

Abstract
Ferroportin and hepcidin are critical proteins for the regulation of systemic iron homeostasis. Ferroportin is the only known mechanism for export of intracellular non–heme-associated iron; its stability is regulated by the hormone hepcidin. Although ferroportin profoundly affects concentrations of intracellular iron in tissues important for systemic iron absorption and trafficking, ferroportin concentrations in breast cancer and their influence on growth and prognosis have not been examined. We demonstrate here that both ferroportin and hepcidin are expressed in cultured human breast epithelial cells and that hepcidin regulates ferroportin in these cells. Further, ferroportin protein is substantially reduced in breast cancer cells compared to nonmalignant breast epithelial cells; ferroportin protein abundance correlates with metabolically available iron. Ferroportin protein is also present in normal human mammary tissue and markedly decreased in breast cancer tissue, with the highest degree of anaplasia associated with lowest ferroportin expression. Transfection of breast cancer cells with ferroportin significantly reduces their growth after orthotopic implantation in the mouse mammary fat pad. Gene expression profiles in breast cancers from >800 women reveal that decreased ferroportin gene expression is associated with a significant reduction in metastasis-free and disease-specific survival that is independent of other breast cancer risk factors. High ferroportin and low hepcidin gene expression identifies an extremely favorable cohort of breast cancer patients who have a 10-year survival of >90%. Ferroportin is a pivotal protein in breast biology and a strong and independent predictor of prognosis in breast cancer.
Introduction
Iron is essential for normal cell function. Many cancers exhibit an increased requirement for iron, presumably because of the need for iron as a cofactor in proteins essential to sustain growth and proliferation (1–3). Modulation of iron-regulatory proteins affects growth of lung tumor xenografts (4, 5), and agents that deplete iron are currently under investigation as anticancer therapies (6–9).
Ferroportin (ferroportin 1, also termed Ireg1, MTP1, and SLC40A1) is a cell surface transmembrane protein and is the only known export protein for nonheme iron (10–12). Ferroportin is expressed at high concentrations on duodenal enterocytes, placenta, hepatocytes, and macrophages (10–12) and is an essential component of systemic iron homeostasis (13). Ferroportin is regulated by at least three mechanisms: transcriptional regulation, which controls levels (14) and splice variants (15) of the messenger RNA (mRNA); translational control, which regulates ferroportin through an iron-regulatory element in the 5′ untranslated region of ferroportin mRNA (16); and organismal iron status, which regulates ferroportin-mediated iron efflux through a direct interaction of ferroportin with the peptide hormone hepcidin (17). Hepcidin is secreted by the liver and binds to a specific extracellular loop domain on ferroportin (18). This results in phosphorylation (19) of ferroportin on the cell surface, which in turn leads to internalization and proteasome-mediated degradation of ferroportin (17).
Ferroportin has not been extensively studied in cancer (20, 21), and only limited examination of ferroportin has been made outside the tissues generally thought to be important in systemic iron homeostasis, such as the intestine, liver, bone marrow, and reticuloendothelial system (22). Because ferroportin has a central role in iron regulation, was among the genes decreased in breast cancer samples in an in silico analysis of the UniGene database (23), and is expressed in rat mammary epithelium (24), we examined ferroportin in human breast tumors. Here, we identify ferroportin as a critical determinant of outcome in breast cancer and propose a mechanistic explanation for its action.
Results
Ferroportin is decreased in breast cancer epithelial cells compared to breast cells with limited or no malignant potential
To explore whether ferroportin is present in normal human breast epithelial cells and whether its concentrations are altered in breast cancer, we compared ferroportin protein abundance in three pairs of mammary epithelial cell types with variable malignant potential: (i) primary normal human mammary epithelial (HME) cells and tumor-forming variants of these cells derived by sequential transformation of HME cells with the catalytic subunit of telomerase, SV40 T antigen, and high levels of oncogenic H-ras (25) (termed R5 cells here); (ii) MCF10A cells, a spontaneously immortalized diploid cell line obtained from reduction mammoplasty (26), and MCF7 (27), a breast cancer cell line established from a pleural effusion in a patient with metastatic breast cancer; and (iii) SUM102 cells, a breast epithelial cell line with a normal karyotype isolated from early-stage breast cancer (28), and SUM149, a cell line developed from an aggressive inflammatory breast cancer (29). Examination of ferroportin in these cells revealed that protein abundance was reduced in all aggressive breast cancer cell lines when compared to their counterparts with little or no malignant potential (Fig. 1A).
Fig. 1.
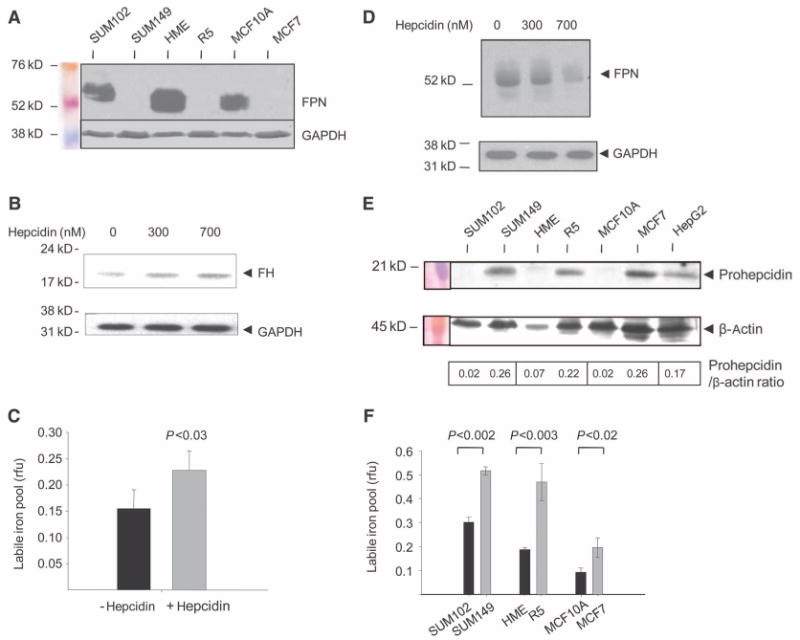
A decrease in ferroportin and increase in hepcidin are associated with an increase in the labile iron pool (LIP) in breast cancer cell lines. (A) Concentrations of ferroportin (FPN) in normal and malignant breast cells. Protein (50 μg) from each cell type was analyzed for ferroportin expression by Western blotting. Loading was assessed with an antibody to GAPDH. (B) Hepcidin treatment increases concentrations of ferritin protein in breast cells. HME cells were treated with vehicle and 300 or 700 nM hepcidin for 6 hours, and ferritin H (FH) was assessed by Western blotting. GAPDH was used as a loading control. The increase in ferritin was about twofold as measured by quantification of ferritin/GAPDH ratios by scanning densitometry. (C) Hepcidin treatment increases the LIP. HME cells were treated with 700 nM hepcidin or vehicle control, and the LIP was measured as described in Materials and Methods. rfu, relative fluorescence units. (D) Ferroportin is degraded in normal mammary epithelial (HME) cells treated with hepcidin. Cells were incubated with vehicle and 300 or 700 nM hepcidin for 6 hours, and ferroportin was measured by Western blotting. GAPDH was used as a loading control. (E) Western blot of prohepcidin protein in normal and malignant breast cells. The HepG2 hepatocellular carcinoma cell line was used as a positive control. (Prohepcidin was detected in all cells on prolonged exposure.) β-Actin was used as a loading control. The calculated ratio of prohepcidin to β-actin signal intensity is shown. (F) LIP in normal and malignant breast cells. Graphs show mean and SD of triplicate determinations.
We next performed real-time reverse transcription polymerase chain reaction (RT-PCR) analysis of ferroportin mRNA in these cells. Consistent with Western blot analysis, ferroportin mRNA levels were lower in malignant R5 and MCF7 cells than in nonmalignant HME and MCF10A cells (fig. S1A). However, ferroportin mRNA was higher in SUM149 breast cancer cells than in nonmalignant SUM102 cells (fig. S1A). No ferroportin splice variants were detected (fig. S1B). These results suggested that posttranscriptional mechanisms might also contribute to observed ferroportin protein abundance.
Hepcidin is expressed and regulates ferroportin in breast cells
A recently discovered mechanism of posttranscriptional regulation of ferroportin involves hepcidin, a peptide hormone that binds to ferroportin and triggers its degradation (17). This regulatory axis has been elucidated in cell types responsible for control of systemic iron, such as the enterocyte, macrophage, and hepatocyte (22). To test whether hepcidin-mediated regulation of ferroportin also occurs in mammary epithelial cells and whether this mechanism of posttranscriptional control might contribute to the decrease in ferroportin protein abundance in breast cancer cells, we assessed prohepcidin expression in our panel of primary breast cells and cell lines. Prohepcidin mRNA was detected in normal breast epithelial cells and in all cancer cell lines tested (fig. S2). Further, hepcidin-mediated degradation of ferroportin was associated with an increase in ferritin and increased labile iron in breast cells (Fig. 1, B and C); in addition, ferroportin expressed in breast epithelial cells was susceptible to degradation in response to treatment with exogenous hepcidin (Fig. 1D). Thus, the entire ferroportin-hepcidin regulatory axis is intact and functional in mammary epithelial cells. We then performed Western blot analysis to test whether hepcidin concentrations differed in our panel of normal and malignant breast cells. Notably, concentrations of pro-hepcidin protein were higher in all breast cancer cells when compared to nonmalignant breast cells (Fig. 1E). Collectively, these results suggest that both transcriptional and posttranscriptional mechanisms contribute to the decrease in ferroportin concentrations in breast cancer cells when compared to their nonmalignant counterparts.
Ferroportin reduction in breast cancer cells is associated with an increase in labile iron
Alterations in iron efflux mediated by changes in concentrations of ferroportin affect the labile iron pool (LIP) as measured by changes in ferritin in transfected cells (17). Similarly, we observed that overexpression of ferroportin in a breast cancer cell line was associated with a reduction in ferritin (fig. S3). Thus, a decrease in ferroportin such as we observed in breast cancer cells might be expected to increase metabolically available iron. To test this, we directly measured the LIP in normal breast epithelial cells and fully transformed breast cancer cells and found that the low concentrations of ferroportin protein expressed in breast cancer cells were indeed associated with higher concentrations of the LIP (Fig. 1F). This suggests that variations in ferroportin expression have functional consequences in cellular iron homeostasis.
Increased concentrations of ferroportin reduce breast tumor growth in mice
To explore the mechanism by which ferroportin affects the behavior of breast cancer cells in vivo and to address whether alterations in ferroportin drive or simply correlate with a more aggressive breast cancer phenotype, we transfected human MDA-MB-231-luc cells, which express low concentrations of ferroportin (fig. S4A), with an expression vector for ferroportin or with a control empty vector. Transfection restored ferroportin to values approximating those in nonmalignant HME cells (fig. S4B). Female nude mice were then injected orthotopi-cally in the mammary fat pad with control or transfected cells, and tumor growth was monitored for 4 weeks, when control tumors reached the predetermined humane size limit. Enhanced expression of ferroportin decreased final tumor weights and the rate of tumor growth (Fig. 2, A to C). Thus, ferroportin overexpression reduces growth of xenografted breast cancer cells in vivo.
Fig. 2.
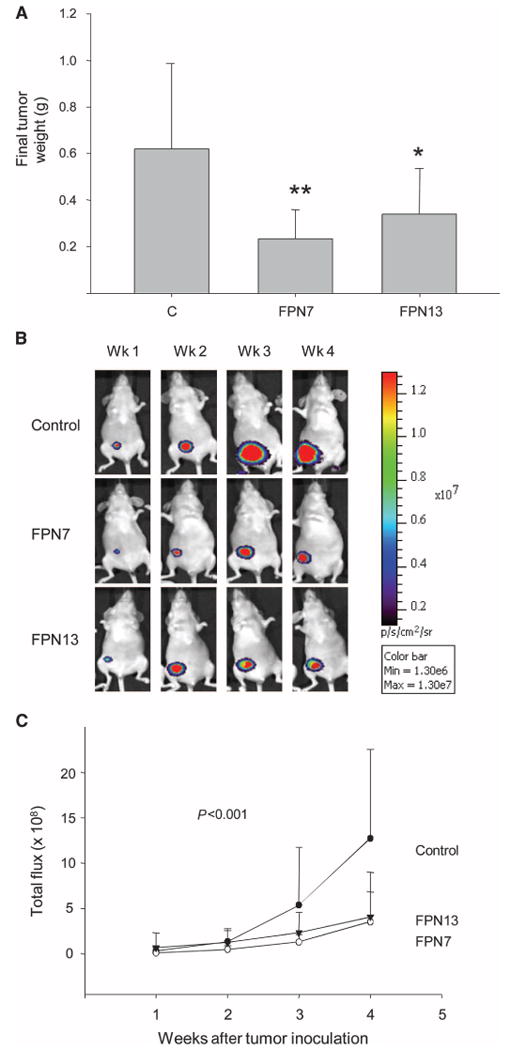
Increased concentrations of ferroportin decrease growth of breast cancer xenografts. MDA-MB-231-luc breast cancer cells were transfected with an expression vector for ferroportin or control empty vector. Two independent ferroportin clones were isolated (FPN7 and FPN13). (A) Final tumor weights [n = 10, 8, and 13 for controls (C), FPN7, and FPN13, respectively]. *P = 0.013, **P = 0.029, difference from controls; Student's t test. (B) Representative bioluminescent images of individual mice within each group. (C) Quantified bioluminescence in control and ferroportin tumors. Means and SDs are plotted. P value represents test for the time by group interaction, indicating a significant difference among the three groups.
Ferroportin is decreased in human breast cancer tissue
To test whether ferroportin concentrations were also altered in the tissue of breast cancer patients, we performed immunohistochemical analysis. Tissue derived from a single patient that contained areas of normal epithelium (Fig. 3A), ductal carcinoma in situ (Fig. 3B), and invasive breast cancer (Fig. 3C) within the same section showed that staining intensity decreases with increasing malignant potential, with highest expression in normal ductal structures and lowest expression in invasive tissue. Immunohistochemical staining of tissue from an additional four patients revealed a similar trend (fig. S5).
Fig. 3.
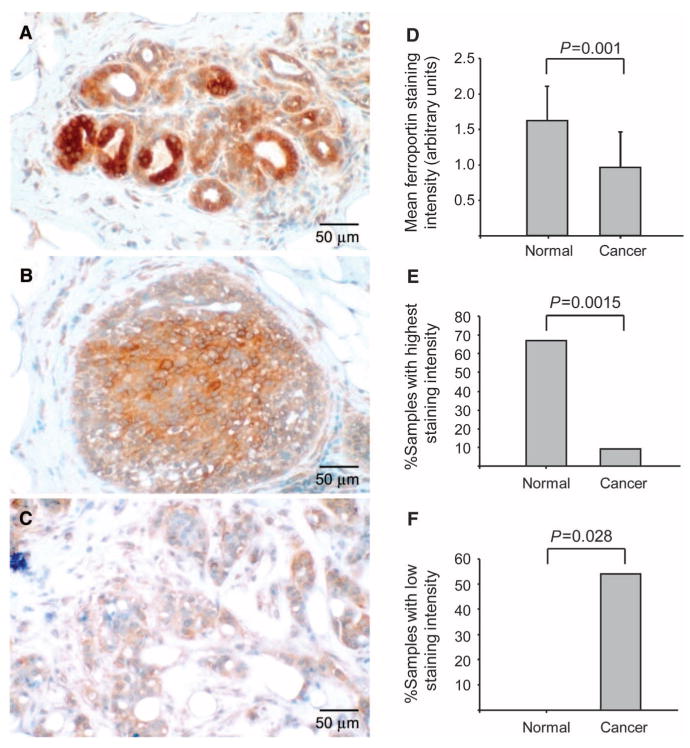
Ferroportin is decreased in human breast cancer tissue. (A to C) Ferroportin staining in tissue. Tissue was isolated from a patient diagnosed with invasive ductal carcinoma. Within this single tissue, normal epithelium, ductal carcinoma in situ, and invasive breast cancer cells were observed. The tissue was stained with antibody to ferroportin 1. Original magnification, ×220. (A) Normal tissue. (B) Ductal carcinoma in situ. (C) Invasive breast cancer. (D to F) Ferroportin staining of breast TMAs. Breast TMAs were stained with antibody to ferroportin, and intensity of staining was scored as described in Materials and Methods. The range of scores was 0 to 2 (low to high). (D) Mean and SD of intensity score of normal breast tissue and cancer tissue. (E) Percentage of cells with a staining intensity of 2. (F) Percentage of tissue specimens with a staining intensity of 1.
Next, tissue microarrays (TMAs) containing 154 samples of breast tissue from breast cancer patients and 6 samples from normal breast were stained with antibody to ferroportin and scored semiquantitatively by two independent blinded observers on a scale of 0 to 2, with 0 representing low or undetectable staining and 2 representing intense staining. The overall intensity of staining in the normal samples was higher than that of the cancer samples (1.63 ± 0.5 compared to 0.96 ± 0.5 in the cancer samples) (mean ± SD, P = 0.001) (Fig. 3D). Seventy percent of the normal samples received the highest staining intensity score of 2.0, whereas only 9% of the cancer samples received this score (P = 0.0015, Fisher's exact test) (Fig. 3E). Conversely, staining in 52% of the cancer samples scored <1.0; this low staining intensity was seen in none of the normal samples (P = 0.028, Fisher's exact test) (Fig. 3F). These observations are consistent with a reduction in ferroportin protein abundances in human breast cancer tissue and in breast cancer cell lines.
Breast cancer molecular subtypes differ in ferroportin expression
Breast cancers have been classified into molecular subtypes with significant outcome differences by hierarchical clustering of microarray expression data: normal-like, luminal A, ERBB2+-like, luminal B, and basal (30). We tested whether these breast cancer subtypes differed in ferroportin gene expression. Tumors from a cohort of 251 consecutive breast cancer patients (31) were assigned molecular subtypes (32) according to computed correlations with subtype centroids. Ferroportin was significantly differentially expressed among the subtypes, with lowest expression in the poor-prognosis subtypes (Fig. 4A). The average ferroportin expression in the subtypes with poorest prognosis— basal, ERBB2+-like, and luminal B—was only 65% of that of the favorable prognosis subtypes luminal A and normal-like (P < 0.0001). The normal-like subtype (with highest ferroportin expression) showed an average of twice as much expression as that observed in the basal subtype (with lowest ferroportin expression) (P < 0.0001). Differences between expression of ferroportin in subtypes with good prognosis (normal and luminal A) and subtypes with poorer prognosis (ERBB2+, luminal B, and basal) were statistically significant in all pairwise comparisons (Fig. 4A). Low ferroportin was also significantly associated with classical prognostic indicators of poor outcome, including high histologic grade (P ≤ 0.0009), absence of estrogen receptor (ER) (P < 0.0001), and spread of disease to lymph nodes (P < 0.027) (Fig. 4, B to D). Thus, lower ferroportin gene expression is associated with poor-prognosis molecular and clinical subtypes.
Fig. 4.

Ferroportin expression is correlated with clinical and molecular features of breast cancer. (A) Ferroportin expression in breast cancer molecular subtypes. Shown are box-and-whisker plots of ferroportin gene expression as a function of molecular subtype in consecutive breast cancer patients from Uppsala, Sweden (31). Shaded rectangles represent interquartile range; line in the middle of each rectangle represents median value. Lines extending from the interquartile range mark the 5th and 95th percentile values, and the individual open circles represent values that are either above the 95th percentile or below the 5th percentile for each distribution. P values are shown above bridges linking the subtypes. LumA, luminal A; LumB, luminal B; ERBB2+, ErbB2/HER2/neu-positive–like. (B) Ferroportin expression is correlated with histologic grade. Shown are box-and-whisker plots of ferroportin gene expression as a function of histologic grade (1, 2, 3) in the Uppsala cohort. P values (Student's t test) are shown above bridges linking grade categories. (C) Ferroportin expression is correlated with breast tumor ER status. Shown are box-and-whisker plots of ferroportin gene expression as a function of ER status (+, −) in the Uppsala cohort. P value (Student's t test) is shown above the bridge linking ER categories. (D) Ferroportin expression is correlated with lymph node (LN) status. Shown are box-and-whisker plots of ferroportin gene expression as a function of lymph node status (+, −) in the Uppsala cohort. P value (Student's t test) is shown above the bridge linking grade categories.
Ferroportin expression predicts clinical outcome in breast cancer
The remarkably consistent decrease in ferroportin protein abundance in malignant breast tissue and the association of decreased ferroportin gene expression with molecular subtypes of breast cancer with poor prognosis led us to test whether ferroportin concentrations were related to breast cancer outcome. To perform this analysis, we leveraged four large patient cohorts with accompanying gene expression profiling data from studies of breast cancer patients with extensive clinical follow-up. These represent four of the largest data sets in the public domain in which microarray profiles and long-term patient outcomes are available: (i) 103 patients from the Norway/Stanford study of response to chemotherapy of locally advanced cancer (33); (ii) 295 consecutive breast cancer patients from the Netherlands Cancer Institute (NKI) (34); (iii) 251 consecutive breast cancer patients from Uppsala, Sweden (35); and (iv) 159 surgically resected breast cancer patients from the Karolinska Institute in Stockholm, Sweden (31). Patient outcomes measured in these studies were either disease-specific survival (DSS) (death due to breast cancer) or distant metastasis–free survival (DMFS) (recurrence of cancer at a distant organ site). In each study, we calculated a mean concentration of ferroportin gene expression. Patient samples with ferroportin expression at or above this cutoff were classified as high expressors, and those below were classified as low expressors. DMFS or DSS of high versus low ferroportin expressors was analyzed by Kaplan-Meier survival analysis.
In all four studies, low ferroportin gene expression was associated with a statistically significant and clinically substantial reduction in metastasis-free survival [P value from log-rank test = 0.003 (Norway/Stanford), 0.0006 (NKI), 0.036 (Uppsala), and 0.007 (Stockholm)] (Fig. 5, A to D). The most pronounced effect was seen in the Norway/Stanford study, where the 8-year disease-free survival rates were separated by >30% (77% for those with high ferroportin compared to 43% for those with low ferroportin). The other three studies showed comparable metastasis-free survival benefits for high ferroportin (89% versus 65% for NKI, 90% versus 76% for Uppsala, and 91% versus 79% for Stockholm).
Fig. 5.

Ferroportin expression in primary breast tumors is prognostic of low risk of recurrence in multiple independent microarray data sets. Breast cancer patients were ranked according to ferroportin expression levels, and DSS or DMFS of patients with below-mean expression was compared to that of patients with above-mean expression. (A to D) Kaplan-Meier plots are shown for (A) the Norway/Stanford cohort (33) (included were 103 tumors with reported expression values for ferroportin; data for 19 tumors were reported as “missing” in the original data set and these were excluded from the analysis), (B) the NKI cohort (34), (C) the Uppsala cohort (35), and (D) the Stockholm cohort (31). Log-rank tests were used to compare the survival curves between groups and to generate the P values for these comparisons.
Hepcidin expression provides incremental predictive value to ferroportin measures in breast cancer patients
Hepcidin-mediated posttranslational modulation of ferroportin activity is not directly assessable through gene expression analysis. However, because protein-level inhibition of ferroportin is linked to high mRNA expression of hepcidin (36), and hepcidin is expressed in breast cells (Fig. 1 and fig. S2), we examined the relationship among hepcidin gene expression, ferroportin gene expression, and disease outcome in breast cancer patients. We selected for this analysis a combined population-based (unselected) cohort in which all patients (n = 504) had been studied with a microarray platform containing probe sets for both ferroportin and hepcidin (31, 37, 38) (see Materials and Methods). The signal intensity for hepcidin was substantially above the negative control and roughly comparable to that of other genes with roles in breast cancer, such as HER2/neu, ERα, VEGF, BRCA1, and Ki-67, confirming the expression of hepcidin in breast tumor tissue (fig. S6).
We observed that ferroportin concentrations can discriminate patient outcomes with statistical significance (Fig. 6A, top left panel) in this combined cohort (P = 0.0004), whereas breast tumor hepcidin mRNA is of borderline significance as a prognostic marker by itself (P = 0.06) (Fig. 6A, top right panel). However, in the presence of high ferroportin (Fig. 6A, left bottom panel), hepcidin expression confers statistically significant prognostic resolution (P = 0.001), with the combination of low hepcidin and high ferroportin having 95% 5-year and 91% 10-year DMFS. Conversely, high ferroportin together with high hepcidin gene expression identifies a patient population with poor prognosis comparable to that of low ferroportin. As predicted from the iron biology, in the presence of low ferroportin (Fig. 6A, right bottom panel), differential hepcidin expression adds no additional prognostic value (P = 0.73), reflecting that if little to no ferroportin is made, its posttranslational regulation has no prognostic consequence.
Fig. 6.

Ferroportin and hepcidin prognostic interactions. (A) Associations between DMFS and high or low ferroportin and hepcidin expression levels (based on mean partitioning) in a combined multi-institutional population-based cohort consisting of 504 breast cancer cases. Kaplan-Meier plots and log-rank P values are shown for (i) ferroportin expression, (ii) hepcidin expression, (iii) high ferroportin dichotomized by low versus high hepcidin, and (iv) low ferroportin dichotomized by low versus high hepcidin. (B) Prognostic value of high ferroportin and low hepcidin expression in a combined multi-institutional cohort of uniformly treated ER+ breast cancer patients. The Kaplan-Meier plot compares the combined effects of ferroportin and low or high hepcidin expression on DMFS in patients treated with tamoxifen monotherapy.
To further assess the prognostic value of ferroportin plus hepcidin gene expression, we used stepwise Cox proportional hazards regression to determine whether ferroportin plus hepcidin expression is an independent predictor of metastasis-free survival after allowing for other conventional prognostic variables (lymph node status, tumor size, grade, age, and ER status) to be considered as covariates. We observed that high ferroportin and low hepcidin remained a significant and independent predictor of metastasis-free survival even in the presence of other traditional risk factors (P = 0.003) (table S1). These results indicate that assessment of ferroportin and hepcidin expression provides additional prognostic power beyond that which can be obtained with conventional clinical prognostic factors.
Given the ability of combined ferroportin plus hepcidin mRNA expression to identify a population of breast cancer patients with a 10-year metastasis-free survival rate of >90%, we sought to further define a clinical context in which this interaction might be useful in therapeutic decision making. ER+ breast cancer is one such context because identifying patients who benefit from tamoxifen alone versus those who will require more aggressive combined tamoxifen plus chemotherapy remains a considerable prognostic challenge. Thus, we assembled a curated collection of ER+ breast tumor expression profiles for which treatment or outcome data and microarray expression measurements inclusive of ferroportin and hepcidin were publicly available. From this collection of patients (n = 518), we selected a subgroup (n = 276) who had all received similar therapy (adjuvant tamoxifen monotherapy). Forty-one percent of the patients in this group were lymph node–positive. The high ferroportin and low hepcidin expressors (n = 76) demonstrated a significantly better metastasis-free survival rate (93% at 5 years and 89% at 10 years; P = 0.0005) than the remaining population (76% at 5 years and 65% at 10 years) (Fig. 6B).
Discussion
Iron availability can be regulated by increased uptake, a shift of iron from storage to active pools (the LIP), or a reduction in cellular iron export. Several of these processes are altered in cancer. For example, an increase in transferrin receptor 1, a cell surface receptor responsible for transferrin-mediated iron uptake, occurs in many cancers, including breast cancer (39–41). Ferritin, an iron storage protein, is decreased by the c-myc (42) and E1a (43) oncogenes; reduced ferritin is thought to shift iron from storage to a labile pool of, metabolically available iron. Similarly, antisense-mediated repression of ferritin increases the LIP (44) and stimulates H-ras–dependent proliferation (45). In principle, a decrease in iron export could also increase labile iron and affect breast cancer phenotype and outcome. However, relatively little is known regarding the role of iron export in cancer.
Here, we observed a marked reduction of ferroportin, the only known exporter of nonheme iron, in breast cancer compared to normal breast epithelium. This reduction of ferroportin protein occurred both in malignant breast cancer cell lines and in breast cancer tissue, particularly in the more aggressive and invasive areas of the cancer. The alteration in ferroportin expression was sufficient to alter the LIP, a key arbiter of iron availability in cells, and to affect growth of tumor xenografts. Our results are concordant with emerging evidence of the importance of ferroportin in iron homeostasis, both at an organismal level in transmitting the signals from hepcidin to the systemic iron-regulatory network (46) and in regulating iron homeostasis in cells (17). These data also suggest that altered iron homeostasis may play a previously unappreciated role in aggressive breast cancer behavior, although additional investigation will be needed to clarify the role of ferroportin and hepcidin in breast cancer biology.
Our data also indicate that ferroportin plays an important role in the clinical behavior of breast cancer. Our results reveal that (i) ferroportin gene expression is a previously unrecognized determinant of outcome that in logistic regression analysis is independent of other prognostic factors; (ii) ferroportin not only equals the best clinical predictors of outcome in breast cancer patients but also tracks with recently identified molecular subtypes of breast cancer that can add significant prognostic and predictive information to standard outcome parameters of breast cancer (47); (iii) the marked decrease in tumor growth in vivo of ferroportin-overexpressing breast cancer cells provides evidence that ferroportin expression not only is a marker of poor prognosis in primary breast cancer but also contributes to a clinically aggressive phenotype; and (iv) the additive value of ferroportin and hepcidin gene expression in separating good- and poor-prognosis patients provides further support for a critical role of iron homeostasis in breast cancer behavior.
Combined ferroportin and hepcidin gene expression identifies a clinical subset of breast cancer patients who should be evaluated in future studies to determine whether they could be spared potentially toxic treatments. The survival of ER+ patients with high ferroportin and low hepcidin gene expression seen in Fig. 6B is comparable to that of ER+, node-negative patients classified into the good outcome group by the Oncotype Dx 21-gene panel (48). Using combined ferroportin and hepcidin gene expression, we identified not only node-negative but also node-positive, ER+ breast cancer patients who exhibit this good outcome (41% of the high ferroportin and low hepcidin expressors in our study were node-positive at diagnosis). Thus, if confirmed in additional patient cohorts, ferroportin activity, as approximated by a two-gene model of ferroportin and hepcidin transcript concentrations, may be clinically useful as a treatment indicator for both node-negative and node-positive, ER+ breast cancer patients.
Ferroportin may be important in other tumor types. Ferroportin was decreased by a factor of 6 in an analysis of global gene expression changes in human hepatocellular carcinoma (49); our inspection of the Oncomine database revealed that decreases in ferroportin are observed in prostate cancer and leukemia, although they are not seen in brain cancer, esophageal cancer, or seminoma (50).
It remains to be determined how the cross talk among ferroportin, hepcidin, and other members of the iron-regulatory pathway is mediated differently in normal and cancer cells, as well as how the complex interplay of cell types in breast tissue may contribute to alterations in iron homeostasis in breast cancer. Finally, the metastasis-free survival advantage to patients whose tumors have increased ferroportin gene expression, and the incremental enhancement in outcome for a group with both increased ferroportin expression and low hepcidin expression, suggest that measures of ferroportin and hepcidin gene expression might help aid prognosis or guide therapy for women with breast cancer, a possibility that is best tested in appropriately designed prospective trials.
Materials and Methods
Cell culture
HME cells were obtained from Lonza. HME cells transduced with h-TERT, SV40 T antigen, and high levels of H-ras are termed R5 cells here and were a gift from the laboratory of R. Weinberg (25). All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. Cells were maintained in Dulbecco's minimal essential medium (DMEM)–F12 (Gibco/BRL) supplemented with L-glutamine, insulin (10 μg/ml), human epidermal growth factor (10 ng/ml), and hydrocortisone (0.5 μg/ml) for 24 hours before harvest. MCF7 and MCF10A cell lines were obtained from the Wake Forest University Comprehensive Cancer Center Tissue Culture Core facility. SUM149 and SUM102 cell lines were a gift of I. Berquin (Wake Forest University School of Medicine). HepG2 and HeLa cells were obtained from the American Type Culture Collection (ATCC) and grown in DMEM. K562 (from ATCC) was cultured in RPMI 1640 medium. Hepcidin was obtained from Peptides International, dissolved in water, and added to cells at a final concentration of 300 or 700 nM. Cells were harvested after 6 hours of treatment.
Western blotting
Cells were washed once in phosphate-buffered saline (PBS) and scraped. Whole cellular protein was extracted with NP-40 lysis buffer [25 mM tris (pH 7.4), 1% Triton X-100, 1% SDS, 1% sodium deoxy-cholate, 150 mM NaCl, aprotinin (2 μg/ml), 1 mM phenylmethylsulfonyl fluoride] containing complete protease inhibitor cocktail (Roche Diagnostics). Samples were separated by SDS–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluo-ride, and blotted with antibodies to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Fitzgerald), β-actin (Chemicon International), ferritin H (49), ferroportin (Alpha Diagnostics or Abcam), and hepcidin (Abcam) (see details for this and other methods in the Supplementary Material). Western blots were quantified with UNSCANIT software.
Measurement of ferroportin mRNA and splice variants
Real-time RT-PCR was performed to measure ferroportin mRNA in different breast cell lines. PCR was carried out on the ABI Prism 7000 sequence detection system (Applied Biosystems). The standard curve method was chosen for quantification. Total RNA was isolated with Trizol reagent (Invitrogen) according to the manufacturer's instructions.
Tissue array staining
Studies on human tissue specimens were conducted with approval from the Wake Forest University Health Sciences Institutional Review Board. Construction of the breast TMA has been described (51). Slides were stained with antibody to ferroportin (Alpha Diagnostics).
Semiquantitative analysis of staining intensity was performed as described (51) by two independent blinded observers, with 0 representing low or undetectable staining, 1 representing intermediate staining, and 2 representing intense staining.
LIP assay
The cellular LIP was measured with fluorescent metallosensor calcein, essentially as described (20).
Transfection and isolation of ferroportin-expressing breast cancer cells
An expression vector encoding a functional ferroportin–green fluorescent protein (GFP) fusion protein was obtained as a gift from J. Kaplan (University of Utah) (17). The ferroportin-GFP cassette was amplified by PCR and subcloned into a lentiviral vector carrying a puromycin resistance marker (gift of G. Sui, Wake Forest University Health Sciences). Plasmids were subsequently purified and sequenced. Lentivirus particles were produced by transient cotransfection of the ferroportin expression vector and packaging vectors (VSVG, pMDLG, and RSV-REV) into 293T cells (52, 53). Viral particles containing control empty vector were prepared similarly. Lentivirus was harvested after 48 hours and used to infect the MDA-MB-231-luc-D3H2LN human breast cancer cell line (Caliper Life Sciences).
Monitoring of tumor growth in vivo
All animal procedures were approved by the Wake Forest University School of Medicine Animal Care and Use Committee. Female athymic nude mice (∼10 weeks of age) were anesthetized by isoflurane inhalation (2% induction, 1 to 2% maintenance) and injected with 60 μl of 2 × 106 MDA-MB-231-luc-D3H2LN-ferroportin or MDA-MB-231-luc-D3H2LN-vector cells suspended in 50% Matrigel–50% Dulbecco's PBS (Invitrogen) into the fourth inguinal mammary fat pad. Tumor growth was monitored weekly by bioluminescent imaging in a subset of animals. Bioluminescent imaging was performed with a cooled charge-coupled device camera mounted in a light-tight specimen box (IVIS, Caliper Life Sciences). Tumors were excised and weighed at the termination of the study.
Microarray data sets
Correlations between ferroportin expression in primary breast tumors and metastatic recurrence in patients were assessed with gene expression profiles from publicly accessible microarray data sets: (i) the Norway/Stanford study (33) (http://genome-www.stanford.edu/breast_cancer/mopo_clinical/data.shtml), (ii) the NKI study (34) (http://www.rii. com/publications/2002/nejm.html), (iii) the Uppsala study (35) [Gene Expression Omnibus (GEO) accession number GSE3494], and (iv) the Stockholm study (31) (GEO accession number GSE1456).
For analyzing ferroportin and hepcidin interactions, two large combined multi-institutional cohorts were used. The first consists of three population-based cohorts totaling 504 breast cancer cases annotated for clinical follow-up: Uppsala (GSE3494) (35), Stockholm (GSE1456) (31), and Singapore (GSE4922) (37, 38). This data set was used because not all the data sets we previously analyzed for ferroportin included information on hepcidin expression. In this instance, each cohort represents an unselected population of patients exhibiting a diverse range of breast cancer phenotypes, and each was profiled on both the Affymetrix U133A and U133B microarray platforms. The ferroportin microarray probe set (233123_at) is found only on the U133B GeneChip, whereas the hepcidin probe set (220491_at) is found exclusively on the U133A GeneChip. This cohort allowed us to investigate the prognostic interaction between ferroportin and hepcidin in unselected patient populations. The second large combined cohort, unlike the first, consists exclusively of ER+ breast cancer cases (n = 518) derived from both unselected and selected patient populations: Uppsala (GSE3494) (35), Stockholm (GSE1456) (31), Singapore (GSE4922) (37, 38), and Oxford (GSE6532) (54). The Oxford collection is a selected cohort composed of only ER+ breast cancer cases treated by adjuvant tamoxifen monotherapy (54). The purpose of this combined cohort was to allow a subset analysis of ER+ breast cancer cases uniformly treated with adjuvant hormonal therapy without chemotherapy (as shown in Fig. 6B).
Assignment of tumors to molecular subtypes
The Uppsala cohort was used (21). Of the 251 tumors in this cohort, 228 showed correlation of >0.1 with at least one subtype; the remaining 23 were classified as “no subtype” and censored. Molecular subtypes were assigned by Calza et al. (32).
Statistical analyses
Statistical analyses were performed in the core biostatistical facility of the Comprehensive Cancer Center of Wake Forest University by one of us (RD.). The significance of LIP values in cancer and noncancer cells was assessed with t tests. The significance of ferroportin in breast cancer versus normal breast epithelial tissue was calculated with Fisher's exact test. The significance of ferroportin in tumor growth was calculated with a two-way repeated-measures analysis of variance (ANOVA), where group, time, and the group by time interaction were included in the model. The significance of ferroportin and/or hepcidin expression in 10-year DMFS was calculated by the Kaplan-Meier method. Cox proportional hazards regression models were used to examine the significance of ferroportin plus hepcidin expression in the presence of established prognostic factors. The significance of ferroportin in breast cancer molecular subtypes was determined with a one-way ANOVA model followed by pairwise comparisons between groups within the ANOVA framework.
Supplementary Material
Supplementary Material
http://www.sciencetranslationalmedicine.org/cgi/content/full/2/43/43ra56/DC1
Materials and Methods
Fig. S1. Ferroportin mRNA and splice variants in breast epithelial cells.
Fig. S2. Assessment of hepcidin mRNA in breast cells by RT-PCR.
Fig. S3. Increased ferroportin expression decreases ferritin in breast cancer cells.
Fig. S4. Relative concentrations of ferroportin protein in MDA-MB-231 cells and transfectants and specific detection of ferroportin by Western blotting.
Fig. S5. Ferroportin protein is decreased in breast cancer.
Fig. S6. Signal intensities of ferroportin and hepcidin relative to control genes and other breast cancer genes in the Uppsala cohort.
Table S1. Ferroportin and hepcidin expression predicts outcome in multivariate analyses.
References
Acknowledgments
We thank I. De Domenico and J. Kaplan for contributing the ferroportin expression clone and J. Buss for assistance in developing the calcein assay.
Funding: Supported in part by grant R37DK42412 from the National Institute of Diabetes and Digestive and Kidney Diseases (F.M.T.), R01 DK071892 (S.V.T.), and by a minority supplement to R37DK42412 (Z.K.P.).
Footnotes
Author contributions: Z.K.P. and L.D.M. performed the research and helped write the paper; W.W., T.K., M.C.W., H.H., L.T., and G.S. performed the research; R.D. analyzed the results; S.V.T. and F.M.T. provided funding, designed the research, and wrote the paper.
Competing interests: The authors declare that they have no competing interests. Wake Forest University Health Sciences is planning to submit a patent application based on this work.
Accession numbers: Accession numbers for the gene expression data are provided in the Microarray data set section of Materials and Methods.
References and Notes
- 1.Thelander L, Gräslund A, Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: Possible regulation mechanism. Biochem Biophys Res Commun. 1983;110:859–865. doi: 10.1016/0006-291x(83)91040-9. [DOI] [PubMed] [Google Scholar]
- 2.Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Curr Med Chem. 2003;10:1021–1034. doi: 10.2174/0929867033457638. [DOI] [PubMed] [Google Scholar]
- 3.Cairo G, Bernuzzi F, Recalcati S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006;1:25–39. doi: 10.1007/BF02829934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maffettone C, Chen G, Drozdov I, Ouzounis C, Pantopoulos K. Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS One. 2010;5:e10163. doi: 10.1371/journal.pone.0010163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis. 2007;28:785–791. doi: 10.1093/carcin/bgl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci USA. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DA, Antholine WE, Wong SJ, Richardson DR, Chitambar CR. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: Effect on the R2 subunit of ribonucleotide reductase. Clin Cancer Res. 2001;7:3574–3579. [PubMed] [Google Scholar]
- 8.Karp JE, Giles FJ, Gojo I, Morris L, Greer J, Johnson B, Thein M, Sznol M, Low J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk Res. 2008;32:71–77. doi: 10.1016/j.leukres.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao V, Klein S, Agama K, Toyoda E, Adachi N, Pommier Y, Shacter E. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIα in breast cancer cells. Cancer Res. 2009;69:948–957. doi: 10.1158/0008-5472.CAN-08-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 11.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 12.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 13.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, Muckenthaler MU. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica. doi: 10.3324/haematol.2009.020123. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009;9:461–473. doi: 10.1016/j.cmet.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39:710–715. doi: 10.1016/s0168-8278(03)00408-2. [DOI] [PubMed] [Google Scholar]
- 17.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 18.De Domenico I, Nemeth E, Nelson JM, Phillips JD, Ajioka RS, Kay MS, Kushner JP, Ganz T, Ward DM, Kaplan J. The hepcidin-binding site on ferroportin is evolutionarily conserved. Cell Metab. 2008;8:146–156. doi: 10.1016/j.cmet.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18:2569–2578. doi: 10.1091/mbc.E07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmström P, Gåfvels M, Eriksson LC, Dzikaite V, Hultcrantz R, Eggertsen G, Stål P. Expression of iron regulatory genes in a rat model of hepatocellular carcinoma. Liver Int. 2006;26:976–985. doi: 10.1111/j.1478-3231.2006.01316.x. [DOI] [PubMed] [Google Scholar]
- 21.Boult J, Roberts K, Brookes MJ, Hughes S, Bury JP, Cross SS, Anderson GJ, Spychal R, Iqbal T, Tselepis C. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin Cancer Res. 2008;14:379–387. doi: 10.1158/1078-0432.CCR-07-1054. [DOI] [PubMed] [Google Scholar]
- 22.McKie AT, Barlow DJ. The SLC40 basolateral iron transporter family (IREG1/ferroportin/MTP1) Pflugers Arch. 2004;447:801–806. doi: 10.1007/s00424-003-1102-3. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Zhu B, Yu L. In silico comparison of gene expression levels in ten human tumor types reveals candidate genes associated with carcinogenesis. Cytogenet Genome Res. 2006;112:53–59. doi: 10.1159/000087513. [DOI] [PubMed] [Google Scholar]
- 24.Leong WI, Lönnerdal B. Iron transporters in rat mammary gland: Effects of different stages of lactation and maternal iron status. Am J Clin Nutr. 2005;81:445–453. doi: 10.1093/ajcn.81.2.445. [DOI] [PubMed] [Google Scholar]
- 25.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 27.Brooks SC, Locke ER, Soule HD. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem. 1973;248:6251–6253. [PubMed] [Google Scholar]
- 28.Sartor CI, Dziubinski ML, Yu CL, Jove R, Ethier SP. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Res. 1997;57:978–987. [PubMed] [Google Scholar]
- 29.Ignatoski KM, Ethier SP. Constitutive activation of pp125fak in newly isolated human breast cancer cell lines. Breast Cancer Res Treat. 1999;54:173–182. doi: 10.1023/a:1006135331912. [DOI] [PubMed] [Google Scholar]
- 30.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lønning P, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall P, Ploner A, Bjöhle J, Huang F, Lin CY, Liu ET, Miller LD, Nordgren H, Pawitan Y, Shaw P, Skoog L, Smeds J, Wedrén S, Ohd J, Bergh J. Hormone-replacement therapy influences gene expression profiles and is associated with breast-cancer prognosis: A cohort study. BMC Med. 2006;4:16. doi: 10.1186/1741-7015-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calza S, Hall P, Auer G, Bjöhle J, Klaar S, Kronenwett U, Liu ET, Miller L, Ploner A, Smeds J, Bergh J, Pawitan Y. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006;8:R34. doi: 10.1186/bcr1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, Brown PO, Børresen-Dale AL, Botstein D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 35.Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee PL, Beutler E. Regulation of hepcidin and iron-overload disease. Annu Rev Pathol. 2009;4:489–515. doi: 10.1146/annurev.pathol.4.110807.092205. [DOI] [PubMed] [Google Scholar]
- 37.Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Liu X, Datta A, Govindarajan K, Tam WL, Han J, George J, Wong C, Ramnarayanan K, Phua TY, Leong WY, Chan YS, Palanisamy N, Liu ET, Karuturi KM, Lim B, Miller LD. RCP is a human breast cancer–promoting gene with Ras-activating function. J Clin Invest. 2009;119:2171–2183. doi: 10.1172/JCI37622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet. 1980;2:390–392. doi: 10.1016/s0140-6736(80)90440-7. [DOI] [PubMed] [Google Scholar]
- 40.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999;283:676–679. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- 43.Tsuji Y, Kwak E, Saika T, Torti SV, Torti FM. Preferential repression of the H subunit of ferritin by adenovirus E1A in NIH-3T3 mouse fibroblasts. J Biol Chem. 1993;268:7270–7275. [PubMed] [Google Scholar]
- 44.Kakhlon O, Cabantchik ZI. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic Biol Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 45.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem J. 2002;363:431–436. doi: 10.1042/0264-6021:3630431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- 47.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 49.Horne DT, Scherf U, Vockley J. U.S. Patent 6,974,667. B2 Gene Logic Inc. 2005
- 50.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winter JL, Stackhouse BL, Russell GB, Kute TE. Measurement of PTEN expression using tissue microarrays to determine a race-specific prognostic marker in breast cancer. Arch Pathol Lab Med. 2007;131:767–772. doi: 10.5858/2007-131-767-MOPEUT. [DOI] [PubMed] [Google Scholar]
- 52.Deng Z, Wan M, Sui G. PIASy-mediated sumoylation of Yin Yang 1 depends on their interaction but not the RING finger. Mol Cell Biol. 2007;27:3780–3792. doi: 10.1128/MCB.01761-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 54.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, Sotiriou C. Definition of clinically distinct molecular subtypes in estrogen receptor–positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
http://www.sciencetranslationalmedicine.org/cgi/content/full/2/43/43ra56/DC1
Materials and Methods
Fig. S1. Ferroportin mRNA and splice variants in breast epithelial cells.
Fig. S2. Assessment of hepcidin mRNA in breast cells by RT-PCR.
Fig. S3. Increased ferroportin expression decreases ferritin in breast cancer cells.
Fig. S4. Relative concentrations of ferroportin protein in MDA-MB-231 cells and transfectants and specific detection of ferroportin by Western blotting.
Fig. S5. Ferroportin protein is decreased in breast cancer.
Fig. S6. Signal intensities of ferroportin and hepcidin relative to control genes and other breast cancer genes in the Uppsala cohort.
Table S1. Ferroportin and hepcidin expression predicts outcome in multivariate analyses.
References