 3d (Panel a) and 3e (Panel b) showing insertion of tethered aryl rings into well formed Pockets (1 and 2). The protein is rendered as an electrostatic surface with red indicating negative and blue positive charge.
The modelling in Figure 3 orients the 5-phenyl-3-isoxazolecarboxylic acid group within the catalytic cleft, while the 5-carboxyindolyl group interacts in a secondary Pocket. It is worth noting that 5-carboxyindolyl-containing motifs have been incorporated into PTP inhibitors as phenylphosphate mimetics designed to interact within the catalytic clefts.[31–33] This could potentially indicate that (in the specific case of 3e) alternate binding modes may be possible in which the 5-carboxyindolyl group rather than the 5-phenyl-3-isoxazolecarboxylic acid group is bound within the catalytic cleft. In fact modelling of such “reversed” orientations does show that they are possible. However, the resulting binding interactions are significantly less favourable than the orientations shown in Figure 3, and they most probably do not contribute significantly to overall binding affinity[34] as well as the dual specificity phosphatases.[35]
Examination of Specificity of 3e
In order to ascertain some measure of enzyme specificity, the inhibitory activity of 3e was measured against a panel of phosphatases. These included the classical tyrosine-specific phosphatases PTP1B and LAR,[36] DUSP14, DUSP22 and the Variola phosphatase VH1 (Table 3). In these assays relative to YopH, 3e showed good selectivity against LAR (45 – fold) and VH1 (> 400 – fold), and less selectivity against other the other phosphatases examined (from 4 – fold to 8 – fold).
Table 3.
Inhibitory potencies of 3e against a panel of phosphatases.
| Phosphatase | IC50 (µM)[a] | Fold change |
|---|---|---|
| YopH | 3.0 | |
| PTP1B | 13.7 | 4.7 |
| LAR | 134.9 | 45 |
| DUSP-14 | 22.5 | 7.5 |
| DUSP-22 | 24.5 | 8.2 |
| Variola | > 1200 | > 400 |
IC50 values were determined as indicated in the Experimental procedures.
Relative to YopH
Conclusion
The focus of the current study was to develop new inhibitors directed against YopH by the replacement of the phosphoryl group in previously identified high affinity substrate 1 with 3-isoxazolecarboxylic acid moiety and appendage of an aminooxy handle to yield a lead inhibitor scaffold that was optimized using oxime-based library diversification. This protocol resulted in the identification of bivalent analogue that showed low micromolar, non-promiscuous inhibition against YopH, with moderate to good enzyme selectivity. Although the specific target of the study was YopH, the approach may be more broadly applicable for the development of inhibitors directed against other phosphatases.
Experimental Section
General
The following reagents used for YopH enzyme assays were obtained from Sigma-Aldrich: pNPP tablets; 30% BSA solution (protease free); 1.0 M HEPES solution (pH 7.0 – 7.6) and dithiotreitol (DTT). Aqueous EDTA (0.5 M, pH 8.0) was obtained from Invitrogen and 96-well clear bottom plates were purchased from FisherSci (Costar 3603). All reactions were carried out under argon unless otherwise stated. All solvents were anhydrous and obtained from Sigma-Aldrich. High pressure liquid chromatography (HPLC) was performed using a Waters Prep-LC 4000 system and Phenomenex Gemini 10 µ, 110 Å C18 columns (250 × 21.20 mm 10 micron) at a flow rate of 10 mL/minute (prep. HPLC) and 1 mL/minute (analytical HPLC) with a mobile phase of A = 0.1% aqueous TFA and B = 0.1% TFA in aqueous acetonitrile. Typical gradients were from 10% B to 100% B over 40 minutes with UV monitoring at 220 nm, 254 nm and 280 nm. NMR spectra were recorded using a Varian 400 MHz spectrometer. Unit mass resolution LC-MS were obtained on synthetic intermediates and high resolution mass spectra (HRMS) were obtained for final products (University of California at Riverside Mass Spectral Facility). Optical densities were measured with Biotek Synergy 2 spectrophotometer at Åabs 405 nm using absolute readout for determination of IC50 values.
Recombinant proteins
The PTPse domain of YopH (residues 164–468) was expressed in Escherichia coli according to the previously published procedure. [3, 24] as were the variola major H1[37] and human DUSP-14 dual specificity phosphatases.[38] Human DUSP-22, PTPase1B and LAR catalytic domains were expressed and purified using generic methodology.[39]
General syntheses of oximes 3 and 16
A solution of 72 mM aminoxy platform (15 µL DMSO) and a solution of 72 mM aldeyde (15 µL DMSO) were placed in 1.5 mL microtube with cap. To this mixture was added 144 mM AcOH (15 µL DMSO). The reaction mixture was then gently agitated overnight at RT and the resultant oximes (24 mM) were directly evaluated in vitro against YopH without any further purification.
Determination of YopH IC50 values
Total reactions volumes of 100 µL/well of reaction volume were used in 96 well plates. Buffer was prepared as above. To each well was added 79 µL of assay buffer, 0.25% BSA (5 µL) followed by 5 µL of inhibitors in DMSO at dilutions of 1200, 480, 192, 77, 31, 25, 12, 5, 2, 0.8, 0.4 and 0 µM. To the reaction mixtures was then added 5µL of YopH in buffer (25 µg/mL) followed by 6 µL of 10 mM pNPP buffer each plate was agitated gently at 25° C for 15 – 20 minutes. Hydrolysis of the substrate was immediately measured. IC50 values were determined by fitting the data with sigmoidal curve generated using the Boltzman equation. A parallel independent assay was performed with 0.01% TritonX-100. Inhibition constants for 3a–e and 16a–e are provided in Table 1 and 2. Data curves are provided in the Supporting Information.
Determination of Ki value
Total reactions volumes of 100 µL/well of reaction volume were used in 96 well plates. Buffer was prepared as above. To each well was added 65 µL of assay buffer, 0.25% BSA (5 µL) followed by 5 µL of inhibitors in DMSO at dilutions 8, 4, 2 µM. To each of the fixed inhibitor concentration was then added 20 µL of pNPP with dilutions ranging from 2000 to 3.9 µM (KM pNPP against YopH with no inhibitor is 600 µM). The reaction was then started by the addition of 5 µL YopH and was immediately monitored at 405 nm. Data were then fitted for the determination of Ki and mechanism of inhibition.
In Silico Studies
Docking of inhibitors 3d and 3e onto YopH was done with ICM Chemist Pro® software running on a MacIntosh computer (OSX v10.5.8) using default parameters and procedures.[22] In summary, modelling started with the X-ray crystal structure of YopH in complex with the peptide Ac-Asp-Ala-Asp-Glu-F2Pmp-Leu-amide (PDB 1QZ0),[23, 24] where “F2Pmp” represents the non-hydrolysable pTyr mimetic, phosphonodiflouoromethylphenylalanine. [25, 26] The “convert PDB” command was used to convert to native ICM format, with optimization of hydrogens. All H2O molecules were removed from the enzyme with the exception of the catalytically-conserved H2O (identified as “cw”). For creation of the ligand, the phosphonodiflouoromethylphenyl portion of the peptide was first isolated, then the phosphonodiflouoromethyl group was replaced by the 3-isoxazolecarboxylic acid moiety and the resulting 5-phenyl-3-isoxazolecarboxylic acid structure was re-docked using the “re-dock” option under the “Ligand” menu. All docking was performed using the standard “re-dock” command, which utilizes a rigid receptor protocol. Docking was performed both in the presence (Figure 2) and absence of the catalytically-conserved H2O. Subsequent docking was conducted with retention of this H2O. Binding scores for best poses were calculated automatically. To the docked 5-phenyl-3-isoxazolecarboxylic acid moiety was added the meta-phenyl ring and the resulting structure was re-docked. Further structural editing provided the final inhibitors, which were re-docked. The best binding poses (lowest binding scores) for 3d and 3e are shown in Figure 3.
Ethyl 5-(3-Bromophenyl)isoxazole-3-carboxylate (5)
To a solution of ((3-bromophenyl)ethynyl)trimethylsilane (1.00 g, 3.95 mmol) in MeOH (5 mL) was added K2CO3 (2.90 g, 15.9 mmol) and the mixture was stirred at room temperature (1.5 h). Solvent was removed and the residue was partitioned (EtOAc : H2O), the organic phase was washed (H2O) and dried (MgSO4) and taken to dryness. Solvent was evaporated at reduced pressure and the resultant crude material was purified by silica gel chromatography (hexanes : EtOAc, 9 : 1) to yield intermediate 1-bromo-3-ethynylbenzene. A solution of this alkyne (0.54 g, 2.97 mmol) and ethylchloroximido acetate (0.76 g, 5.05 mmol) in anhydrous THF (5 mL) was purged with argon, then Et3N (0.70 mL, 5.04 mmol) was via syringe over 30 minutes and the mixture was stirred at 50° C (overnight). After cooling to room temperature the mixture was partitioned (1 N HCl : EtOAc) and the aqueous phase was extracted (EtOAc) and the combined organic extract was dried and taken to dryness. Purification by silica column chromatography (hexanes : EtOAc, from 3 : 1 to 1 : 1) to provide 6 as a white amorphous solid (0.74 g, 84% yield). 1H NMR (400 MHz, CDCl3): δ = 7.95 (t, J = 2.0 Hz, 1H), 7.73 (m, 1H), 7.59 (m, 1H), 7.37 (t, J = 8.0 Hz, 1H), 6.95 (s, 1H), 4.48 (q, J = 7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (400 MHz, CDCl3): δ = 170.13 (1C), 159.87 (1C), 157.16 (1C), 133.82 (1C), 130.82 (1C), 128.94 (1C), 128.50 (1C), 124.57 (1C), 123.34 (1C), 100.87 (1C), 62.45 (1C), 14.29 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C12H10BrNO3, 318.0 and 320.0; found, 318.1 and 320.0.
Ethyl 5-(4'-(Hydroxymethyl)-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylate (6)
A mixture of 5 (500 mg, 1.67 mmol), (4-(hydroxymethyl)phenyl)boronic acid (385 mg, 2.55 mmol), Pd(PPh3)4 (96 mg, 0.08 mmol), saturated aqueous K2CO3 (10 mL), toluene (10 mL) and EtOH (4 mL) was purged with argon and stirred at reflux (overnight). The reaction mixture was partitioned (H2O : EtOAc) and the organic phase was dried, taken to dryness and the residue was purified by silica column chromatography (hexanes : EtOAc, from 3 : 1 to 1 : 1) to yield 6 as a white amorphous solid (372 mg, 69% yield). 1H NMR (400 MHz, CDCl3): δ = 8.01 (t, J = 2.0 Hz, 1H), 7.77 (m, 1H), 7.69 (m, 1H), 7.65 (m, 2H), 7.56 (t, J = 8.0 Hz, 1H), 7.48 (m, 2H), 6.98 (s, 1H), 4.77 (d, J = 2.8 Hz, 2H), 4.48 (q, J = 7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (400 MHz, CDCl3): δ = 171.77 (1C), 160.16 (1C), 157.14 (1C), 146.40 (1C), 142.07 (1C), 140.82 (1C), 139.43 (1C), 132.45 (1C), 130.46 (1C), 129.74 (1C), 127.73 (1C), 127.47 (1C), 127.27 (1C), 127.15 (1C), 124.78 (1C), 100.35 (1C), 64.71 (1C), 62.47 (1C), 14.33 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C19H17NO4, 346.1; found, 346.1.
Ethyl 5-(4'-(((1,3-Dioxoisoindolin-2-yl)oxy)methyl)-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylate (7)
To a mixture of 6 (350 mg, 1.08 mmol), N-hydroxyphtalimide (212 mg, 1.30 mmol), and PPh3 (368 mg, 1.41 mmol) in anhydrous THF (10 mL) was added diisopropyl azodicarboxylate (DIAD) (0.28 mL, 1.41 mmol) and the mixture was stirred at room temperature (overnight). The mixture was partitioned (H2O : CH2Cl2) and the organic extracts were dried, taken to dryness and the residue was purified by precipitation from EtOAc to give 7 as a white solid amorphous solid (463 mg, 91% yield). 1H NMR (400 MHz, CDCl3): δ = 8.02 (m, 1H), 7.84-7.62 (m, 10H), 7.57 (t, J = 8.0 Hz, 1H), 6.99 (s, 1H), 5.28 (s, 2H), 4.49 (q, J = 7.2 Hz, 2H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (400 MHz, CDCl3): δ = 171.69 (1C), 163.68 (1C), 160.15 (1C), 157.20 (1C), 141.82 (1C), 141.07 (1C), 134.66 (2C), 133.67 (1C), 130.63 (2C), 129.85 (1C), 129.72 (1C), 129.16 (1C), 129.06 (1C), 127.49 (2C), 127.45 (1C), 127.36 (1C), 125.16 (1C), 124.80 (1C), 123.72 (2C), 100.42 (1C), 79.60 (1C), 62.45 (1C), 14.36 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C27H20N2O6, 491.1; found, 491.0.
5-(4'-((Aminooxy)methyl)-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylic Acid (2)
To a solution of 7 (200 mg, 0.49 mmol) in EtOH (5 mL) was added 50% aqueous hydrazine hydrate (0.13 mL, 1.96 mmol) and the reaction mixture was stirred at room temperature (3 h). The resulting precipitate was removed by filtration and the filtrate was taken to dryness to yield a white solid, which was stirred with a mixture of THF (2 mL), H2O (5 mL) and 1 N NaOH (2.0 mL) (1.5 h). Volatile organics were removed under reduced pressure and the resulting aqueous phase was subjected directly to HPLC purification as described above in the General Synthetic methods (retention time = 17.7 minutes) to yield 2 with amorphous solid (79 mg, 52% yield; 100% purity by analytical HPLC). 1H NMR (400 MHz, CD3OD): δ = 8.87 (s, 1H), 8.26 (m, 1H), 7.96 (m, 1H), 7.87 (m, 3H), 7.66 (t, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.53 (m, 2H), 4.96 (s, 2H), 2.51 (t, J = 2.0 Hz,2H). 13C NMR (400 MHz, CD3OD): δ = 171.06 (1C), 161.22 (1C), 158.28 (1C), 140.96 (1C), 139.73 (1C), 134.87 (1C), 130.50 (1C), 129.98 (2C), 129.46 (1C), 127.47 (2C), 127.39 (1C), 125.25 (1C), 124.41 (1C), 101.85 (1C), 76.15 (1C). HRMS-ESI (m/z): [M + H]+ calcd for C17H15N2O4, 311.1026; found, 311.1025.
4'-(Hydroxymethyl)-6-nitro-[1,1'-biphenyl]-3-ol (9)
Mixture of 3-bromo-4-nitrophenol (1.5 g, 6.88 mmol), (4-(hydroxymethyl)phenyl)boronic acid (1.5 g, 10.32 mmol) and Pd(PPh3) (0.40 g, 0.34 mmol) in 10 mL satd solution of K2CO3, 4 mL EtOH and 10 mL toluene was purged with Argon and stirred at 70°C overnight. Water was added upon cooling and aqueous layer was extracted by EtOAc. Organic extract was dried over MgSO4, filtered and solvent was removed. The crude material was purified via silica gel chromatography (CH2Cl2 : EtOAc 4:1) to give 9 as a yellow solid product (1.3 g, 76%). 1H NMR (400 MHz, CDCl3): δ = 7.88 (d, J = 9.2 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 6.91 (dd, J = 2.4 Hz, J = 8.8 Hz, 1H), 6.80 (d, J = 4.0 Hz, 1H), 4.62 (s, 1H). 13C NMR (400 MHz, CDCl3): δ = 161.59 (1C), 143.09 (1C), 140.12 (1C), 137.98 (1C), 129.52 (1C), 128.70 (1C), 128.28 (1C), 127.81 (1C), 119.28 (1C), 115.95 (1C), 115.58 (1C), 64.41 (1C). ESI-MS (m/z): [M - H]− calcd. for C13H11NO4, 244.1; found, 244.1.
4'-(hydroxymethyl)-6-nitro-[1,1'-biphenyl]-3-yl trifluoromethanesulfonate (10)
To a solution of 9 (1.0 g, 4.08 mmol) and N-phenyl-bis(trifluoromethanesulfonamide) (1.6 g, 4.40 mmol) in 12 mL CH3CN was added DIPEA (0.78 mL, 0.59 mmol) and the reaction mixture was stirred at room temperature for 2 h. TLC analysis showed completion of reaction and 10 mL EtOAc was added. The resultant mixture was washed with water, and organic layer was dried over MgSO4, filtered and solvent removed. Purification via silica gel chromatography (1:1 Hex : EtOAc) gave 10 as a pure yellow oil product (1.49 g, 97%). 1H NMR (400 MHz, CDCl3): δ = 7.93 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.38 (dd, J = 2.4 Hz, J = 8.8 Hz, 1H), 7.33 (d, J = 2.8 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 4.74 (s, 2H). 13C NMR (400 MHz, CDCl3): δ = 150.87 (1C), 148.09 (1C), 142.26 (1C), 139.12 (1C), 134.76 (1C), 129.70 (1C), 128.12 (1C), 127.58 (1C), 126.58 (1C), 124.97 (1C), 121.24 (1C), 121.58 (1C), 117.28 (1C), 64.89 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C14H10F3NO6S, 400.0; found, 399.9.
(2'-Nitro-5'-((trimethylsilyl)ethynyl)-[1,1'-biphenyl]-4-yl)methanol (11)
To a solution of 10 (1.5 g, 3.98 mmol), Pd(PPh3)2Cl2 (0.28 g, 0.40 mmol), and CuI (0.15 g, 0.80 mmol) in 20 mL CH3CN purged with Argon was added DIPEA (1.87 mL, 13.9 mmol) followed by dropwise addition of TMS-acetylene (2.25 mL, 15.9 mmol). The reaction mixture was stirred at room temperature for 2days. Upon completion 15mL EtOAc was added for dilution and was washed with satd solution of NH4Cl. Organic layer was dried over MgSO4, filtered and solvent removed. Purification via silica gel chromatography (Hex : EtOAc 2 : 1) gave 11 as a brown oil product (1.3 g, 98%). 1H NMR (400 MHz, CDCl3): δ = 7.54 (d, J = 8.0 Hz, 1H), 7.25 (m, 2H), 7.14 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 6.4 Hz, 2H), 4.47 (s, 2H), 0.00 (s, 9H). 13C NMR (400 MHz, CDCl3): δ = 148.05 (1C), 141.43 (1C), 136.63 (1C), 136.28 (1C), 135.62 (1C), 131.57 (1C), 129.83 (1C), 128.30 (1C), 128.06 (1C), 127.46 (1C), 124.59 (1C), 123.72 (1C), 102.83 (1C), 99.73 (1C), 65.12 (1C), 0.00 (3C). ESI-MS (m/z): [M + Na]+ calcd. for C18H19NO3Si, 348.1; found, 348.1.
(5'-Ethynyl-2'-nitro-[1,1'-biphenyl]-4-yl)methanol (12)
Potassium carbonate (1.0 g, 7.37 mmol) was added to a solution of 11 in 10 mL MeOH and 5 mL CH2Cl2. The reaction mixture was stirred for 4h at room temperature. Solvent was evaporated and 10 mL water and 10 mL EtOAc were added. The aqueous layer was extracted with EtOAc and the organic extract was dried over MgSO4, filtered and solvent removed. The crude product was purified via silica gel chromatography (Hex : EtOAc 2 : 1) to give 12 as a pale yellow solid product (0.64 g, 69 %). 1H NMR (400 MHz, CDCl3): δ = 7.79 (d, J = 8.0 Hz, 1H), 7.52 (m, 2H), 7.39 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 6.4 Hz, 2H), 4.71 (s, 2H), 3.26 (s, 1H), 1.80 (s, 1H). 13C NMR (400 MHz, CDCl3): δ = 148.05 (1C), 141.28 (1C), 136.32 (1C), 135.71 (1C), 135.44 (1C), 131.55 (1C), 127.97 (2C), 127.18 (2C), 126.64 (1C), 124.28 (1C), 81.38 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C15H11NO3, 246.1; found, 246.1.
Ethyl 5-(4'-(hydroxymethyl)-6-nitro-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylate (13)
Mixture of 12 (445.3 mg, 1.76 mmol) and ethyl 2-chloro-2-(hydroxyimino)acetate (553.0 mg, 3.52 mmol in 10 mL THF was purged with Argon. To this mixture triethylamine (0.49 mL, 3.52 mmol) was added slowly over 30 min and stirred at room temperature overnight. A mixture of 5 mL 1N HCl and 5 mL EtOAc was added. Organic layer was dried over MgSO4, filtered and solvent removed. The crude product was purified via silica gel chromatography (Hex : EtOAc 2 : 1) to give pale 13 as a yellow solid product (385.1 mg, 59%). 1H NMR (400 MHz, CD3CN): δ = 8.01 (m, 2H), 7.98 (m, 1H), 7.45 (m, 2H), 7.37 (m, 2H), 7.28 (s, 1H), 4.64 (d, J = 5.6 Hz, 2H), 4.10 (q, J = 7.2 Hz, 2H), 3.27 (t, J = 5.6 Hz, 1H), 1.37 (t, J = 7.2 Hz, 3H). 13C NMR (400 MHz, CD3CN): δ = 170.58 (1C), 160.85 (1C), 158.94 (1C), 144.31 (1C), 138.05 (1C), 136.48 (1C), 131.42 (1C), 130.81 (2C), 129.27 (2C), 128.40 (2C), 127.03 (1C), 126.55 (1C), 104.12 (1C), 64.63 (1C), 63.60 (1C), 14.75 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C19H16N2O6, 391.1; found, 391.0.
Ethyl 5-(4'-(((1,3-dioxoisoindolin-2-yl)oxy)methyl)-6-nitro-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylate (14)
Mixture of 13 (123.7 mg, 0.34 mmol), N-hydroxyphtalimide (65.7 mg, 0.40 mmol) and PPh3 (115.0 mg, 0.44was dissolved in anhydrous THF and was purged with Argon. Diisopropyl diazo-1,2-dicarboxylate (56.0 µL, 0.44 mmol) was slowly added to the above solution. The reaction mixture was stirred at room temperature overnight. The reaction mixture was cooled to 5–10 °C to precipitate the product and 10 mL water and 10 mL Et2O were added. The aqueous layer was separated and the organic phase with the precipitate was filtered by washing with cold Et2O. The filtrate was washed into a new flask by passing CH2Cl2 through the filter. Solvent was evaporated to give 14 as pure white solid product (104.0 mg, 60%) that was used without further purification. 1H NMR (400 MHz, CDCl3): δ = 7.97 (d, J = 8.4 Hz, 1H), 7.89 (dd, J = 8.4 Hz, J = 1.6 Hz, 1H), 7.83 (d, J = 1.6 Hz, 1H), 7.79-7.82 (m, 2H), 7.71-7.73 (m, 2H), 7.63 (d, J = 6.4 Hz, 2H), 7.35 (d, J = 6.4 Hz, 2H), 7.05 (s, 1H), 5.25 (s, 2H), 4.45 (q, J = 6.8 Hz, 2H), 1.42 (t, J = 6.8 Hz, 3H). 13C NMR (400 MHz, CDCl3): δ = 169.91 (1C), 163.41 (2C), 159.43 (1C), 157.74 (1C), 149.73 (1C), 137.25 (1C), 137.05 (1C), 134.46 (1C), 134.43 (1C), 130.17 (2C), 129.97 (1C), 129.25 (1C), 128.81 (1C), 128.03 (2C), 125.57 (1C), 125.22, (1C) 123.54 (4C), 102.22 (1C), 79.23 (1C), 62.48 (1C), 14.11 (1C). ESI-MS (m/z): [M + Na]+ calcd. for C27H19N3O8, 536.1; found, 536.1.
5-(4'-((Aminooxy)methyl)-6-nitro-[1,1'-biphenyl]-3-yl)isoxazole-3-carboxylic acid (15)
To a solution of 14 (95.0 mg, 0.19 mmol) in CH2Cl2 (5 mL) was added 50% aqueous hydrazine hydrate (23.0 µL, 0.37 mmol) and the reaction mixture was stirred at room temperature (3 h). The solvent was removed and the resultant crude mixture was redissolved in EtOAc and filtered to remove solid byproduct. Solvent was removed under reduced pressure and the white solid product was stirred with a mixture of THF (2 mL), H2O (5 mL) and 1 N NaOH (1.5 mL) (1.5 h). Volatile organics were removed under reduced pressure and the resulting aqueous phase was subjected directly to HPLC purification as described above in the General Synthetic methods (retention time = 19.5 minutes) to yield 15 as amorphous white solid product (33.1 mg, 49% yield; 99% purity by analytical HPLC). 1H NMR (400 MHz, DMSO-d6): δ = 8.19 (m, 2H), 8.13 (m, 1H), 7.76 (s, 1H), 7.45-7.51 (m, 4H). MALDI-MS (m/z): [M + Na]+ calcd. for C17H13N3O6, 378.0702; found, 378.0712.
Supplementary Material
Acknowledgements
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Hinnebusch BJ. J. Mol. Med. 1997;75:645. doi: 10.1007/s001090050148. [DOI] [PubMed] [Google Scholar]
- 2.Titball RW, Leary SE. Br. Med. Bull. 1998;54:625. doi: 10.1093/oxfordjournals.bmb.a011715. [DOI] [PubMed] [Google Scholar]
- 3.Zhang ZY, Clemens JC, Schubert HL, Stuckey JA, Fischer MW, Hume DM, Saper MA, Dixon JE. J. Biol. Chem. 1992;267:23759. [PubMed] [Google Scholar]
- 4.Zhang ZY. J. Biol. Chem. 1995;270:11199. doi: 10.1074/jbc.270.19.11199. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Montserat J, Lawrence DS, Zhang ZY. Biochemistry. 1996;35:9349. doi: 10.1021/bi960700+. [DOI] [PubMed] [Google Scholar]
- 6.Montserat J, Chen L, Lawrence DS, Zhang ZY. J. Biol. Chem. 1996;271:7868. doi: 10.1074/jbc.271.13.7868. [DOI] [PubMed] [Google Scholar]
- 7.Taylor SD, Kotoris CC, Dinaut AN, Wang Q, Ramachandran C, Huang Z. Bioorg. Med. Chem. 1998;6:1457. doi: 10.1016/s0968-0896(98)00075-3. [DOI] [PubMed] [Google Scholar]
- 8.Soellner MB, Rawls KA, Grundner C, Alber T, Ellman JA. J. Am. Chem. Soc. 2007;129:9613. doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- 9.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. J. Med. Chem. 2002;45:1712. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 10.McGovern SL, Helfand BT, Feng B, Shoichet BK. J. Med. Chem. 2003;46:4265. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 11.Bahta M, Lountos GT, Dyas B, Kim SE, Ulrich RG, Waugh DS, Burke TR. J. Med. Chem. 2011;54:2933. doi: 10.1021/jm200022g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu G, Xin Z, Pei Z, Hajduk PJ, Abad-Zapatero C, Hutchins CW, Zhao H, Lubben TH, Ballaron SJ, Haasch DL, Kaszubska W, Rondinone CM, Trevillyan JM, Jirousek MR. J. Med. Chem. 2003;46:4232. doi: 10.1021/jm034122o. [DOI] [PubMed] [Google Scholar]
- 13.Srinivasan R, Uttamchandani M, Yao SQ. Org. Lett. 2006;8:713. doi: 10.1021/ol052895w. [DOI] [PubMed] [Google Scholar]
- 14.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. J. Med. Chem. 2008;51:4370. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, Hakami RM, Dyas B, Bahta M, Lountos GT, Waugh DS, Ulrich RG, Burke TR., Jr Bioorg. Med. Chem. Lett. 2010;20:2813. doi: 10.1016/j.bmcl.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Structures of aldehydes examined as part of the current study are provided in the Supporting Information.
- 17.Ryan AJ, Gray NM, Lowe PN, Chung CW. J. Med. Chem. 2003;46:3448. doi: 10.1021/jm0340896. [DOI] [PubMed] [Google Scholar]
- 18.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. Nat. Chem. Biol. 2005;1:146. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 19.Grillo MP. Drug Metab. Handb. 2009:655. [Google Scholar]
- 20.Zhou Z, Fahrni CJ. J. Am. Chem. Soc. 2004;126:8862. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
- 21.CM Chemist Pro Software v 3.7-1f/MacOSX. La Jolla, CA: MolSoft LLC; 2009. [ https://molsoft.com/icmchemist-pro-html]. [Google Scholar]
- 22.Abagyan R, Orry A, Raush E, Totrov M. User Guide 3.6. La Jolla, CA: Molsoft LLC; 2009. [ https://www.molsoft.com/icm/index.html]. [Google Scholar]
- 23.Burke TR, Jr, Kole HK, Roller PP. Biochem. Biophys. Res. Commun. 1994;204:129. doi: 10.1006/bbrc.1994.2435. [DOI] [PubMed] [Google Scholar]
- 24.Phan J, Lee K, Cherry S, Tropea JE, Burke TR, Jr, Waugh DS. Biochemistry. 2003;42:13113. doi: 10.1021/bi030156m. [DOI] [PubMed] [Google Scholar]
- 25.Burke TR, Smyth MS, Nomizu M, Otaka A, Roller PP. J. Org. Chem. 1993;58:1336. [Google Scholar]
- 26.Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Biochemistry. 1994;33:6490. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
- 27.Combs AP, Yue EW, Bower M, Ala PJ, Wayland B, Douty B, Takvorian A, Polam P, Wasserman Z, Zhu W, Crawley ML, Pruitt J, Sparks R, Glass B, Modi D, McLaughlin E, Bostrom L, Li M, Galya L, Blom K, Hillman M, Gonneville L, Reid BG, Wei M, Becker-Pasha M, Klabe R, Huber R, Li Y, Hollis G, Burn TC, Wynn R, Liu P, Metcalf B. J. Med. Chem. 2005;48:6544. doi: 10.1021/jm0504555. [DOI] [PubMed] [Google Scholar]
- 28.Hu X, Vujanac M, Stebbins CE. J. Mol. Graph Model. 2004;23:175. doi: 10.1016/j.jmgm.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Tautz L, Bruckner S, Sareth S, Alonso A, Bogetz J, Bottini N, Pellecchia M, Mustelin T. J. Biol. Chem. 2005;280:9400. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- 30.Leone M, Barile E, Vazquez J, Mei A, Guiney D, Dahl R, Pellecchia M. Chem. Biol. Drug. Des. 2010;76:10. doi: 10.1111/j.1747-0285.2010.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman HE, Blair ER, Johndrow JE, Bishop AC. J. Am. Chem. Soc. 2005;127:2824. doi: 10.1021/ja043378w. [DOI] [PubMed] [Google Scholar]
- 32.Blair ER, Hoffman HE, Bishop AC. Bioorg. Med. Chem. 2006;14:464. doi: 10.1016/j.bmc.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, He Y, Liu S, Yu Z, Jiang ZX, Yang Z, Dong Y, Nabinger SC, Wu L, Gunawan AM, Wang L, Chan RJ, Zhang ZY. J. Med. Chem. 2010;53:2482. doi: 10.1021/jm901645u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The focus of the current work is not to validate modeling predictions and the central methodology of the paper does not depend on how accurately in silico results may reflect actual protein-ligand interactions
- 35.Patterson KI, Brummer T, O'Brien PM, Daly RJ. Biochem. J. 2009;418:475. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- 36.Streuli M, Krueger NX, Hall LR, Schlossman SF, Saito H. J. Exp. Med. 1988;168:1523. doi: 10.1084/jem.168.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tropea JE, Phan J, Waugh DS. Protein Expr. Purif. 2006;50:31. doi: 10.1016/j.pep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Lountos GT, Tropea JE, Cherry S, Waugh DS. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1013. doi: 10.1107/S0907444909023762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tropea JE, Cherry S, Nallamsetty S, Bignon C, Waugh DS. Methods Mol. Biol. 2007;363:1. doi: 10.1007/978-1-59745-209-0_1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.