

Abstract
Spermatogonial stem cells (SSCs) divide continuously to support spermatogenesis throughout postnatal life and transmit genetic information to the next generation. Here, we report the successful establishment of the method for the isolation and identification of human SSCs from testicular tissue, and to determine the culture conditions required to expand SSCs on human embryonic stem cell-derived fibroblast-like cells (hdFs). Large-scale cultures of SSCs were maintained on hdF feeder layers and expanded in the presence of a combination of cytokines and glial cell line-derived neurotrophic factor for at least 2 months. Cell surface marker analysis showed that SSCs retained high levels of alkaline phosphatase activity and stained strongly for anti-stage-specific embryonic antigen (SSEA)-1, OCT4 and CD49f. They also expressed the genes OCT4, SOX3 and STRA8 as detected by reverse transcription polymerase chain reaction (RT-PCR) analysis. These data clearly illustrate a novel approach for the growth of human SSCs using hdFs as feeder cells, potentially eliminating xenogeneic contaminants. This system provides a new opportunity for the study of the regulatory mechanism of the 'niche' that governs SSC self-renewal, and will be a valuable source of SSCs for potential clinical applications.
Keywords: human embryonic stem cell-derived fibroblast-like cells (hdFs), spermatogonial stem cells (SSCs), xeno-free culture
Introduction
Male germ stem cells or spermatogonial stem cells (SSCs), are a unique population of cells in postnatal mammals that undergo self-renewal and transmit genes to subsequent generations. Human SSCs could be valuable for the treatment of male infertility and human genetic disorders 1. However, SSCs are rare in the testis, constituting around 1 in 3 000–4 000 testicular cells. The development of techniques for the growth of SSCs and their differentiation into functional spermatozoa in vitro is crucial for clinical application. Although earlier studies have shown that SSCs in other animal species, such as the mouse and hamster, can survive and proliferate for a long time 2, 3, little is known regarding the culture and growth requirements of human SSCs. Generally, stem cells reside within a special microenvironment or 'niche', which provides factors that regulate the proliferation and differentiation of the stem cell population 4. The strongest evidence for niche-based regulation in mammalian tissue probably comes from studies of spermatogenesis. Similar studies in other self-renewing tissues have revealed a close interaction of stem cells and stromal cells that constitute the niche 5. SSCs expand in the presence of feeder layer cells such as mouse embryonic fibroblast (MEF) cells, SIM mouse embryo-derived thioguanine and ouabain resistant STO cells and human embryonic cell-derived fibroblast-like cells (hEFs) 6. However, the use of xenogeneic or allogeneic feeder cells for culturing human SSCs is associated with risks such as pathogen transmission and viral infection. Moreover, the availability of hEFs from aborted foetuses is relatively low, and Sertoli cells cannot support the culture of undifferentiated SSCs equally well. All of these factors limit further application of human SSCs for therapy. Therefore, it is necessary to develop an improved culture system that can support the growth of human SSCs.
Recent studies have shown that using human embryonic stem cell-derived fibroblast-like cells (hdFs) as a feeder layer could efficiently and securely support the growth and maintenance of pluripotency of both autogeneic and allogeneic undifferentiated human embryonic stem cells (hESCs) 7. Bendall et al. 8 reported that insulin-like growth factor and fibroblast growth factor (FGF) could cooperatively establish the regulatory stem cell niche of pluripotent human cells defined by hdFs in vitro. SSCs originating from primordial germ cells (PGCs) and from neonatal or adult mouse testis have pluripotent properties similar to those of embryonic stem cells 9, 10. More recently, mouse embryonic stem cells have been shown to differentiate into sperm cells that can then be used to create healthy baby mice 11.
Therefore, it is possible that similar growth conditions might exist in the spermatogenic system that could be replicated to influence initial colony growth in vitro. This leads to the notion that the stem cell niches of ESCs and SSCs are similar. Here, we assume that an hdF feeder layer creates a unique environment in which stem cells exist and this contributes to their self-renewal and differentiation. In this study, we offer an alternative and secure xeno-free culture system for the continuous growth of undifferentiated human SSCs on the basis of hdF feeder layers.
Materials and methods
Isolation and purification of human SSCs
Human fetal testes (> 6-month-old) were obtained from aborted foetuses in Renji Hospital (Shanghai, China) with signed, informed consent from parents and the approval of the Ethics Committee of Shanghai Jiao Tong University. Testicular single cell suspensions were collected by a two-step enzymatic digestion and then used for sorting. Briefly, testicular cells were obtained by digestion with 1 mg mL−1 collagenase IV (Sigma Chemical Co., St. Louis, MO, USA) at 37°C for 15 min, followed by digestion with 0.25% trypsin-EDTA (trypsin solution containing 0.25 mg mL−1 trypsin and 1 mmol L−1 EDTA; Gibco, Gaithersburg, MD, USA) at 37°C for 10 min. CD49f is a component of the laminin receptor expressed by SSCs and can be used for purifying SSCs 12. The dissociated cells were incubated with fluorescein isothiocyanate (FITC)-conjugated rat anti-CD49f monoclonal antibody (BD Biosciences, Franklin Lakes, NJ, USA), 20 μL per 106 cells at 4°C in the dark for 20 min, and then washed thrice with phosphate-buffered solution (PBS). Next, magnetic-activated cell sorting (MACS) buffer (PBS supplemented with 0.5% [w/v] bovine serum albumin and 2 mmol L−1 EDTA), 80 μL per 107 cells and anti-FITC MACS Microbeads (Miltenyi Biotec, Auburn, CA, USA), 20 μL per 107 cells were added to the cell pellet. After being mixed, the cells were incubated in the dark at 4°C for 15 min. The cells were washed with MACS buffer and then resuspended in 500 μL MACS buffer per 107 cells. MACS was carried out through the LS-column in a VarioMACS separator (Miltenyi Biotec) following the manufacturer's protocol to enrich for the CD49f+ cells in the suspension. Samples of the cells before and after MACS were analyzed by flow cytometry to determine the expression of CD49f to assess the enrichment ratio.
Culture of hESCs and production of feeder layers
The hESC line H1 (NIH code: WA01) was maintained on irradiated primary murine embryonic fibroblasts as described earlier 13, 14. The hESCs were harvested after incubation with 1 mg mL−1 collagenase IV (Sigma) at 37°C for 10 min. The cells were dissociated into small clusters and transferred into six-well plates pre-coated with matrigel (BD Biosciences) and cultured in 80% (v/v) knockout Dulbecco's modified Eagle's medium (KO-DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 15% (v/v) fetal bovine serum (FBS, HyClone, Logan, UT, USA), 2 mmol L−1 L-glutamine, 10 μmol L−1 β-mercaptoethanol, 1% (w/v) nonessential amino acids (Invitrogen) and 10 ng mL−1 recombinant human basic fibroblast growth factor (hbFGF; Pepro tech) at 37°C in a 5% (v/v) CO2 atmosphere. After 9 days, cells with fibroblast-like morphology were transferred to non-coated flasks and cultured in hdF medium consisting of DMEM (Invitrogen), 1 mmol L−1 L-glutamine, 1% nonessential amino acids and 10% (v/v) heat-inactivated bovine serum (Hyclone). The cells were cultured to produce a confluent primary monolayer of hdFs (Figure 2). The hdFs were passaged and cultured every 3–5 days for an additional 6 weeks in hdF medium. These cells were analyzed by flow cytometry. Cell proliferation was inhibited by mitomycin C (Sigma). Passages 3–12 of the hdFs, after 2 h of mitotic inactivation using 12.5 mg mL−1 mitomycin C, were used as feeder layer cells for the growth of human SSCs.
Figure 2.
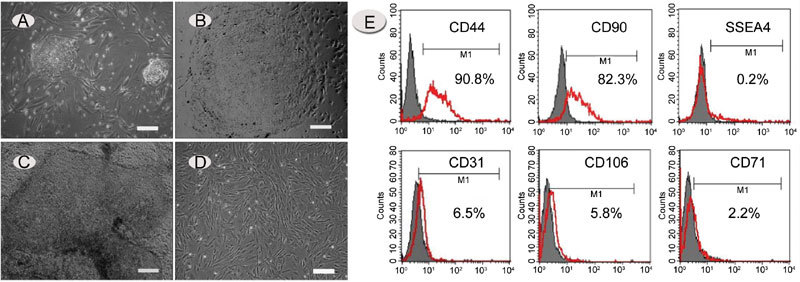
Growth and characterization of hdFs. (A) – (D): Recovery of human embryonic stem cell-derived fibroblast-like cells (hdFs). (A): Morphology of human embryonic stem (hES) cell colonies after 4 days of co-culture with an MEF feeder layer. (B) and (C): Morphology of human embryonic stem cells (hESCs) maintained on matrigel without feeder layer support. (D): Morphology of isolated hdFs. Images were captured with an inverted Leica microscope using a 5 × objective with numerical aperture 0.12 and acquired through Spot camera and Spot software. (E): Flow-cytometric analysis of hdFs (passage 3) for the presence of CD44, CD90, SSEA-4, CD31, CD106 and CD71. The red line seems to be the positive signals from the antibodies and gray is the non-specific controls. Horizontal bars indicate mean percentages of positive cells from three independent experiments. (A) and (D): bars = 25 μm; (B) and (C), bars = 50 μm.
Growth of human SSCs on hdFs
Purified CD49f+ germ cells (human SSCs) were maintained on a feeder layer of mitomycin C-treated hdFs in a six-well culture plate. The culture medium consisted of DMEM supplemented with 15% (v/v) FBS, 1% (w/v) nonessential amino acids, 1 mmol L−1 L-glutamine, 0.1 mmol L−1 β-mercaptoethanol, 1% (w/v) penicillin–streptomycin (Invitrogen), 1 mmol L−1 sodium pyruvate, 100 μg mL−1 transferrin (Sigma), 4 ng mL−1 hbFGF, 10 ng mL−1 recombinant human glial cell line-derived neurotrophic factor (GDNF) and 1 500 IU mL−1 recombinant human leukemia inhibitory factor LIF (Peprotech Inc., Rocky Hill, NJ, USA). Prolonged culture of human SSCs was carried out at 37°C in a 5% (v/v) CO2 atmosphere and the culture medium was changed every 1–2 days. After the formation of germ-cell colonies, cells were passaged by incubation in cell dissociation buffer or trypsin dissociated, and then seeded. Under such culture conditions, human SSCs were passaged every 4–5 days. In order to study the effects of hdFs and GDNF on the growth of human SSCs, purified CD49f+germ cells were also cultured in feeder-free and non-coated flasks or in culture medium not supplemented with GDNF. For cryopreservation, cells were resuspended in FBS supplemented with 10% (v/v) DMSO at 1.0 × 106 cells mL−1, chilled at approximately 1°C per minute to −196°C and stored in liquid nitrogen.
Flow-cytometric analysis
To identify the nature of feeder layer cells, hdFs derived from hESCs were analyzed by flow cytometry using CD106, CD31, CD44, CD90, CD71 and SSEA-4 markers. Likewise, samples of the testicular cells before MACS and CD49f+ germ cells obtained after MACS were analyzed by flow cytometry using CD49f (α6-integrin), CD117 (C-KIT), CD90 (THY-1), β1-integrin and EE2 markers. Operation procedures were performed as described earlier 15. Briefly, cells were harvested in 0.25% (w/v) trypsin-EDTA, suspended in staining buffer (PBS + 5% (v/v) FBS) at a concentration of 106 cells per mL per 100 μL and stained with primary antibodies (BD Biosciences) at 4°C for 20 min. After being washed thrice with staining buffer, cells were stained with secondary antibody (FITC-conjugated mouse-IgG2a and PE-conjugated rat-IgG2a [Sigma]) at 4°C for 20 min. Isotype-matched control antibodies (BD PharMingen) were used to determine the background staining. Cells were again washed thrice and resuspended in staining buffer and then analyzed by flow cytometry. The samples were analyzed using a FACSCalibur (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) machine with CellQuest acquisition software. Data analysis was carried out using Cell Quest (BD Bioscience, CA, USA) or FlowJo Software (Tree Star Inc, OR, USA).
Immunocytochemistry and alkaline phosphatase (AKP) staining
Expression of stem cell markers was examined by immunocytochemistry and alkaline phosphatase (AKP) staining. Immunocytochemistry and AKP staining procedures were carried out as described earlier 16. Briefly, human SSC colonies were fixed with 4% paraformaldehyde for 30 min. Fixed cells were first blocked in blocking buffer (5% [v/v] goat serum, 1% [w/v] bovine serum albumin and 0.1% [v/v] Triton X-100) for 1 h, followed by incubation with the primary antibody at 4°C overnight. Appropriately, coupled secondary antibodies (Molecular Probes Inc., Eugene, OR, USA) were used for single and double labeling. All secondary antibodies were tested for cross-reactivity and non-specific immunoreactivity. The following antibodies were used: anti-Oct-4 antibody, anti-stage-specific embryonic antigen (SSEA)-1 antibody and anti-CD49f antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). AKP activity was determined using the Vector Blue Alkaline Phosphatase Substrate Kit (Invitrogen).
Reverse transcription polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from CD49f+ germ cells and human SSCs cultured on hdF feeder layers using Trizol (Invitrogen) according to the manufacturer's instructions, and RT-PCR was performed as described earlier 17. Genomic DNA was degraded by incubation with DNAse I for 15 min at 37°C. RNA was reverse transcribed using M-MLV reverse transcriptase and random hexamers following the manufacturer's instructions. PCR was carried out using Taq polymerase (Invitrogen). Primer sequences (Table 1) were designed by Primer Express 2.0 Software, verified to be gene-specific with BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) and synthesized by the Invitrogen company. PCR products were run on 2% (w/v) agarose gels and stained with ethidium bromide. Expression of the housekeeping gene β-actin was used to normalize the PCR reactions. Results were assessed by the presence or absence of PCR products of appropriate size.
Table 1. Polymerase chain reaction (PCR) primers used for the amplification of reference gene candidates used in this study.
| Gene | Genebank accession | Forward primer (5′–3′) | Reverse primer (5′–3′) | Size (bp) |
|---|---|---|---|---|
| OCT4 | NM_002701.4 | CTGGGTTGATCCTCGGACCT | CCATCGGAGTTGCTCTCCA | 243 |
| SOX3 | NM_005634.2 | CGCGGGTTCCTGCTGATTT | CGGGGTTCTTGAGTTCAGTCT | 222 |
| STRA8 | NM_182489.1 | CTGTGGCAGAACCTCTCGG | GAACCTCACTTTTGTCCAGGAA | 238 |
| NGN3 | NM_020999.2 | ACTGTCCAAGTGACCCGTGA | TCAGTGCCAACTCGCTCTTAG | 205 |
| DAZL | NM_001351.2 | ACACTGAAACTTATATGCAGCC | CGGAGGTACAACATAGCTCCTT | 182 |
| NOTCH1 | NM_017617.3 | CGCTGACGGAGTACAAGTG | GGTAGGAGCCGACCTCGTT | 246 |
| C-KIT | NM_000222.2 | GTTCTGCTCCTACTGCTTCGC | TAACAGCCTAATCTCGTCGCC | 135 |
| β-actin | NM_001101.2 | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT | 250 |
Statistics
Statistical analysis was carried out using the Paired t-test. The mean and SD are given in the figures, as described in the legends.
Results
Sorting of human SSCs and flow-cytometric analysis
Testicular cells consist of spermatogenic cells and somatic cells (mainly Sertoli cells and Leydig cells), and MACS was used to remove testicular somatic cells and to enrich the suspension with SSCs (CD49f+ germ cells). To test the efficiency of MACS and to characterize SSCs, flow-cytometric analysis was carried out on unsorted testicular cells and sorted CD49f+ germ cells obtained after MACS. Figure 1 shows that there was an obvious enrichment of CD49f+ cells from 9% to 94% after MACS. Data are representative of three independent experiments. The CD49f+ cells comprised a single phenotypic population characterized by surface antigens detected by cytometric analysis (Figure 1A). They were strongly positive for CD49f, β1-integrin (SSC marker), CD90 (undifferentiated spermatogonia marker) and EE2 (spermatogonia marker), and most cells expressed C-KIT weakly (differentiated spermatogonia marker) 4, 18. In contrast, for non-enriched testicular cells, only weak expression of these markers was observed, confirming that CD49f+ germ cells are phenotypically distinct from other testicular and somatic cells. These results suggest that it is reasonable to use CD49f as a marker for the purification of human SSCs.
Figure 1.
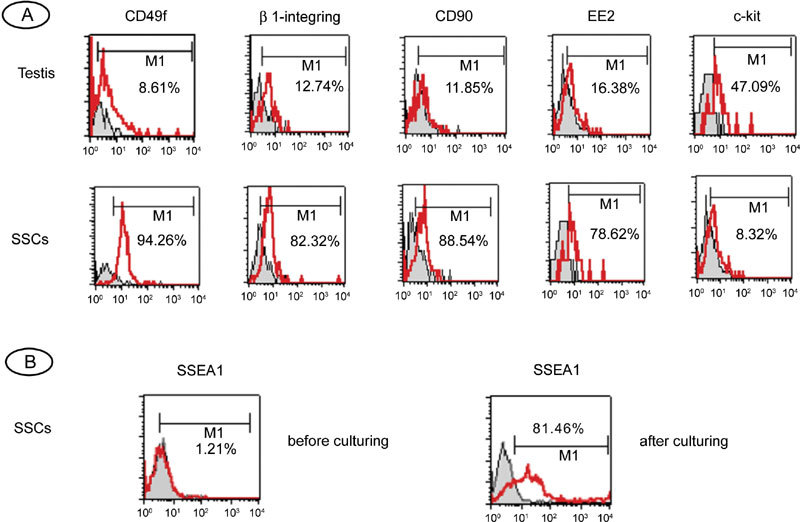
Flow-cytometric characterization of human fetal testicular cells and human spermatogonial stem cells (SSCs). (A): Flow-cytometric characterization of human fetal testicular cells before and after magnetic cell sorting (MACS). The cells were stained with antibodies against CD49f, β1-integrin, CD90, EE2 and c-kit. The histograms are overlaid on relative isotype-matched control antibody histograms. Non-specific isotype staining controls are shown as shaded histograms; positively stained cells are shown as open histograms in red. Horizontal bars indicate mean percentages of positive cells from three independent experiments. (B): Flow-cytometric analysis of SSEA-1 expression in human SSCs before and after culture.
Growth and characterization of hdFs
The hESCs were induced to differentiate into hdFs by culturing on matrigel-coated plates (Figure 1A–C). When colonies of hESCs were transferred and cultured in the absence of a feeder layer, some colonies attached to the plate and showed early signs of spontaneous differentiation. Most cells showed a fibroblast-like or mesenchymal-like morphology appearing as long, flat cells with an elongated, condensed nucleus (Figure 2D). The colonies with fibroblast-like cells were further used for the establishment of hdFs. Flow cytometry (Figure 2E) revealed that most cells stained positively for CD44 (differentiating hESC marker) and CD90 (mesenchymal marker), but not for SSEA-4 (undifferentiated hESC marker) and CD31 (endothelial-specific cell marker), and very few cells showed expression of CD106 (mesenchymal cell-specific marker) and CD71 (transferring receptor). When hdFs were processed with three different concentrations of mitomycin C (10 mg L−1, 12.5 mg L−1 and 15 mg L−1), proliferation was inhibited, but viability was maintained. After treatment with 12.5 mg L-1 mitomycin C for 2 h, the mitosis of hdFs was significantly inhibited (data not shown).
SSCs growth and morphology
The isolated CD49f+ cells (human SSCs) were cultured on hdF feeder layers in a medium containing GDNF. After 48 h, the transferred cells proliferated and spread on the bottom of the well. Within 1 week, the germ cells started to form colonies of tightly compacted cells (Figure 3A and B). In the steady phase, cells were passaged every 5–6 days to fresh hdF feeder layers at 1:3 to 1:4 dilutions. Human SSCs maintained on hdF feeder layers were cultivated for over 10 passages and still remained in a relatively steady state (Figure 3C–F). In addition, cryopreserved cells recovered quickly, within 2–3 days, after thawing from frozen vials, and an undifferentiated state could be easily and reliably maintained (data not shown). When human SSCs were transferred to hdF feeder-free plates or in a medium lacking GDNF, the cells did not proliferate and could not form compact colonies (Figure 3G and H). After surviving for several days, most of the germ cells eventually disappeared from the culture within 7–10 days.
Figure 3.
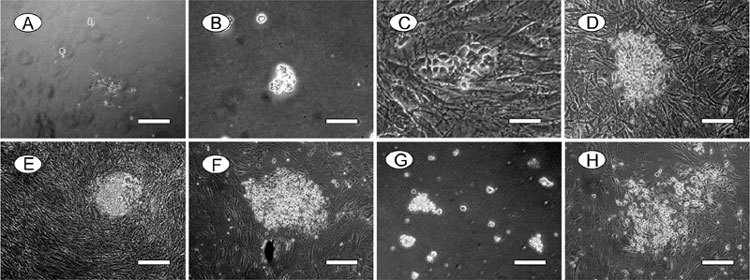
In vitro growth and morphology of human spermatogonial stem cells (SSCs). (A) – (F): Human SSCs were cultured on human embryonic stem cell-derived fibroblast-like cells (hdFs) with the addition of Glial cell line-derived neurotrophic fator (GDNF). Note that human SSCs form compact colonies. Human SSCs at 1 day in vitro (A), 3 days (B), 5 days (C), 10 days (D), 1 month (E) and 2 months (F). (G): Human SSCs cultured under feeder-free conditions on day 3. Note that human SSCs cannot form compact colonies without hdF feeder layer support. (H): Human SSCs cultured on hdFs without the addition of GDNF on day 3. Note that human SSC colonies cannot be maintained in the absence of GDNF. Bars = 50 μm.
Characterization of human SSCs
Immunocytochemistry was used to analyze whether the hdF feeder layer-cultured human SSCs were similar to other SSCs and hESCs in their expression of cell surface markers that characterize undifferentiated, pluripotent stem cells. These include SSEA-1, OCT4 and CD49f. The human SSC colonies grown on hdFs were strongly positive for these surface markers (Figure 4B–D). SSC colonies were also strongly positive for AKP, which is characteristic of ES and EG cells.
Figure 4.
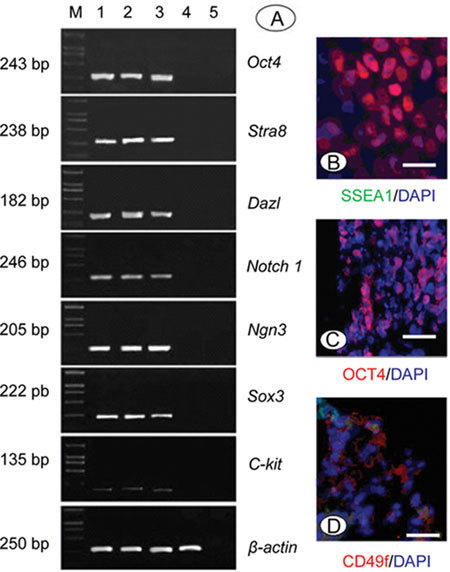
Phenotypic characterization of human SSCs. (A): RT-PCR analysis of several spermatogonial stem cell (SSC) genes before culture (lane 1), after culture for 2 weeks (lane 2) and 1 month (lane 3), and on hdF (lane 4). Negative control (lane 5). Marker (lane M). (B)–(D): Immunocytological analysis of human SSC colonies grown on hdF for surface markers SSEA-1 (B), CD49f (D) and the nuclear marker OCT4 (C). Bars = 500 m.
RT-PCR was used to confirm some of the markers analyzed by immunochemistry and to examine additional markers associated with stem cells. Several genes such as OCT4, SOX3, NGN3 and NOTCH1 are expressed in As, Apr and Aal spermatogonia and were shown to play an important role in the regulation of the self-renewal of SSCs 20, 21, 22. DAZL is a surface marker of germ cells 23. Stra8 is expressed in pre-meiotic germ cells and C-KIT is expressed in embryonic and pre- and post-meiotic germ cells and in Leydig cells in the testis 18, 24. These markers were used to identify SSCs. RT-PCR analysis of human SSCs before and after culture were positive for the expression of OCT4, SOX3, NGN3, NOTCH1, STRA8 and DAZL, but C-KIT was only weakly expressed or was not expressed at all (Figure 4A).
To confirm the phenotype of cultured cells, SSCs cultured on hdFs for 2 months were analyzed by flow cytometry. These cells remained CD49f+, similar to cells before the culturing procedure (data not shown). However, fewer SSEA-1-positive cells were found in CD49f+ germ cells before culture, and cells from SSC colonies were strongly positive for SSEA-1 (ES cell marker and PGC marker) (Figure 1B).
Discussion
SSCs are capable of self-renewal and the production of a large number of committed progenitors that are destined to differentiate into spermatozoa throughout life 25. Isolation and culture of SSCs has become an approach to study the microenvironment of these cells and factors controlling their expansion and differentiation. The isolation and culture of SSCs from genetically engineered mice has been fairly successful, but little is known regarding the culture and growth requirements of human SSCs. Mammalian SSC culture experiments have proved that cultured SSCs require a feeder layer, but the specific roles of feeder layer cells in the initial and prolonged culture of stem cells are not yet clear. Feeder layer cells provide an ideal environment for the growth of stem cells, including detoxification of the culture medium, the secretion of many unique proteins that participate in cell growth and extracellular matrix formation and remodeling 26. There are several potential advantages of using hdFs as feeder layer cells to support the growth of hESCs 7, and the growth of human SSCs with hdFs as the feeder layer may prevent the interspecies transfer of viruses, but these advantages are rarely studied in human SSC culture. GDNF is a member of the transforming growth factor superfamily and was originally identified as a factor promoting the survival of midbrain dopaminergic neurons. GDNF is also a key factor in the developmental behavior of SSCs 19. Earlier studies showed that GDNF seemed to have a beneficial effect on the maintenance of SSCs 3, 19. It might also provide an optimal environment for the clonal expansion of human SSCs. However, growth requirements for human SSCs remain unclear. To address these problems, we have developed a culture system that supports the replication and maintenance of human SSCs. In the presence of GDNF and using hdFs as feeder layer cells, human CD49f+ cells isolated from fetal testis proliferated over a 2-month period. This system showed that the hdFs in the feeder layer could support the growth of human SSCs, demonstrated the feasibility of the culture of human SSCs and suggested that the colony formation of human SSCs is critically dependent on GDNF. Although the concept that the niche creates an essential environment for the growth of stem cells has been well established in several tissue systems, our study provides the first evidence that hESCs possess the ability to generate autologous hdFs that can serve as the feeder layer for the growth of human SSCs in vitro.
As active growth of somatic cells interferes with germ-cell proliferation, the initiation of the culture was facilitated by the enrichment of germ-cell populations. To isolate SSCs and eliminate somatic cells from testicular tissue, several approaches are available, such as gravity sedimentation to separate cells of different sizes, fluorescence-activated cell sorting or MACS. The MACS system is fast and causes only minimal stress to the SSCs during the process of enrichment. One of the most crucial steps for the enrichment of SSCs is the availability of highly specific markers. Signaling pathway proteins or receptors expressed exclusively on the surface of spermatogonia can be used for cell separation by MACS. To isolate the undifferentiated human spermatogonia, marker proteins such as CD49f, β1-integrin and Thy-1 (glycosyl phosphatidylinositol-anchored surface antigen) could be used 27. The marker CD49f was described earlier in the literature for mouse and human spermatogonical stem cell enrichment 28. In our study, SSCs were enriched using CD49f as a specific surface marker and MACS as a separation approach. The phenotype and number of SSCs obtained after MACS were assessed by flow-cytometric analysis. By implementing these methods, human SSCs could be successfully established and were able to proliferate for 1 month.
In this study, we illustrated the spontaneous differentiation of hESCs into fibroblast-like cells. The nature of these cells was supported by the expression of certain characteristic markers, in particular CD44 and CD90, which are both present in mesenchymal and fibroblast cells. During the cultivation of SSCs, feeder layer cells should lose their proliferative capacity while maintaining their self-secretory function to avoid growth competition between SSCs and feeder layer cells. Therefore, hdFs should be preprocessed before being used as feeder layer cells. In our study, we used mitomycin C to treat hdFs. Mitomycin C inhibits the duplication of DNA by cross-linking with DNA through alkanization and causing the depolymerization of DNA double strands. The time and dosage of mitomycin C should be carefully controlled 29. Undertreatment would not inhibit proliferation effectively, whereas overtreatment would cause the death of hdFs, with the DNA fragments and lysosomal enzymes released by dead hdFs interfering with the growth and maintenance of SSCs. After experimentation, we found that treatment with 12.5 mg L−1 mitomycin C for 2 h resulted in the inhibition of hdF proliferation and the maintenance of viability for as long as 15 days. These processed hdFs could be employed as the feed layer cells.
Development of a culture system that supports self-renewal and proliferation of SSCs in vitro is enormously valuable for understanding their biological characteristics and the possible modification of their genes. The most striking result of our experiments is that the culture system consisting of an inactivated hdF feeder layer and medium supplemented with GDNF can support the growth of human SSCs. Using the culture system optimized for SSCs, we showed that human SSCs could continuously proliferate for over 1 month and retain all the characteristics of stem cells. The expression of specific cell surface markers was determined by immunocytochemical analysis, RT-PCR and flow cytometry 6, 12. Human SSC colonies cultivated on an hdF feeder layer showed typical expression patterns, such as being positive for OCT4, SSEA-1, CD49f and AKP. In addition, human SSCs also expressed SSC-associated genes such as OCT4, SOX3, NGN3 and NOTCH1, but not C-KIT. Furthermore, we have analyzed the supporting role of GDNF. In the presence of GDNF, human SSCs formed densely packed clumps of cells and continuously proliferated. This suggested that GDNF induced cell signaling that played a central role in the self-renewal of human SSCs. Overall, these data showed that the culture system using hdFs as feeder layer could support the growth of human SSCs and maintain all of the features of SSCs after continuous culture.
In conclusion, we have developed a novel method using hdFs as a specific and unique 'niche' (feed layer) for the growth of human SSCs. This system can support the growth of human SSCs and eliminates the risks and concerns associated with the use of xenogeneic feeders.
Acknowledgments
This study was supported by grants from the Shanghai Municipal Population and Family Planning Commission, China (No. 2007JG06) and the Shanghai Leading Academic Discipline Project, China (No. Y0205).
References
- Kubota H, Brinster RL. Technology insight: in vitro culture of spermatogonial stem cells and their potential therapeutic uses. Nat Clin Pract Endocrinol Metab. 2006;2:99–108. doi: 10.1038/ncpendmet0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong D, Mclean DJ, Griswold MD. Long-term culture and transplantation of murine testicular germ cells. J Androl. 2003;24:661–9. doi: 10.1002/j.1939-4640.2003.tb02724.x. [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Muneto T, Lee J, Takenaka M, Chuma S, et al. Long-term culture of male germline stem cells from hamster testes. Biol Reprod. 2008;78:611–7. doi: 10.1095/biolreprod.107.065615. [DOI] [PubMed] [Google Scholar]
- Kateri A, Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–5. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- Dao MA, Creer MH, Nolta JA, Verfaillie CM. Biology of umbilical cord blood progenitors in bone marrow niches. Blood. 2007;110:74–81. doi: 10.1182/blood-2006-08-034447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte PM, van Bragt MP, de Rooij DG, van Pelt AM. Spermatogonial stem cells: characteristics and experimental possibilities. APMIS. 2005;113:727–42. doi: 10.1111/j.1600-0463.2005.apm_302.x. [DOI] [PubMed] [Google Scholar]
- Stojkovic P, Lako M, Stewart R, Przyborski S, Armstrong L, et al. An autogeneic feeder cell system that efficiently supports growth of undifferentiated human embryonic stem cells. Stem Cells. 2005;23:306–14. doi: 10.1634/stemcells.2004-0137. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448:1015–21. doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119:1001–12. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Guan K, Nayernia K, Maier MS, Wagner S, Dressel R, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature. 2006;440:1199–203. doi: 10.1038/nature04697. [DOI] [PubMed] [Google Scholar]
- Kerkis A, Fonseca SA, Serafim RC, Lavagnolli TM, Abdelmassih S, et al. In vitro differentiation of male mouse embryonic stem cells into both presumptive sperm cells and oocytes. Cloning Stem Cells. 2007;9:535–48. doi: 10.1089/clo.2007.0031. [DOI] [PubMed] [Google Scholar]
- Shinohara T, Brinster RL. Enrichment and transplantation of spermatogonial stem cells. Int J Androl. 2000;23 Suppl 2:89–91. doi: 10.1046/j.1365-2605.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- Reubinoff BE, Pera MF, Fong CY, Trounson A, Bonso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Sinha S, Wamhoff BR, Hoofnagle MH, Thomas J, Neppl RL, et al. Assessment of contractility of purified smooth muscle cells derived from embryonic stem cells. Stem Cells. 2006;24:1678–88. doi: 10.1634/stemcells.2006-0002. [DOI] [PubMed] [Google Scholar]
- Suemori H, Yasuchika K, Hasegawa K, Fujioka T, Tsuneyoshi N, et al. Efficient establishment of new human embryonic stem cell lines and long term maintenance with stable karyotype by enzymatic bulk passage. Biochem Biophys Res Commun. 2006;345:926–32. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- Joannides A, Fiore-Hériché C, Westmore K, Caldwell M, Compston A, et al. Automated mechanical passaging: a novel and efficient method for human embryonic stem cell expansion. Stem Cells. 2006;24:230–5. doi: 10.1634/stemcells.2005-0243. [DOI] [PubMed] [Google Scholar]
- Sandlow JI, Feng HL, Cohen MB, Sandra A. Expression of c-KIT and its ligand, stem cell factor, in normal and subfertile human testicular tissue. J Androl. 1996;17:403–8. [PubMed] [Google Scholar]
- Hayashi T, Kageyama Y, Ishizaka K, Xia G, Kihara K, et al. Requirement of Notch 1 and its ligand jagged 2 expressions for spermatogenesis in rat and human testes. J Androl. 2001;22:999–1011. doi: 10.1002/j.1939-4640.2001.tb03441.x. [DOI] [PubMed] [Google Scholar]
- von Schonfeldt V, Wistuba J, Schlatt S. Notch-1, c-kit and GFRalpha-1 are developmentally regulated markers for premeiotic germ cells. Cytogenet Genome Res. 2004;105:235–9. doi: 10.1159/000078194. [DOI] [PubMed] [Google Scholar]
- Kubota H, Avarbock MR, Brinster RL. Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc Natl Acad Sci USA. 2003;100:6487–92. doi: 10.1073/pnas.0631767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RA, Fulton N, Cowan G, Coutts S, Saunders PT. Conserved and divergent patterns of expression of DAZL, VASA and OCT4 in the germ cells of the human fetal ovary and testis. BMC Dev Biol. 2007;7:136. doi: 10.1186/1471-213X-7-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Engel W, Nayernia K. Stem cell protein Piwil2 modulates expression of murine spermatogonial stem cell expressed genes. Mol Reprod Dev. 2006;73:173–9. doi: 10.1002/mrd.20391. [DOI] [PubMed] [Google Scholar]
- Wang YB, Chen B, Wang Z. Transplantation of spermatogonial stem cells and prospects of its application. Zhonghua Nan Ke Xue. 2008;14:635–9. [PubMed] [Google Scholar]
- Amit M. Feeder-layer free culture system for human embryonic stem cells. Methods Mol Biol. 2007;407:11–20. doi: 10.1007/978-1-59745-536-7_2. [DOI] [PubMed] [Google Scholar]
- Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc Natl Acad Sci USA. 2006;103:9524–9. doi: 10.1073/pnas.0603332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Guo YL, Li XH, Jin FS, Sun ZY.Long-term survival of human spermatogonia stem cells in vitro and its functional identification Zhonghua Nan Ke Xue 200511886–90.894. [PubMed] [Google Scholar]
- Conrad S, Renninger M, Hennenlotter J, Wiesner T, Just L, et al. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456:344–9. doi: 10.1038/nature07404. [DOI] [PubMed] [Google Scholar]
- Nieto A, Cabrera CM, Catalina P, Cobo F, Barnie A, et al. Effect of mitomycin-C on human foreskin fibroblasts used as feeders in human embryonic stem cells: immunocytochemistry MIB1 score and DNA ploidy and apoptosis evaluated by flow cytometry. Cell Biol Int. 2007;31:269–78. doi: 10.1016/j.cellbi.2006.11.006. [DOI] [PubMed] [Google Scholar]