

Abstract
IRF4, a member of the interferon regulatory factor (IRF) family, was previously shown to be restricted in the immune system and involved in the differentiation of immune cells. However, we interestingly observed that IRF4 was also highly expressed in both human and animal hearts. Given that several transcription factors have been shown to regulate the pathological cardiac hypertrophy, we then ask whether IRF4, as a new transcription factor, plays a critical role in pressure overload–elicited cardiac remodeling. A transgenic mouse model with cardiac-specific overexpression of IRF4 was generated and subjected to an aortic banding for 4 to 8 weeks. Our results demonstrated that overexpression of IRF4 aggravated pressure overload–triggered cardiac hypertrophy, fibrosis, and dysfunction. Conversely, IRF4 knockout mice showed an attenuated hypertrophic response to chronic pressure overload. Mechanistically, we discovered that the expression and activation of cAMP response element–binding protein (CREB) were significantly increased in IRF4-overexpressing hearts, while being greatly reduced in IRF4-KO hearts on aortic banding, compared with control hearts, respectively. Similar results were observed in ex vivo cultured neonatal rat cardiomyocytes on the treatment with angiotensin II. Inactivation of CREB by dominant-negative mutation (dnCREB) offset the IRF4-mediated hypertrophic response in angiotensin II–treated myocytes. Furthermore, we identified that the promoter region of CREB contains 3 IRF4 binding sites. Altogether, these data indicate that IRF4 functions as a necessary modulator of hypertrophic response by activating the transcription of CREB in hearts. Thus, our study suggests that IRF4 might be a novel target for the treatment of pathological cardiac hypertrophy and failure.
Keywords: cardiac hypertrophy, CREB, IRF4, pressure overload, transcription factor
Cardiac hypertrophy, an increase in heart muscle mass, is the remodeling of the myocardium in response to a variety of extrinsic and intrinsic stimuli.1–3 Although hypertrophic response is initially compensatory for an increased workload, long-standing hypertrophy eventually leads to congestive heart failure, arrhythmia, and sudden death.1–5 Currently, numerous molecules and signaling pathways have been identified to regulate the process of cardiac hypertrophy and heart failure. In particular, the provocation of these complex sets of interwoven signaling pathways culminate in the nucleus to activate a diversified panel of transcription factors, including myocyte enhancer factor-2 (MEF2), nuclear factor of activated T cells (NFAT), activator protein-1 (AP-1), and nuclear factor κB (NF-κB), which induce the activation of the fetal cardiac gene program leading to pathological hypertrophy.6,7 Nevertheless, how to coordinate these transcriptional regulations in the heart on increased biomechanical stress remains incompletely understood.
The interferon regulatory factor (IRF) family is a group of transcription factors, which comprises 9 members (IRF1–9) in both humans and mice.8–10 IRFs are originally described as downstream regulators of interferon signaling and play important roles in the regulation of innate and adaptive immune response.8–10 Recent studies further demonstrate that IRFs are involved in multiple biological processes, such as cell growth, apoptosis, and oncogenesis.10,11 IRF4 (also known as PU.1 interaction partner [Pip], multiple myeloma oncogene 1 [MUM1], lymphocyte-specific interferon regulatory factor [LSIRF], NFEM5, and Interferon Consensus Sequence binding protein for Activated T cells [ICSAT]), as a member of IRF family, was initially identified in B and T lymphocytes.12 Like all other IRF members, IRF4 contains a highly conserved N-terminal DNA-binding domain with a signature tryptophan pentad and a carboxy-terminal portion of an IRF-association domain, which regulates the transcriptional activity of IRFs by mediating protein–protein interactions.12 Although it is well recognized that IRF4 plays an essential role in the regulation of lymphoid-, myeloid-, and dendritic cell differentiations, recent evidence has pointed out that IRF4 to be a tumor suppressor.11,13,14 Moreover, Eguchi et al8 reported that IRF4 is a critical determinant of the transcriptional response to nutrient availability in adipocytes. However, whether IRF4, as a new transcription factor, plays a role in the heart, especially response to chronic pressure overload, has never been examined thus far. Hence, it will be very interesting to determine the consequence of overexpression and ablation of IRF4 in the myocardium after chronic aortic banding (AB).
In the present study, we first discovered that IRF4 was downregulated in heart samples collected from both human patients with dilated cardiomyopathy and animals after AB. Next, we used IRF4 transgenic (TG) and knockout (KO) mouse models, which underwent chronic pressure overload via the AB approach. Our results revealed that either overexpression or deletion of IRF4 did not substantially affect cardiac phenotype under basal conditions. Remarkably, however, elevation of IRF4 did promote, and absence of IRF4 did prevent, the development of pressure overload–induced cardiac hypertrophy and heart failure. Mechanistically, we identified that cAMP response element–binding protein (CREB) is an important target of IRF4 in the regulation of cardiac hypertrophy.
Methods and Materials
The animal protocol was approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. An expanded Methods section is available in the online-only Data Supplement, which includes detailed methods on the following: reagents, human heart samples, experimental animal models and AB surgery, echocardiography and hemodynamics evaluation, histological analysis, Western blotting and quantitative real-time polymerase chain reaction, CREB-luciferase reporter assays, cardiomyocyte culture and infection with recombinant adenoviral vectors, immunofluorescence staining, and statistical analysis.
Results
Expression of IRF4 Is Downregulated in Human Dilated Cardiomyopathic Hearts and Murine Hypertrophic Hearts
IRF4 is well known to be expressed in immune cells and adipocytes.8–10 Whether it is detectable in hearts, however, has never been examined thus far. Hence, our initial experiments were performed to determine the tissue distribution of IRF4 in mice, using Western blotting. We observed that IRF4 was highly expressed in brain, heart, liver, lung, kidney, and skeletal muscles, and less expressed in spleen (Figure 1A), which was confirmed by real-time polymerase chain reaction (Figure S1A in the online-only Data Supplement). Next, we examined whether expression levels of IRF4 were altered in diseased hearts. Human dilated cardiomyopathic samples were collected and revealed that protein levels of IRF4 were reduced by ≈67%, whereas β–myosin heavy chain (MHC) and atrial natriuretic factor (2 markers of the hypertrophic heart) were markedly increased, compared with healthy donor hearts (Figure 1B). Similarly, in an experimental mouse model with AB-induced cardiac hypertrophy (evidenced by elevation of β-MHC and atrial natriuretic factor levels, Figure 1C), we observed that the expression of IRF4 was downregulated by ≈36% in mouse hearts at week 4 after AB, and by ≈65% at week 8 after AB, compared with sham-operated hearts (Figure 1C; P<0.01 versus shams). The real-time polymerase chain reaction and immunofluorescence staining results also demonstrated that IRF4 was expressed in the nuclei of cardiomyocytes under normal conditions and downregulated in hypertrophic hearts (Figure S1B and S1C). Furthermore, using ex vivo cultured neonatal cardiomyocytes treated with either Ang II (1 μmol/L) or phenylephrine (100 μmol/L) for 48 hours to induce hypertrophy, we found that the levels of IRF4 were reduced by ≈56% in these hypertrophic cardiomyocytes (Figure 1D; P<0.01 versus PBS). Together, these data indicate that IRF4 is actually expressed in hearts, and its protein levels are significantly decreased in human dilated cardiomyopathy hearts and pressure overload–induced hypertrophic mouse hearts, as well as ex vivo Ang II/phenylephrine-treated cardiomyocytes.
Figure 1.

Interferon regulatory factor 4 (IRF4) is expressed and downregulated in the heart on hypertrophic stimuli. A, Western blot analysis of the IRF4 expression in brain, heart, liver, spleen, lung, kidney, and skeletal muscles collected from wild-type mice (n=4). B, Western blot analysis of β–myosin heavy chain (MHC), atrial natriuretic peptide (ANP), and IRF4 in normal donor hearts and dilated cardiomyopathy (DCM) hearts collected from human patients (n=4, *P<0.01 vs donor hearts). C, Western blot analysis of β-MHC, ANP, and IRF4 in an experimental mouse model with aortic banding (AB)-induced cardiac hypertrophy (n=4, *P<0.01 vs shams). D, Western blot analysis of β-MHC, ANP, and IRF4 in cardiomyocytes stimulated by angiotensin (Ang) II (1 μmol/L) and phenylephrine (PE; 100 μmol/L); n=4 independent experiments, *P<0.01 vs PBS. Top, Representative blots. Bottom, Bar graphs are quantitative results.
Absence of IRF4 in the Hearts Attenuates Pressure Overload–Induced Hypertrophy
After having determined the alterations of IRF4 levels in diseased hearts, 1 question would be asked: what is the role of reduced IRF4 expression in the development of cardiac hypertrophy and failure? To address this concern, we used an IRF4-KO mouse model (Jackson Laboratory, Figure 2A) and subjected them to AB or sham operation for 8 weeks, and IRF4 KO did not affect blood pressure before or 1 week after AB (Figure S2). It is important to note here that under basal conditions, the IRF4-KO mice were viable, fertile, and had no pathological alterations in cardiac structure and function at 10 weeks of age (Table S1). However, after 8-week AB, IRF4-null hearts remarkably exhibited an attenuation of hypertrophy, as evidenced by the following: (1) shrunk gross heart (Figure 2B); (2) decreased cardiomyocyte cross-sectional area (CSA, wheat germ agglutinin [WGA] staining; Figure 2B and 2C); and (3) reduced ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) (Figure 2D), compared with AB-treated wild-type hearts (IRF4+/+). Similar decreases were also observed in the left ventricular (LV) chamber dimension or wall thickness, as assessed by echocardiography (left ventricular end-diastolic dimension: wild-type [WT]/5.30±0.15 versus KO/4.58±0.05; left ventricular end-systolic dimension: WT/4.15±0.18 versus KO/3.21±0.07; P<0.01; Figure 2E and Table S2). Accordingly, LV contraction was significantly improved in IRF4-KO hearts (fractional shortening [FS]: 30%), compared with WTs (FS: 22%; P<0.01; Figure 2E) on chronic pressure overload. Moreover, the mRNA expression levels of several hypertrophic markers, including atrial natriuretic peptide, brain natriuretic peptide, and β-MHC, were much lower in the IRF4-KO mice than those of WT mice after 8-week AB (Figure 2F).
Figure 2.

Absence of interferon regulatory factor 4 (IRF4) attenuates pressure overload–induced cardiac hypertrophy. A, Deletion of IRF4 was confirmed by Western blotting (n=4). IRF4+/+ stands for wild-type (WT) hearts, and IRF4−/− represents knockout (KO) hearts. B, Histological analyses of the hematoxylin and eosin (HE) staining and the wheat germ agglutinin (WGA) staining of WT and IRF4-KO hearts 8 weeks after aortic banding (AB) surgery (n=5–6). C, Statistical results for the cell-sectional area (CSA, n=100+ cells per group). D, Statistical results for the ratios of heart weight (HW)/body weight (BW), lung weight (LW)/BW, and HW/tibia length (TL) in the indicated groups (n=12 for WTs and n=14 for KOs). E, Parameters of echocardiographic results for WT and KO mice (n=6–10 per experimental group). FS indicates fractional shortening; LVEDD, left ventricular end-diastolic dimension; and LVESD, left ventricular end-systolic dimension. F, Real-time polymerase chain reaction (PCR) analyses of hypertrophic markers (atrial natriuretic peptide [ANP], brain natriuretic peptide [BNP], and β–myosin heavy chain [MHC]) induced by AB in the indicated mice (n=4 per experimental group). G, Picrosirius red (PSR) staining on histological sections of the left ventricle (LV) in the indicated groups 8 weeks after AB (n=5–6 per experimental group, scale bar =50 μm). H, Quantification of the total collagen volume in WT and IRF4-KO mice after AB (n=25+ fields per experimental group). I, Real-time PCR analyses of the fibrotic markers (collagen I, collagen III, and connective tissue growth factor [CTGF]) in the indicated groups (n=4). *P<0.05 vs WT/sham; #P<0.05 vs WT/AB.
To further define the effect of IRF4 deficiency on maladaptive cardiac remodeling, we evaluated fibrosis, a classical feature of pathological cardiac hypertrophy. The extent of fibrosis was quantified by collagen volume through the visualization of the total amount of collagen present in the interstitial and perivascular spaces. Our results showed that both interstitial and perivascular fibrosis were dramatically increased in WT hearts subjected to chronic AB but markedly limited in KO hearts (Figure 2G and 2H). Finally, we measured the synthesis of collagen by analyzing the mRNA levels of fibrotic markers, such as collagen I, collagen III, and connective tissue growth factor. Our results consistently revealed a decreased fibrotic response in KO mice (Figure 2I). Collectively, all these data indicate that deletion of IRF4 blocks pathological cardiac hypotrophy induced by chronic AB. This suggests that reduced IRF4 expression observed in human dilated cardiomyopathy hearts may play compensatory role in defending various stress conditions.
Overexpression of IRF4 Aggravates Pressure Overload–Induced Cardiac Hypertrophy
Next, we were curious about whether increased IRF4 expression in hearts has any effects on chronic pressure overload. To this end, we generated a TG mouse model with cardiac-specific overexpression of IRF4. Given that function domains are highly conserved between human and mouse IRF4 (human IRF4 protein shares 92% homology with mouse IRF4 protein),8–10 we cloned a human IRF4 cDNA into the downstream of a cardiac-specific promoter, α-MHC promoter (Figure 3A), to distinguish mouse endogenous IRF4 from transgene. Four germ lines of IRF4-TG mice were generated and verified by Western blotting (Figure 3B). Notably, the expression of mouse endogenous IRF4 was not affected by the overexpression of human IRF4 gene (Figure 3B), and transgene IRF4 was specially expressed in the heart (Figure 3C). All IRF4-TG mice were healthy and showed no apparent cardiac morphological or pathological abnormalities (Table S1). We then selected the TG line 8 (Tg8) with the highest level of IRF4 for following experiments. To define the consequence of increased IRF4 expression in hearts on chronic pressure overload, IRF4-TG mice and their non-transgene (NTG) littermates were subjected to AB for 4 weeks. In contrast to the results observed in IRF4-KO mice, overexpression of IRF4 aggravated pathological cardiac hypertrophy induced by chronic AB compared with NTG mice, as determined by examinations of hematoxylin and eosin and WGA–fluorescein isothiocyanate staining (Figure 3D). The cardiomyocyte CSA and ratios of HW/BW, HW/TL, and LW/BW were also significantly increased in TG hearts, compared with NTG hearts on 4-week AB (Figure 3E and 3F). Accordingly, the cardiac structure and function, as measured by left ventricular end-diastolic dimension, left ventricular end-systolic dimension, and LVFS, were significantly aggravated (Figure S3A and Table S3), and the markers of cardiac hypertrophy (atrial natriuretic peptide, brain natriuretic peptide, and β-MHC) were also dramatically upregulated in TG mice compared with NTG mice (Figure 3G). We finally examined the effect of IRF4 overexpression on cardiac fibrosis and found that the collagen present in both interstitial and perivascular spaces (picrosirius red [PSR] staining), the extent of cardiac fibrosis quantified by LV collagen volume, and the expression of fibrosis markers (collagen I, collagen III, and connective tissue growth factor) consistently increased in TG mice, compared with NTG mice on chronic AB (Figure S3B–S3D). Together, these gain-of-function data indicate that overexpression of IRF4 promotes cardiac hypertrophy and fibrosis in response to chronic pressure overload.
Figure 3.
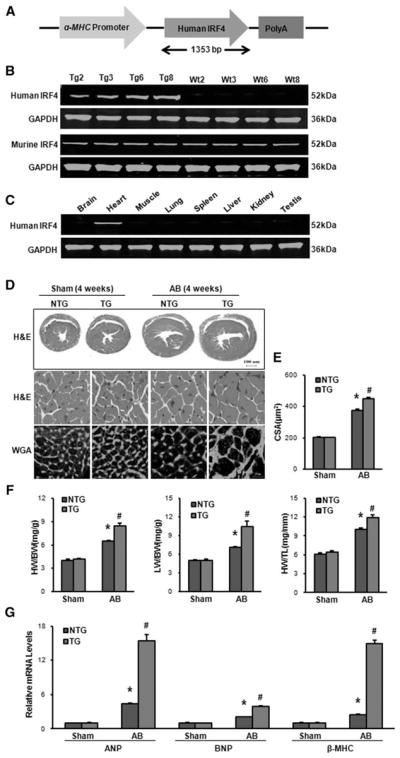
Overexpression of interferon regulatory factor 4 (IRF4) aggravates pressure overload–induced cardiac hypertrophy. A, Schematic diagram of the construction of transgenic (TG) mice with full-length human IRF4 cDNA under the control of α–myosin heavy chain (MHC) promoter. B, Representative blots for determination of TG IRF4 and endogenous IRF4 levels expressed in the heart tissue from 4 lines of TG and their control WT mice (n=5). C, Representative Western blots for evaluation of transgene expression in different tissues from TG mice as indicated (n=5). D, Histological analyses of the hematoxylin and eosin (HE) staining and the wheat germ agglutinin (WGA) staining of NTG and IRF4-TG mice 4 weeks after the aortic banding (AB) surgery (n=5). E, Statistical results for the cell-sectional area (n=100+ cells per group). F, The ratios of heart weight (HW)/body weight (BW), lung weight (LW)/ BW, and HW/tibia length (TL) in the indicated groups (n=11–13). G, Real-time polymerase chain reaction analyses of the hypertrophic markers atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β–myosin heavy chain (MHC) induced by AB in NTG and IRF4-TG mice (n=4). *P<0.05 vs WT/sham; #P<0.05 vs WT/AB.
IRF4 Regulates Angiotensin II–Induced Cardiomyocyte Hypertrophy Ex Vivo
Considering that chronic overexpression or deletion of IRF4 in vivo may have compensatory effects, which influence the consequence of IRF4 in pressure overload–induced cardiac hypertrophy, we then performed ex vivo studies using cultured neonatal rat cardiomyocytes, a well-controlled experimental setting. Primary cultured cardiomyocytes were infected with either AdshIRF4 to knockdown or AdIRF4 to overexpress IRF4 (Figure 4A). Subsequently, these infected cardiomyocytes were exposed to either angiotensin II (Ang II, 1 μmol/L) or PBS control for 48 hours, followed by immunostaining with α-actinin to measure the size of cells. Notably, either knockdown (AdshIRF4) or overexpression of IRF4 (AdIRF4) did not alter the size of cultured neonatal cardiomyocytes under basal PBS treatments, compared with AdshRNA and AdGFP cells (Figure 4C and 4D). However, in response to Ang II–induced cell hypertrophy, knockdown of IRF4 significantly reduced cell size (CSA) by 21.6%, compared with shRNA-infected controls (Figure 4B and 4C). Conversely, Ang II–induced cell hypertrophy was greatly enhanced by 64.2% in IRF4-overexpressing neonatal cardiomyocytes, compared with AdGFP cells (Figure 4B and 4D). Accordingly, the ratio of protein/DNA and expression of hypertrophy markers (atrial natriuretic peptide and β-MHC) were dramatically suppressed in AdshIRF4-infected cardiomyocytes (Figure 4E and Figure S4A), whereas remarkably aggravated in AdIRF4-infected cardiomyocytes (Figure 4F and Figure S4B), compared with controls, respectively. These ex vivo data, consistent with in vivo results (Figures 2 and 3, above), suggest that downregulation of IRF4 mitigates pathological cardiac hypertrophy, whereas upregulation of IRF4 promotes pathological cardiac hypertrophy.
Figure 4.

Interferon regulatory factor 4 (IRF4) regulates angiotensin II-induced cardiomyocyte hypertrophy ex vivo. A, IRF4 was knocked down in AdshIRF4-infected cardiomyocytes, whereas IRF4 was increased by 1.7-fold in AdIRF4-infected myocytes (n=3 independent experiments). Left, Representative blots. Right, Quantitative results. B, Representative images of cardiomyocytes infected with AdshIRF4 or AdIRF4 in response to angiotensin (Ang) II (1 μmol/L). C, Quantitative results of the cell-sectional area showing that knockdown of IRF4 significantly attenuated Ang II–triggered hypertrophy, compared with shRNA-cells (n=50+ cells per group, *P<0.05 vs AdshRNA/PBS; #P<0.05 vs AdshRNA/Ang II). D, Quantitative results of the cell-sectional area showing that overexpression of IRF4 greatly increased Ang II–triggered hypertrophy, compared with control green fluorescent protein (GFP) cells (n=50+ cells per group, *P<0.05 vs AdGFP/PBS; #P<0.05 vs AdGFP/Ang II). E, Real-time polymerase chain reaction (PCR) analysis of hypertrophy markers (atrial natriuretic peptide [ANP] and β–myosin heavy chain [MHC]) in AdshIRF4 cells and control AdshRNA cells after the treatment with PBS and Ang II for 48 hours (n=4 independent experiments, *P<0.05 vs AdshRNA/PBS; #P<0.05 vs AdshRNA/Ang II). F, Real-time PCR assays for evaluation of mRNA levels of ANP and β-MHC in IRF4-overexpressing cells and control GFP cells on the treatment with either PBS or Ang II for 48 hours (n=4 independent experiments, *P<0.05 vs AdGFP/PBS; #P<0.05 vs AdGFP/Ang II).
IRF4 Regulates the Expression of CREB in Mouse Hypertrophic Hearts
Accumulating evidence has indicated that numerous transcription factors (ie, serum response factor [SRF], CREB, MEF2C, myocardin, GATA binding protein 4 [GATA4], and NFATc1) are involved in the development of pathological cardiac hypertrophy.4–7 Therefore, it would be very intriguing to examine whether the expressions/ activities of such transcription factors are interfered by up- and downregulation of IRF4 in hearts on chronic pressure overload. Using Western blotting, we determined the expression levels of SRF, CREB, MEF2C, myocardin, GATA4, and NFATc1 in AB-induced hypertrophic hearts collected from both IRF4-overexpressing and -absent mice. Interestingly, we observed that, with the exception of CREB (Figure 5A–5D), there were no changes in other transcription factors (data not shown). Specifically, the total and phosphorylated protein levels of CREB were decreased by 45% and 40%, respectively, in AB-induced hypertrophic hearts of IRF4-null mice, compared with those of WT controls (IRF4+/+; Figure 5A and 5B). By contrast, the levels of total and phosphorylated CREB were more pronounced in AB-treated hearts of IRF4-TG mice than those hearts of NTG mice (Figure 5C and 5D). To further confirm these findings, we decreased or increased IRF4 expression ex vivo by infecting cultured neonatal rat cardiomyocytes with either AdshIRF4 or AdIRF4, followed by treatment with 1 μmol/L Ang II for 48 hours. Similar to in vivo observations, the levels of total and phosphorylated CREB were reduced in AdshIRF4 cells by 39% and 31%, respectively, compared with control AdshRNA cells (Figure 5E and 5F). Conversely, overexpression of IRF4 enhanced not only the phosphorylation but also total levels of CREB, comparable with control cells (Figure 5G and 5H). Together, both in vivo and ex vivo results consistently indicate that IRF4-mediated pathological cardiac hypertrophy is associated with CREB.
Figure 5.

Interferon regulatory factor 4 (IRF4) regulates the expression of cAMP response element–binding protein (CREB) in mouse hypertrophic hearts. A and B, Representative Western blots and quantitative results of the CREB phosphorylation and total levels in the IRF4 knockout (KO) mice 8 weeks after aortic banding (AB; n=6 hearts, *P<0.05 vs wild-type [WT]/sham; #P<0.05 vs WT/AB). C and D, Representative Western blots and quantitative results of the CREB phosphorylation and total levels in the IRF4 transgenic (TG) mice 4 weeks after AB (n=6 hearts, *P<0.05 vs NTG/sham; #P<0.05 vs NTG/AB). E and F, Representative Western blots and quantitative results of the CREB phosphorylation and total levels in AdshIRF4 or AdshRNA adenoviral vector-infected cardiomyocytes on addition of angiotensin (Ang) II (1 μmol/L) for 48 hours (n=3 independent experiments; *P<0.05 vs AdshRNA/PBS; #P<0.05 vs AdshRNA/Ang II). G and H, Representative Western blots and quantitative results of the CREB phosphorylation and total levels in AdIRF4 or AdGFP adenoviral vector-infected cardiomyocytes on addition of Ang II (1 μmol/L) for 48 hours (n=3 independent experiments; *P<0.05 vs AdGFP/PBS; #P<0.05 vs AdGFP/Ang II).
Regulation of Cardiac Hypertrophy by IRF4 Is Largely Dependent on the Activation of CREB
To determine whether IRF4-regulated cardiac hypertrophy is directly or indirectly associated with CREB, we coinfected neonatal rat cardiomyocytes with AdshIRF4 plus AdCREB, or AdIRF4 plus AddnCREB (dominant negative mutation of CREB to inhibit CREB activity), respectively, followed by the addition of Ang II for 48 hours. Our results of cell size analysis show that, among shRNA-control groups (Figure 6A, blue bars), Ang II–induced myocyte hypertrophy was more pronounced in AdCREB cells than control cells. These results are consistent with previous reports showing that activation of CREB promotes cardiac hypertrophy.15,16 Interestingly, we observed that suppression of Ang II–induced cell hypertrophy by knockdown of IRF4 (Figure 6A, red bars) was released by increased CREB expression (AdCREB cells). In the set of gain-of-function experiments, we observed that Ang II–induced cell hypertrophy was increased by overexpression of IRF4 (Figure 6B, red bars). However, accelerative effects of IRF4 on Ang II–induced cell hypertrophy were dramatically dampened down by overexpression of dominant negative CREB (Figure 6B). Collectively, these data suggest that detrimental role of IRF4 in pathological cardiac hypertrophy is dependent, at least partly, on the activation of CREB.
Figure 6.
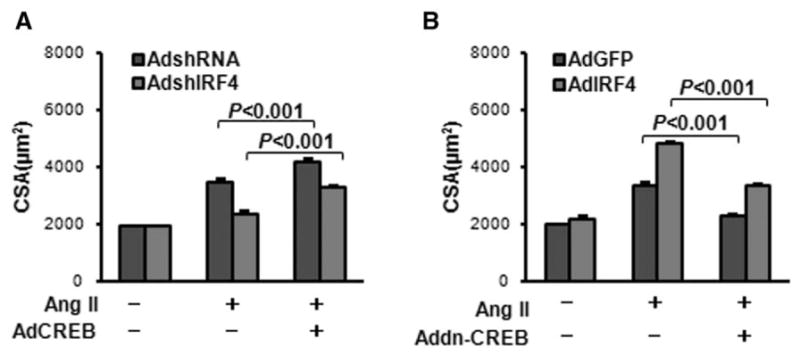
Regulation of interferon regulatory factor 4 (IRF4)-mediated cardiac hypertrophy is largely dependent on the activation of cAMP response element–binding protein (CREB). A, Neonatal rat cardiomyocytes were infected with AdshIRF4 or AdCREB, followed by the treatment with 1 μmol/L of angiotensin (Ang) II for 48 hours. Cell-sectional areas (CSA) were then measured (see Methods) and revealed that inhibition of myocyte hypertrophy by knockdown of IRF4 was released by overexpression of CREB. B, IRF4-mediated promotion of myocyte hypertrophy (middle red bar) was attenuated by inactivation of CREB (AddnCREB, far right red bar). n=50+ cells per experimental group.
Discussion
An increasing number of studies have demonstrated that transcription factors are profoundly involved in the development of cardiac hypertrophy.4,17,18 Accordingly, understanding of transcriptional regulation mechanisms will provide a new strategy for the prevention and treatment of cardiac hypertrophy and heart failure. In the present study, we identified that IRF4, as a transcription factor known in immune cells, was also highly expressed in human and mouse cardiomyocytes. Intriguingly, the expression of IRF4 was downregulated in human dilated cardiomyopathy hearts and mouse hypertrophic hearts, suggesting that IRF4 may be involved in the process of cardiac hypertrophy and failure. Indeed, using loss-of-function and gain-of-function approaches, we discovered that IRF4 was a necessary modulator in the development of pathological cardiac hypertrophy. Absence of IRF4 in the heart attenuated AB-induced hypertrophic response, whereas TG mice with cardiac-specific overexpression of IRF4 showed aggravated cardiac hypertrophy, fibrosis, and dysfunction in response to pressure overload. Therefore, our study presented here for the first time suggests that blockade of IRF4 transcriptional function might be a novel remedy for the treatment of pathological cardiac hypertrophy.
The possible mechanisms by which IRF4 regulates cardiac hypertrophy may be associated with its downstream targets. IRF4 contains a highly conserved N-terminal DNA-binding domain and the carboxy-terminal portion of an IRF-association domain, which modulate the transcriptional activity of IRFs by mediating protein–protein interactions.13 In immune cell system, it has been reported that IRF4 functions as a positive regulator of many downstream transcription factors (ie, NFAT, signal transducer and activator of transcription 3 [STAT3/6], and forkhead box P3 [FOXP3]) by interacting with its C-terminal transactivation domain.19 However, IRF4-controlled transcription factors involving in cardiac hypertrophy have never been investigated thus far. In the present study, we first examined the alterations of hypertrophy-related transcription factors, including SRF, CREB, MEF2C, myocardin, GATA4, and NFATc116,20–24 in IRF4-overexpressing and –KO hearts on chronic pressure overload. Interestingly, CREB was the only one to be regulated in hypertrophic hearts by IRF4. In fact, using a bio-informatics approach, we identified that there are 3 putative IRF4 binding sites located in the promoter region of CREB (Figure S5). The luciferase-reporter assays further showed that IRF4 increased the transcriptional activity of CREB by binding with the CREB promoter via the N-terminal DNA-binding domain (Figure S6). Thus, IRF4-mediated regulation of cardiac hypotrophy may be directly associated with its downstream target, CREB. Notably, under basal condition, either overexpression or ablation of IRF4 in mouse hearts did not alter the total levels of CREB; however, on pressure overload, both total and phosphorylated CREB were increased in IRF4-TG mice, while being decreased in IRF4-KO mice. These data suggest that IRF4-mediated regulation of CREB might be stress-dependent. Indeed, this is supported by our data showing that blockade of CREB activation offset the IRF4-elicited hypertrophic response (Figure 6). Currently, it is well documented that increased CREB expression results in the activation of fetal gene program (ie, atrial natriuretic peptide, brain natriuretic peptide, and β-MHC), leading to cardiac hypertrophy.15 In addition, several previous studies also suggested that the activation of CREB was essential for cardiomyocyte hypertrophy25 and Ang II–induced smooth muscle hypertrophy.26,27 Therefore, we believe that the detrimental role of IRF4 in pathological cardiac hypertrophy is dependent, at least in part, on the activation of CREB.
It is important to note here, numerous studies have shown that IRF4 is closely related to inflammation.28,29 For example, Mudter et al28 implicated IRF4 as a key regulator of mucosal interleukin-6 production in T cell–dependent experimental colitis. Lassen et al29 demonstrated that loss of IRF4 aggravated postischemic renal failure, which is mediated by tumor necrosis factor-α. Therefore, we further examined the status of inflammation/inflammatory cell infiltration in AB-treated IRF4-KO and WT hearts. Our results showed that mRNA levels of interleukin-1β, interleukin-6, tumor necrosis factor-α, and CD68 were significantly increased in hearts from both groups (WT and IRF4-KO) on 1-week AB and decreased to basal levels after 4-week AB, but there were no significant differences between WT and IRF4-KO hearts on AB surgery (Figure S7). These data suggest that IRF4-KO may not alter the status of inflammation in AB-treated murine hearts.
In conclusion, the present study demonstrates that upregulation of IRF4 aggravates pressure overload–induced cardiac hypertrophy, fibrosis, and failure by targeting the CREB transcription factor. Conversely, knockdown/out of IRF4 mitigates the development of pathological cardiac hypertrophy. These findings suggest that IRF4 might be a novel therapeutic target for the prevention of cardiac hypertrophy and failure.
Perspectives
The current study provides in vivo and in vitro evidence that IRF4, a member of IRF transcription factor family, functions as a positive regulator of the hypertrophic response in the heart through physically interacting with the CREB promoter, and thereby activating the myocardial fetal gene program. These observations suggest that blockade of IRF4 expression/activity would be beneficial to the heart in prevention/attenuation of pathological hypertrophy and failure.
Supplementary Material
Novelty and Significance.
What Is New?
Interferon regulatory factor 4 (IRF4) is identified to be expressed in human and mouse heart tissues, and IRF4 is downregulated in the heart on hypertrophic stimuli.
IRF4 is a necessary modulator responsible for the development of pressure overload–induced cardiac hypertrophy, fibrosis, and cardiac dys-function in vivo and Ang II–induced cardiomyocyte hypertrophy in vitro through regulating the transcription and phosphorylation of cAMP response element–binding protein.
What Is Relevant?
Several transcription factors have been elucidated to contribute to cardiac hypertrophy.
Effects of IRF4, as an immune transcription factor, on the development of cardiac hypertrophy and its underlying mechanisms are not known.
This study advances our understanding the role of transcription factors in the process of cardiac hypertrophy and provides a basis for searching novel therapeutic targets to prevent cardiac hypertrophy and heart failure.
Summary
Our findings demonstrate that absence of IRF4 protects hearts against chronic pressure overload–induced cardiac hypertrophy, fibrosis, and cardiac dysfunction, whereas overexpression of myocardial IRF4 aggravates pathological hypertrophic response by regulating the transcription and phosphorylation of cAMP response element–binding protein. These observations suggest that IRF4 might be a novel target for the treatment of cardiac hypertrophy and heart failure.
Acknowledgments
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81100230, No. 81070089, and No. 81200071), the National Science and Technology Support Project (No. 2011BAI15B02 and No. 2012BAI39B05), and the National Basic Research Program of China (No. 2011CB503902).
Footnotes
Disclosures
None.
References
- 1.Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z, Bai X, Zhou H, Jiang H, Heximer SP, Qin M, Huang H, Liu PP, Huang C. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA. 2010;107:13818–13823. doi: 10.1073/pnas.1008397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu J, Bian ZY, Zhang R, et al. Interferon regulatory factor 3 is a negative regulator of pathological cardiac hypertrophy. Basic Res Cardiol. 2013;108:326. doi: 10.1007/s00395-012-0326-9. [DOI] [PubMed] [Google Scholar]
- 3.Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C, Jiang H, Yang Q, Bian ZY, Bai X, Zhu LH, Wang L, Li H. Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-beta-activated kinase 1-dependent signaling. Hypertension. 2010;56:232–239. doi: 10.1161/HYPERTENSIONAHA.110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akazawa H, Komuro I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res. 2003;92:1079–1088. doi: 10.1161/01.RES.0000072977.86706.23. [DOI] [PubMed] [Google Scholar]
- 5.Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. doi: 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124–132. doi: 10.1172/JCI33255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest. 2005;115:538–546. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 10.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 11.Acquaviva J, Chen X, Ren R. IRF-4 functions as a tumor suppressor in early B-cell development. Blood. 2008;112:3798–3806. doi: 10.1182/blood-2007-10-117838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas PS, Bhagat G, Pernis AB. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol Rev. 2010;233:79–96. doi: 10.1111/j.0105-2896.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, Belz GT, Smyth GK, Busslinger M, Nutt SL, Kallies A. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol. 2011;12:304–311. doi: 10.1038/ni.2006. [DOI] [PubMed] [Google Scholar]
- 14.Veldhoen M. Interferon regulatory factor 4: combinational control of lymphocyte differentiation. Immunity. 2010;33:141–143. doi: 10.1016/j.immuni.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Huggins GS, Lepore JJ, Greytak S, Patten R, McNamee R, Aronovitz M, Wang PJ, Reed GL. The CREB leucine zipper regulates CREB phosphorylation, cardiomyopathy, and lethality in a transgenic model of heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H1877–H1882. doi: 10.1152/ajpheart.00516.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson PA, Reusch JE, McCune SA, Leinwand LA, Luckey SW, Konhilas JP, Brown DA, Chicco AJ, Sparagna GC, Long CS, Moore RL. Restoration of CREB function is linked to completion and stabilization of adaptive cardiac hypertrophy in response to exercise. Am J Physiol Heart Circ Physiol. 2007;293:H246–H259. doi: 10.1152/ajpheart.00734.2006. [DOI] [PubMed] [Google Scholar]
- 17.Tshori S, Gilon D, Beeri R, Nechushtan H, Kaluzhny D, Pikarsky E, Razin E. Transcription factor MITF regulates cardiac growth and hypertrophy. J Clin Invest. 2006;116:2673–2681. doi: 10.1172/JCI27643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H, Shen DF, Bian ZY, Zong J, Deng W, Zhang Y, Guo YY, Li H, Tang QZ. Activating transcription factor 3 deficiency promotes cardiac hypertrophy, dysfunction, and fibrosis induced by pressure overload. PLoS ONE. 2011;6:e26744. doi: 10.1371/journal.pone.0026744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brass AL, Kehrli E, Eisenbeis CF, Storb U, Singh H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev. 1996;10:2335–2347. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- 20.Nelson TJ, Balza R, Jr, Xiao Q, Misra RP. SRF-dependent gene expression in isolated cardiomyocytes: regulation of genes involved in cardiac hypertrophy. J Mol Cell Cardiol. 2005;39:479–489. doi: 10.1016/j.yjmcc.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Kontaraki JE, Parthenakis FI, Patrianakos AP, Karalis IK, Vardas PE. Myocardin gene regulatory variants as surrogate markers of cardiac hypertrophy - study in a genetically homogeneous population. Clin Genet. 2008;73:71–78. doi: 10.1111/j.1399-0004.2007.00932.x. [DOI] [PubMed] [Google Scholar]
- 22.van Berlo JH, Elrod JW, Aronow BJ, Pu WT, Molkentin JD. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2011;108:12331–12336. doi: 10.1073/pnas.1104499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFκB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang ZY, Liu XH, Hu WC, Rong F, Wu XD. The calcineurin-myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;298:H1499–H1509. doi: 10.1152/ajpheart.00980.2009. [DOI] [PubMed] [Google Scholar]
- 25.El Jamali A, Freund C, Rechner C, Scheidereit C, Dietz R, Bergmann MW. Reoxygenation after severe hypoxia induces cardiomyocyte hypertrophy in vitro: activation of CREB downstream of GSK3beta. FASEB J. 2004;18:1096–1098. doi: 10.1096/fj.03-1054fje. [DOI] [PubMed] [Google Scholar]
- 26.Funakoshi Y, Ichiki T, Takeda K, Tokuno T, Iino N, Takeshita A. Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem. 2002;277:18710–18717. doi: 10.1074/jbc.M110430200. [DOI] [PubMed] [Google Scholar]
- 27.Fukuyama K, Ichiki T, Ono H, Tokunou T, Iino N, Masuda S, Ohtsubo H, Takeshita A. cAMP-response element-binding protein mediates prostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells. Biochem Biophys Res Commun. 2005;338:910–918. doi: 10.1016/j.bbrc.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Mudter J, Amoussina L, Schenk M, et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J Clin Invest. 2008;118:2415–2426. doi: 10.1172/JCI33227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lassen S, Lech M, Römmele C, Mittruecker HW, Mak TW, Anders HJ. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol. 2010;185:1976–1983. doi: 10.4049/jimmunol.0904207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.