

Abstract
In flowering plants, the embryo develops within a nourishing tissue - the endosperm - surrounded by the maternal seed integuments (or seed coat). As a consequence, the isolation of plant embryos at early stages (1 cell to globular stage) is technically challenging due to their relative inaccessibility. Efficient manual dissection at early stages is strongly impaired by the small size of young Arabidopsis seeds and the adhesiveness of the embryo to the surrounding tissues. Here, we describe a method that allows the efficient isolation of young Arabidopsis embryos, yielding up to 40 embryos in 1 hr to 4 hr, depending on the downstream application. Embryos are released into isolation buffer by slightly crushing 250-750 seeds with a plastic pestle in an Eppendorf tube. A glass microcapillary attached to either a standard laboratory pipette (via a rubber tube) or a hydraulically controlled microinjector is used to collect embryos from droplets placed on a multi-well slide on an inverted light microscope. The technical skills required are simple and easily transferable, and the basic setup does not require costly equipment. Collected embryos are suitable for a variety of downstream applications such as RT-PCR, RNA sequencing, DNA methylation analyses, fluorescence in situ hybridization (FISH), immunostaining, and reporter gene assays.
Keywords: Plant Biology, Issue 76, Cellular Biology, Developmental Biology, Molecular Biology, Genetics, Embryology, Embryo isolation, Arabidopsis thaliana, RNA amplification, transcriptomics, DNA methylation profiling, FISH, reporter assays
Introduction
The embryo of flowering plants is surrounded by the endosperm, a nutritive tissue derived from a second fertilization event. Both embryo and endosperm are surrounded by several cell layers of the seed coat. Collectively these tissues form a seed, which develop inside the fruit. Thus, tissue- and cell-specific analyses of Arabidopsis embryos are strongly impaired due their inaccessibility. Nevertheless, embryos at the late-globular or later stages are relatively well amenable to manual dissection by using fine tungsten needles under the stereomicroscope, or by applying slight pressure on the seed using forceps to extract them. Such techniques were successfully used for transcriptome or epigenome profiling analyses such as microarray hybridization, bisulfite sequencing, or RNA sequencing (e.g. 1-3). In contrast, studies of embryos at the zygote to early globular stage remain technically challenging. To date, only a few studies have reported transcriptome analyses on young embryos using either laser-capture microdissection (LCM) of embryonic tissues from fixed seed sections 4 or manual extraction of individual embryos from within seeds using fine tools 5. However, LCM equipment is not commonly available and manual embryo extraction at early stages is time consuming and requiring excellent dissection skills that are not easily transferable. In addition to genome-wide analyses, in situ gene expression analyses are also difficult to perform on young, whole-mount embryos of Arabidopsis. To some extent, young embryos can be released on microscope slides by gentle pressure on the seeds and used for reporter gene assays or protein detection by immunostaining (for example see 6,7). This technique, however, does not allow high-throughput embryo isolation, thus hindering quantitative analyses.
Therefore, we developed an efficient and rapid protocol for early embryo isolation from Arabidopsis seeds that is simple to set up, easily transferable, and suitable for a variety of downstream applications. The basic principle is to gently crush seeds - dissected from young siliques in an Eppendorf tube using a plastic pestle in an appropriate isolation buffer. The seed extract is placed in droplets on a multi-well slide and is screened for the presence of released embryos at the desired stage using an inverted microscope. Embryos are collected using a glass microcapillary attached to a microinjector or a standard laboratory pipette. For molecular applications, embryos are washed twice by repeated release into drops of new isolation buffer before transferring them to the destination buffer in a minimal volume. For cytological applications (reporter assays, immunostaining, FISH), washing steps can be omitted.
The method offers several advantages: (i) it yields 25-40 embryos in ~45 min for cytological applications or in 3-4 hr for molecular applications (including the washing steps), (ii) it allows isolation of specific embryonic stages, (iii) it is easily transferable to other persons and laboratories due to its simple setup, (iv) it requires affordable equipment for the basic setup which is amenable to upgrades, and (v) it was successfully used for various downstream applications such as RNA sequencing 8, gene-specific DNA-methylation analysis 9, reporter assays (10 and Raissig et al., in prep.), and FISH (J. Jaenisch, U. Grossniklaus, C. Baroux unpublished, see Figure 5).
Protocol
The procedure is summarized in the flowchart shown in Figure 1. The microcapillaries and the instrumental setup are shown in Figure 2 and Figure 3, and typical steps of embryo isolation are shown in Figure 4.
1. Material and Buffer Preparation
1.1 Silicon coating of glass microcapillaries
Place the microcapillaries in a 15 ml Falcon tube with ~5 ml of Sigmacote (Sigma) and invert several times.
Remove the solution, place the Falcon tube containing the capillaries in an aluminum foil and bake them for 3 hr at 60 °C. Store at room temperature.
1.2 Obtain ~50-100 μm-diameter microcapillary tips
Pull 1 mm-diameter glass capillaries either manually over a Bunsen burner or by using a commercial puller (vertical filament puller or micropipette puller).
Use a diamond-tip pen or blade to cut the tip of the pulled capillary to create the desired opening. Select the best-shaped capillaries under a stereo microscope. The opening should be 50-100 μm (Figure 2).
1.3 Slide preparation
Siliconize clean slides by covering all the wells with Sigmacote (~1 ml/slide) for 5 min, remove. Bake 3 hr in aluminum foil. Store at room temperature.
Wash the multi-well glass microscopic slides for 10 min in 10% SDS, 2 x 2 min in nuclease-free water (autoclaved DEPC-ddH2O), 2 min in 70% ethanol, 2 min in 100% ethanol, air-dry. All steps are done in autoclaved Coplin jars. Slides can be re-used multiple times providing thorough cleaning between each usage.
Just prior to embryo isolation spread ~0.5 μl of 10 mg/ml bovine serum albumin (BSA) with a pipette over the whole surface of each well and air-dry.
1.4 Microscope and capillary setup
Use an inverted microscope with a 10x and 20x magnification objective. Optimize the light contrast (embryos are quite transparent).
Place a micromanipulator to hold the glass capillary beside the microscope. The glass capillary is connected to a microinjector (Figure 3A) or to a regular P-200 pipette via a rubber tube (Figure 3B, see Discussion for a detailed description of this setup).
Place the capillary above the microscope slide at a ~70° angle (Figure 3) and adjust the position to have the opening in the field of view.
Just prior to embryo isolation take up and release ~5-10 μl BSA (1 mg/ml) to coat the capillary.
1.5 Buffers
Table 1 lists the isolation and destination buffers depending on the downstream applications.
Prepare ~1 ml isolation buffer per sample freshly before use and keep it on ice.
Prepare the destination buffer in a 0.5 ml Eppendorf low-binding tube on ice (molecular applications) or on a microscope slide (cytological applications) in a humid chamber.
2. Seed Dissection and Embryo Extraction
2.1 Synchronisation of Seed Development
Emasculate flowers and keep them 2 days in the growth chamber while avoiding contact of the exposed pistils with other flowers, then pollinate them (e.g. 11).
Test the stage of development under your growth conditions by microscopic investigation of cleared seeds. With our growth conditions (16 hr light at 21 °C, 8 hr dark at 18 °C and 70% humidity) seeds collected 2.5 days after pollination (DAP) yielded mainly 2-4 cell embryos and seeds collected 3.5 - 4 DAP yielded globular embryos.
2.2 Seed Dissection and Rupture
Remove the seeds from 10-15 siliques (~2.5 DAP) under a stereomicroscope with forceps and insulin needles.
Immerse the seeds in 20 μl isolation buffer in a 2 ml round-bottom Eppendorf tube placed on ice.
Gently crush the seeds with a plastic pestle (pre-cleaned with 10% SDS, rinsed with DEPC-ddH2O and washed with 70% ethanol) to release the embryos until the seed extract is cloudy. The force to apply is to be determined by every user upon trial.
Rinse the pestle with 300 μl of isolation buffer to wash the pestle and dilute the sample.
Spin-down the extract at 5,000 x g for 5 sec. Gently resuspend the pelleted extract by pipetting up-and-down 2-3 times.
Filter the extract with a 30 μm nylon mesh (mounted on tube adaptors, e.g. from PartecCelltricks). Rinse the mesh with an additional 200 μl isolation buffer.
3. Embryo Isolation
3.1 Slide preparation
Place a clean, BSA-coated and siliconized multi-well slide on the stage of the inverted microscope, resuspend the filtered seed extract by pipetting gently up and down and pipette 2 droplets of 40-50 μl seed extract into 1 or 2 wells. Screening only 1 or 2 drops at a time prevents evaporation of the sample.
Place 50 μl of fresh isolation buffer (1st wash drop) in a well of a different slide prepared as before. Keep this slide in a covered, humid chamber to prevent evaporation.
3.2 Screen, clean, collect
Screen the droplets of seed extract for embryos at the desired stage with the 10x magnification objective. If necessary, confirm the stage with the 20x magnification. The embryos usually sink to the bottom of the slide.
Manually remove debris around the embryo with a tungsten needle, an insulin needle or similar equipment.
Move the glass capillary near the embryo using the micromanipulator, take up the embryo with as little solution as possible.
Collect several embryos (e.g. all of one droplet) and release them in the 1st wash drop(molecular application) or in destination buffer (cytological applications). Each collecting round should be kept within 5-10 min and embryos should be collected in a minimal volume (the total volume of all collecting rounds should be <5 μl).
Repeat the screening and collection until the desired amount of embryos is gathered in the 1st wash drop (centrally, if possible, to facilitate recollection).
Recollect all embryos at once from the wash drop (if debris are carried over, remove them with a needle before recollection).
Release the embryos into a 2nd wash drop of 50 μl. Repeat 3.2.6.
Release the embryos in the destination buffer. The transfer should involve only a contact, and not immersion, of the capillary tip.
Replace the microcapillary for the next sample.
Representative Results
Our embryo isolation procedure (Figure 1) allows isolation of up to 40 embryos in 4 hr if washes are performed, e.g. for molecular applications, or in less than an hour if washes are omitted, e.g. for cytological applications. Figure 2 displays high and low quality microcapillary tips and Figure 3 shows the setup of the embryo isolation machine. Figure 4 displays the process of embryo isolation on the inverted microscope.
We successfully applied our procedure for various applications published in several recent articles. The method was originally developed to analyze the parental contribution to the early embryonic transcriptome. Hybrid embryos generated through crosses between two different Arabidopsis accessions (Landsbergerecta (Ler) and Columbia (Col-0)) were isolated at the 2-4 cell stage. Total RNA was extracted, cDNA libraries were produced using linear amplification and sequenced on a SOLiD platform. The allele-specific transcriptomes were generated based on SNP analysis 8. To control our embryonic cDNA libraries prior to sequencing we amplified embryo-specific transcripts, WUSCHEL-HOMEOBOX 2, 8 and 9 (WOX2, WOX8, WOX9) and ACTIN 11 (13,14, Figure 5A).
Additionally, this method was used to isolate young embryos to analyze the embryonic DNA-methylation patterns at specific loci in the genome. Embryos were isolated and washed in 1x TE-buffer. Small-scale bisulfite sequencing was performed as described 9,15.
Similarly, embryo isolation has proved very useful in reporter gene assays with low embryonic expression, and where the maternal seed coat confounds detection or is masked by reporter expression in tissues surrounding the embryo (endosperm, seed coat). Embryos carrying the reporter transgene are stained (ß-glucuronidase; GUS) or directly analyzed (Green or Red Florescent Protein; GFP, RFP) following isolation. An example is given in Figure 5B for embryos expressing a GUS reporter gene under the control of the MEDEA promoter (pMEA) 10. The relatively ease with which many embryos can be isolated also allows for quantitative analyses (e.g. number of embryos stained in different genetic backgrounds, Raissig, Grossniklaus et al. in preparation).
Finally, we successfully applied Fluorescent In Situ Hybridization (FISH) and immunostaining techniques to study the nuclear architecture in isolated embryos embedded in acrylamide pads on slides. An example of FISH using probes against the centromere repeats and nucleolar organizing regions is shown in Figure 5C.
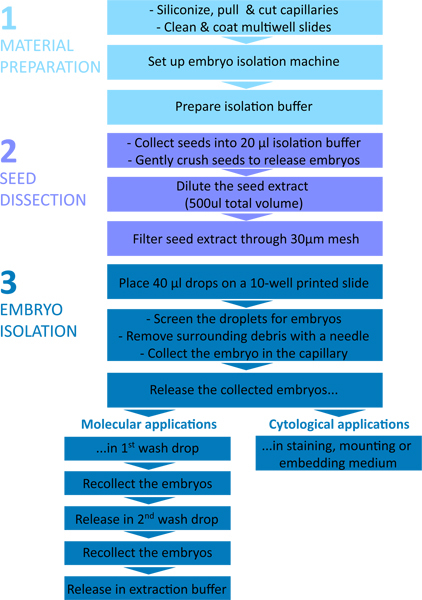 Figure 1. Flow chart of the embryo isolation procedure. The protocol is divided in three parts: 1 - Material preparation; 2 - Seed dissection; 3 - Embryo isolation.
Figure 1. Flow chart of the embryo isolation procedure. The protocol is divided in three parts: 1 - Material preparation; 2 - Seed dissection; 3 - Embryo isolation.
 Figure 2. Microcapillary tips.A. High-quality microcapillary tips with a smooth opening of around 100 μm. B. Low-quality microcapillary tips with wider opening and irregular contour (those tips are acceptable for cytological applications only). Scale bar: 2 mm.
Figure 2. Microcapillary tips.A. High-quality microcapillary tips with a smooth opening of around 100 μm. B. Low-quality microcapillary tips with wider opening and irregular contour (those tips are acceptable for cytological applications only). Scale bar: 2 mm.
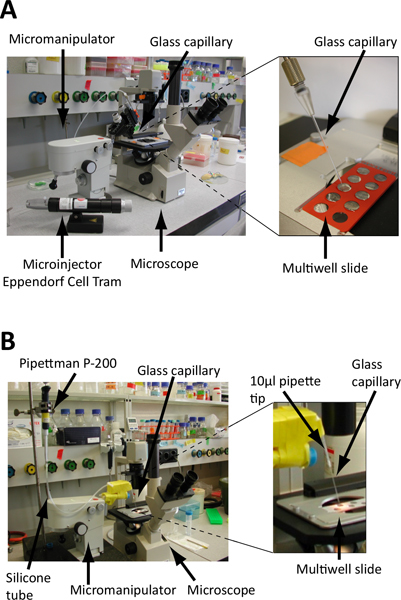 Figure 3. Set up of the embryo isolation microscope.A. and B. Screening is performed on microscope slides under an inverted microscope. Embryos are collected using a glass microcapillary fixed on a micromanipulator to precisely control its position (see also Figure 4) and connected to either a microinjector (A) or standard laboratory pipette (B). A. The glass microcapillary is linked to a hydraulically controlled microinjector (e.g. Eppendorf Cell Tram Vario) for a precise control during embryo collection B. The glass microcapillary is attached to a standard P-200 pipette via a rubber flex tube. Embryo collection and release is controlled by turning the calibration wheel of the pipette. Cut-end pipette tips are used as connectors and the junctions are sealed with parafilm. The microcapillary is fixed on the micromanipulator via polystyrene blocks, Falcon tubes and tape.
Figure 3. Set up of the embryo isolation microscope.A. and B. Screening is performed on microscope slides under an inverted microscope. Embryos are collected using a glass microcapillary fixed on a micromanipulator to precisely control its position (see also Figure 4) and connected to either a microinjector (A) or standard laboratory pipette (B). A. The glass microcapillary is linked to a hydraulically controlled microinjector (e.g. Eppendorf Cell Tram Vario) for a precise control during embryo collection B. The glass microcapillary is attached to a standard P-200 pipette via a rubber flex tube. Embryo collection and release is controlled by turning the calibration wheel of the pipette. Cut-end pipette tips are used as connectors and the junctions are sealed with parafilm. The microcapillary is fixed on the micromanipulator via polystyrene blocks, Falcon tubes and tape.
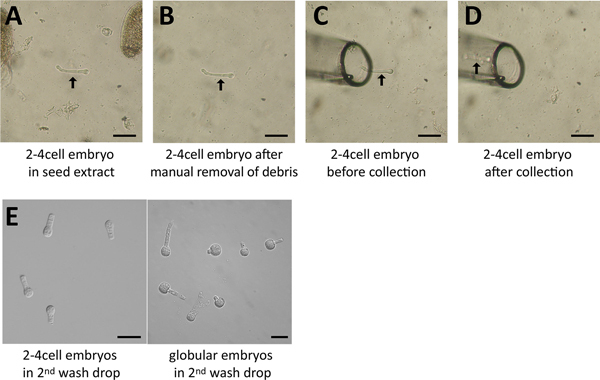 Figure 4. Embryo isolation process.A. A 2-4 cell embryo (arrow) was identified in the seed extract (screening droplet) and is surrounded by debris (seed coat fragments). B. The surrounding debris was removed manually using a needle. C. The glass capillary was moved right beside the embryo (arrow). D. The embryo was collected and is now within the capillary (arrow). E. Several embryos in the last drop following washes. Scale = 50 μm.
Figure 4. Embryo isolation process.A. A 2-4 cell embryo (arrow) was identified in the seed extract (screening droplet) and is surrounded by debris (seed coat fragments). B. The surrounding debris was removed manually using a needle. C. The glass capillary was moved right beside the embryo (arrow). D. The embryo was collected and is now within the capillary (arrow). E. Several embryos in the last drop following washes. Scale = 50 μm.
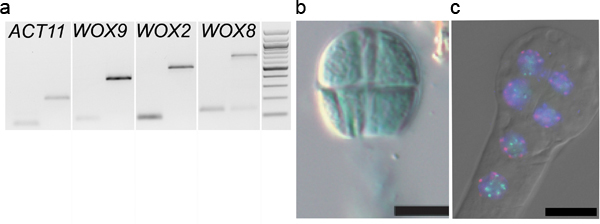 Figure 5. Downstream applications of embryo isolation.A. Gene expression analyses: PCR amplification of WOX9, WOX2, WOX8 and ACTIN11 13,14 on cDNA libraries (generated as described 8) from 2-4 cell embryos washed 1x times (1st lane) and on genomic DNA (2nd lane). B. Reporter assay: An 8-cell embryo isolated from plants carrying a pMEA::GUS construct was stained on a slide in a standard GUS staining solution (reproduced from Raissig et al., 2011 after permission from The Plant Cell, ASPB copyright). C. Fluorescent In-Situ Hybridization (FISH): an 8-cell embryo was hybridized with probes against centromeric repeats (red), and 45S rDNA repeats (green) before indirect immunodetection 16. The dual-color FISH images were overlaid with the DAPI counterstaining and DIC images (DM6000B epifluorescence microscope, Leica, Germany).
Figure 5. Downstream applications of embryo isolation.A. Gene expression analyses: PCR amplification of WOX9, WOX2, WOX8 and ACTIN11 13,14 on cDNA libraries (generated as described 8) from 2-4 cell embryos washed 1x times (1st lane) and on genomic DNA (2nd lane). B. Reporter assay: An 8-cell embryo isolated from plants carrying a pMEA::GUS construct was stained on a slide in a standard GUS staining solution (reproduced from Raissig et al., 2011 after permission from The Plant Cell, ASPB copyright). C. Fluorescent In-Situ Hybridization (FISH): an 8-cell embryo was hybridized with probes against centromeric repeats (red), and 45S rDNA repeats (green) before indirect immunodetection 16. The dual-color FISH images were overlaid with the DAPI counterstaining and DIC images (DM6000B epifluorescence microscope, Leica, Germany).
| Application | Isolation buffer | Destination buffer |
| RNA extraction (e.g. for amplification and transcriptomics) | 1x First-Strand buffer (Invitrogen), 1 mM DTT, 4% RNAseOUT | RNA extraction buffer |
| DNA extraction (e.g. for bisulfite sequencing) | 100 mM Tris, 10 mM EDTA, pH 8 (TE) | DNA extraction buffer or 1x TE buffer |
| GFP reporter analysis | 1 M Glycin in 0.5x MS1 | 1 M Glycin in 0.5x MS |
| GUS reporter analysis | staining buffer2 without X-Gluc (substrate) | staining buffer with X-Gluc (substrate) |
| FISH & Immunostaining | 100 mM Phosphate Buffered Saline (PBS) | activated Acryl:Bisacryl mix (33:3) on a Superfrost Plus slide -polymerize for 30 min |
Table 1. Isolation and destination buffers for different downstream applications. The buffers can be adapted to specific experimental requirements. 1 Murashig & Skoog medium. 2 e.g. Jefferson et al., EMBO J. 6 (13), 3901-3907 (1987).
Discussion
We developed an embryo isolation protocol that is rapid, effective, and can be easily transferred to other laboratories.
The equipment described here consists of an inverted microscope, a micromanipulator, glass microcapillaries, a vertical filament puller and a microinjector (Figure 3A). The setup is similar to the one described for single animal cell isolation for transcriptomics analyses 17. We also successfully worked with a more basic setup where glass microcapillaries were manually stretched over a flame (and cut with a diamond-blade) and operated via a standard laboratory pipette (Pipetman P-200) instead of a microinjector (Figure 3B). In that case the microcapillary was attached to the pipette via a rubber tube. 10 μl filtertips were used to make the junctions, which were wrapped with parafilm to make them air-tight. The capillary - held by the pipette tip - was fixed on the micromanipulator arm using polystyrene blocks and tape (Figure 3B). This basic setup proved to be efficient and reliable 8. Nevertheless, one of the main difficulties was the maintaining of air-tight junctions between the microcapillary and the pipette, especially when changing the capillary. Verifying and adjusting the pressure in the capillary - to ensure a fine-tuned embryo collection in a minimal volume - can be time-consuming using this basic setup. The improved setup with a hydraulically controlled micromanipulator and a vertical filament puller is a considerable improvement and saves time.
The skills required for the successful application of this method are easily transferable. Five users in our laboratory successfully learned the method in a relatively short time. Some simplifications of the protocol are possible depending on the user's ease in manipulation. For instance, it is possible to skip the steps 2.2.5 and 2.2.6 while dissecting only 5 siliques (instead of 15) and cleaning the embryo surroundings thoroughly from debris with a tungsten needle (this procedure was applied in 8). Synchronisation of seed development through emasculation and delayed pollination is facultative but recommended to increase the number of embryos at the same developmental stage, particularly at early stages. In addition, embryo isolation can be expedited if washing steps are omitted, e.g. for cytological applications. Furthermore, slide siliconization (step 1.3.2) is not necessary for users working quickly, such that embryos which stick to the glass surface over time is not a problem.
Whether filtering or manual cleaning is applied, it is extremely important to avoid carry-over of tissue debris that creates potential contamination for downstream RNA or DNA applications. This issue was raised recently in a transcriptome profiling study using a different method of embryo isolation 18. By contrast to the bulk embryo isolation protocol described here, the authors dissected Arabidopsis embryos manually from within individual seeds using fine needles, a lengthy procedure requiring excellent dissection skills. This procedure apparently required 2 or more washes to avoid carry-over of possible contaminating cells from surrounding seed tissue, a situation likely encountered when using this method given the small size of the embryos, the difficulties in accessing them with dissection needles, and the adhesiveness of surrounding cells. By contrast, our procedure allows (i) the dilution of the embryos -extruded from the seeds upon crushing- and surrounding debris in a large volume (500 μl) before embryo collection, (ii) the visual selection of embryos devoid of adhesive, contaminating debris, and (iii) the dilution of possible RNA contamination -leaking from ruptured cells-by the washing steps. While it is possible to use only one wash step if the embryos appear very clean and are collected in less than 5 μl 8, a second wash step is recommended, especially for first-time users that may collect the embryos in several rounds (i.e. with additive volume). Embryos should be collected in as little solution as possible, allowing the maximal dilution of potential contaminants in every washing step. Each collecting round should be kept short (within 5-10 min) to allow maximum recovery upon release into the wash drop (longer presence in the capillary tends to reduce the recovery rate). Furthermore, the transfer of the collected embryos into the extraction medium (following the wash steps) should only involve a contact with the opening of the microcapillary tip and not immersion of the tip, as this might as well transfer debris sticking on the microcapillary wall above the opening. Finally, the capillary should be changed between each new seed extract.
Our protocol allows for several downstream applications by simply using an appropriate isolation buffer that preserve molecular integrity of RNA, DNA, chromatin, enzyme or fluorescent reporter. We successfully isolated embryos in RNA-protective isolation buffer for RNA extraction and transcriptomics analyses 8, 1x TE-buffer for DNA methylation analyses 9, 100 mM phosphate buffered saline (PBS) for FISH and immunostaining (Jaenisch, Grossniklaus and Baroux, unpublished, Figure 5C), and staining solution without substrate for GUS reporter assays (19, Figure 5B). The application most sensitive to isolation conditions/embryo quality is certainly RNA extraction and amplification. The quantity of extracted RNA from isolated embryos was rather low. From ~30 embryos at the 2-4 cell stage (thus 60-120 cells in total) we estimated an amount of ~1ng of total RNA based on both Agilent 2100 Bioanalyzer (Agilent Technologies) and QUBiT (Invitrogen) measurements. Following RNA extraction, RNA quality was verified on an Agilent RNA 6000 Pico Chip (Agilent Technologies). However, the low amount of RNA extracted is not always sufficient to produce a reliable profile. Thus, further tests must be done on the amplified material (cDNA) (e.g. PCR detecting low-expressed, embryo-specific genes and/or a Bioanalyzer profile).
Finally, the ability to isolate embryos by visual criteria in our protocol is a great asset. Embryos from the same silique (Arabidopsis fruit) do not develop synchronously. Thus, working on whole silique extracts (e.g. for RT-PCR analyses or quantitative reporter assays) does not allow a perfect correlation with a given developmental stage. Isolating embryos at a specific developmental stage solves this problem. In addition, a similar problem presents when working with heterozygous mutants where siliques contain different embryo genotypes. Isolating embryos based on visual criteria to distinguish e.g. wild-type from mutant embryo phenotypes allows correlating downstream analyses with the genotype. We have done this successfully to analyze DNA methylation at specific loci in different genotypes 20.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We would like to thank Tal Nawy and Martin Bayer for their advice on embryo isolation. MTR, VG, UG and CB devised the embryo isolation equipment. MTR, VG and CB developed the embryo isolation protocol. MTR, VG and CB established the protocol, isolated the embryos, and generated embryo cDNA, VG performed the PCR, MTR the GUS staining, JJ the FISH experiments. MTR, VG, CG and UG wrote the manuscript. This work was funded by the University of Zürich, a Fellowship of the Roche Research Foundation (to MTR), and grants from the Swiss National Foundation (to UG and CB).
References
- Gehring M, Bubb KL, Henikoff S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science. 2009;324:1447–1451. doi: 10.1126/science.1171609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller B, Sheen J. Cytokinin and auxin interaction in root stem-cell specification during early embryogenesis. Nature. 2008;453:1094–1097. doi: 10.1038/nature06943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Missirian V, Henikoff S. Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS One. 2011;6:23687–23. doi: 10.1371/journal.pone.0023687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NSF-Funded Seed Project of Goldberg & Harada Laboratories [Internet] 2006. Available from: http://estdb.biology.ucla.edu/seed/
- Xiang D, et al. Genome-wide analysis reveals gene expression and metabolic network dynamics during embryo development in Arabidopsis. Plant Physiol. 2011;156:346–356. doi: 10.1104/pp.110.171702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy T, et al. The GATA factor HANABA TARANU is required to position the proembryo boundary in the early Arabidopsis embryo. Dev Cell. 2010;19:103–113. doi: 10.1016/j.devcel.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Baroux C, Pecinka A, Fuchs J, Schubert I, Grossniklaus U. The triploid endosperm genome of Arabidopsis adopts a peculiar, parental-dosage-dependent chromatin organization. Plant Cell. 2007;19:1782–1794. doi: 10.1105/tpc.106.046235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autran D, et al. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell. 2011;145:707–719. doi: 10.1016/j.cell.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Wohrmann HJ, et al. Identification of a DNA methylation-independent imprinting control region at the Arabidopsis MEDEA locus. Genes Dev. 2012;26:1837–1850. doi: 10.1101/gad.195123.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raissig MT, Baroux C, Grossniklaus U. Regulation and Flexibility of Genomic Imprinting during Seed Development. Plant Cell. 2011;23:16–26. doi: 10.1105/tpc.110.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea M, et al. Determination of DNA methylation of imprinted genes in Arabidopsis endosperm. J. Vis Exp. 2011;47(47):e2327. doi: 10.3791/2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SeedGenes Tutorial [Internet] 2010. Available from: http://www.seedgenes.org/Tutorial.html#Nomarski_Optics.
- Breuninger H, Rikirsch E, Hermann M, Ueda M, Laux T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev Cell. 2008;14:867–876. doi: 10.1016/j.devcel.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Kohler C, Page DR, Gagliardini V, Grossniklaus U. The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nature. 2005;37:28–30. doi: 10.1038/ng1495. [DOI] [PubMed] [Google Scholar]
- Epigenome NoE - protocol: Bisulfite sequencing of small DNA/cell samples [Internet] 2007. Available from: http://www.epigenesys.eu/images/stories/protocols/pdf/20111026124522_p35.pdf.
- Fransz P, De Jong JH, Lysak M, Castiglione MR, Schubert I. Interphase chromosomes in Arabidopsis are organized as well defined chromocenters from which euchromatin loops emanate. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14584–14589. doi: 10.1073/pnas.212325299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Singh JM, Eberwine JH. Transcriptome analysis of single cells. J. Vis. Exp. 2011. p. e2634. [DOI] [PMC free article] [PubMed]
- Nodine MD, Bartel DP. Maternal and paternal genomes contribute equally to the transcriptome of early plant embryos. Nature. 2012;482(7383) doi: 10.1038/nature10756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson RA, Kavanagh TA, Bevan MWGUS. fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987;6:3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Wöhrmann H, Raissig MT, Arand J, Gheyselinck J, Gagliardini V, Heichinger C, Walter J, Grossniklaus U. The Polycomb group protein MEDEA and the DNA methyltransferase MET1 interact in Arabidopsis to repress autonomous endosperm development. Plant J. 1111;73(5) doi: 10.1111/tpj.12070. In Press. [DOI] [PubMed] [Google Scholar]