

Abstract
The faithful expression of genes requires that cellular machinery select substrates with high specificity at each step in gene expression. High specificity is particularly important at the stage of nuclear pre-mRNA splicing, during which the spliceosome selects splice sites and excises intervening introns. With low specificity, the usage of alternative sites would yield insertions, deletions and frame shifts in messenger RNA. Recently, biochemical, genetic and genome-wide approaches have significantly advanced our understanding of splicing fidelity. In particular, we have learned that DExD/H-box ATPases play a general role in rejecting and discarding suboptimal substrates and that these factors serve as a paradigm for proofreading NTPases in other systems. Recent advances have also defined fundamental questions for future investigations.
Establishing specificity in pre-mRNA splicing
To maximize the fidelity in the inheritance and expression of genetic information, a cell minimizes errors. In DNA replication, errors have lasting consequences; mistakes are consequently reduced to a frequency of less than one in a billion [1]. In transcription and translation even transient errors present risks to the cell; mistakes are consequently reduced to roughly one in ten thousand [2, 3]. Similarly, in the removal of introns from primary nuclear transcripts, mistakes are minimized to prevent deleterious alterations to the message during the two catalytic steps in splicing: 5' splice site cleavage and exon ligation (Box 1). Given the dominance of introns over exons in eukaryotic genes, establishing fidelity in this process of pre-mRNA splicing is critical. Indeed, recent estimates of the error rate for splicing ranges from one in one hundred to one in one hundred thousand [4–6]. Fidelity mechanisms in splicing were first discovered nearly twenty years ago [7], but our understanding of fidelity in splicing has lagged behind that of DNA replication, transcription and translation. Nevertheless, our understanding of splicing fidelity mechanisms, particularly kinetic proofreading, has deepened considerably in recent years primarily through studies in the genetically tractable model organism Saccaromyces cerevisiae. In a short time, we have gained insights into the mechanisms for rejecting and discarding suboptimal substrates and the generality of these mechanisms across diverse cellular processes. In this review, we focus on these advances, with an emphasis on the general role of DExD/H-box ATPases in fidelity.
DExD/H-box ATPases Antagonize Splicing of Suboptimal Substrates
To rationalize how specificity could be established in translation, Hopfield and Ninio proposed a model nearly forty years ago, termed kinetic proofreading [8, 9]. This general model has been verified for translation and functionally validated in other systems [1–3, 10, 11] including splicing [7, 12–15], although in splicing, kinetic proofreading manifests in a unique and important way.
A central feature of kinetically proofread pathways is a branch in the pathway that competes with a productive step and specifically antagonizes suboptimal substrates [8, 9, 16, 17]. Importantly, a kinetically competing branch can affect the efficiency of a process at any step, independent of whether the competing, on-pathway step is rate-limiting. This concept was recently nicely illustrated for alternative splicing [18]. A role for a splicing factor in establishing a proofreading branch was first implicated by a genetic screen in the yeast S. cerevisiae, which revealed that the splicing factor Prp16 preferentially represses suboptimal substrates that have branch sites that deviate from the consensus [7, 19] (Box 1, Figure I). The DEAH-box ATPase Prp16 is one of eight spliceosomal ATPases that are conserved across eukaryota and belong to the ubiquitous SF2 superfamily of ATPases, which includes the DExD/H-box families of RNA helicases (Box 2). Recent genetic studies have firmly established the importance of spliceosomal DExD/H-box ATPases in antagonizing suboptimal splicing substrates in vivo (Figure 1). First, Prp5, a DEAD-box ATPase that is conserved from budding yeast to humans, was shown to promote branch site fidelity by interrogating U2 snRNA recognition of the branch site [12, 13]. Further, during exon ligation the conserved DEAH-box ATPase Prp22 antagonizes primarily suboptimal 3' splice sites but also suboptimal 5' splice sites and branch sites [14], underscoring the multiple stages at which a substrate feature is inspected. Indeed, in mammals the 3' splice site is also proofread at the earliest stages of intron recognition [20, 21].
Figure I (Box 1).

Splicing involves two sequential transesterification reactions. (a) The reactive groups in each reaction are specified by splice site consensus sequences, shown for metazoa and budding yeast [88]. (b) In the first step of splicing, known as 5' splice site cleavage, the 2' hydroxyl of a conserved branch site adenosine attacks the 5' splice site phosphate forming a branched, lariat intermediate and free 5' exon. In the second step, the 3' hydroxyl of the cleaved 5' exon attacks the 3' splice site phosphate, ligating the exons and excising the lariat intron. N, any nucleotide; R, purine; Y, pyrimidine.
Figure 1.

Functions of known proofreading DExD/H-box ATPases in the splicing pathway. During assembly, small nuclear ribonucleoproteins (snRNPs) (small blue circles) assemble on a pre-mRNA intron (black line) flanked by exons (yellow rectangles) in a stepwise manner. Following engagement of pre-mRNA by U2 snRNP (a), Prp5-dependent rejection (b) competes with branch site recognition by U2 (c) to antagonize splicing at introns with suboptimal branch site sequences [12, 13]. After formation of optimal branch site-U2 interactions, Prp5 activity stabilizes U2 association with the substrate (d) [13, 28]. Following addition of the U4/U6•U5 tri-snRNP, U1 and U4 are released (e), leading to formation of a catalytically active spliceosome (large blue oval). For substrates with suboptimal branch site sequences, Prp16-dependent rejection (f) competes with 5' splice site cleavage (g), resulting in formation of an intermediate that can lead to Prp43-mediated discard of the pre-mRNA substrate (h) [7, 15, 22, 23]. After 5' splice site cleavage of an optimal substrate, Prp16 promotes a transition to the exon ligation conformation of the spliceosome (i) [27]. For suboptimal substrates, Prp22-dedpendent rejection (j) competes with exon ligation (k), resulting in formation of an intermediate that can lead to discard of the 5' exon and lariat intermediate (l), again mediated by Prp43 [14, 38]. After exon ligation of an optimal substrate, Prp22 promotes release of the nascent mRNA from the spliceosome (m) [29, 30]. Following mRNA release, Prp43 promotes release of the excised intron and dissociation of the spliceosome into its component snRNPs (n) [55–58]. The canonical splicing pathway is indicated by heavy arrows.
The fidelity functions of each of these factors require DExD/H-box ATPase function. Mutant alleles of PRP5, PRP16 or PRP22 that compromise ATPase activity permit increased utilization of suboptimal substrates in vivo [7, 12, 14]. Further, prp5 alleles with a range of ATPase activity deficiency show proportional deficiencies in the antagonization of suboptimal branch sites, demonstrating the importance of the rate of rejection in kinetic proofreading [12]. In addition, mutations that do not compromise ATPase activity but compromise RNA unwinding activity also compromise fidelity in vitro [14], highlighting the role of RNP rearrangements in proofreading.
Biochemical studies of DExD/H-box ATPases have confirmed the general roles for these factors in splicing fidelity and have provided further insight into their mechanisms. Prp16 and Prp22, in particular, have been shown to proofread the chemical steps of splicing by parallel mechanisms [14, 15]. Recent genetic studies suggested that Prp16 proofreads by destabilizing the 5' splice site cleavage conformation of the spliceosome, either before or after 5' splice site cleavage [22, 23]. A subsequent in vitro study established that Prp16 proofreads by competing directly with 5' splice site cleavage [15]. Similarly, in vitro studies have established that Prp22 proofreads by competing directly with exon ligation [14]. Genetic studies have revealed that suboptimal substrates are also disfavored because the spliceosome sequesters the substrate away from the relevant catalytic conformation in favor of competing conformations [22, 24–26] (Box 3). Prp16- and Prp22-mediated rejection likely plays a role in driving this sequestration. Additionally, Prp16- and Prp22-mediated rejection can lead to discard of rejected substrates (see below). The similarity between the mechanisms of Prp16-dependent proofreading at 5' splice site cleavage and Prp22-dependent proofreading at exon ligation suggests a general role for DExD/H-box ATPases in proofreading by competing with productive events.
Proofreading DExD/H-box ATPases also promote optimal substrates
Over twenty years ago Prp16 was found not only to antagonize suboptimal branch site substrates but also to promote optimal substrates by enabling spliceosome rearrangements after 5' splice site cleavage [27]. The generality of such a dual role has recently been established (Figure 1). Prp5 not only antagonizes substrates with suboptimal branch sites but also promotes stable binding of U2 snRNP to substrates with optimal branch sites [13, 28]. Further, Prp22 not only antagonizes suboptimal substrates but also promotes optimal substrates by catalyzing release of the mRNA product [29, 30].
Although proofreading DExD/H-box ATPases both antagonize and promote substrates, these opposing activities appear highly related. Recent genetic data indicate that Prp5 both promotes and antagonizes substrates by destabilizing the stem of the U2 branchpoint-interacting stem loop (BSL); the loop is thought to allow sampling of candidate branch sites through base pairing [13]. Therefore, mutations that destabilize this stem suppress a prp5 mutant, relieving stalled optimal substrates, but these same mutations also enhance Prp5-mediated rejection of suboptimal substrates.
Similarly, genetic experiments indicate that Prp16 both rejects suboptimal substrates and promotes optimal substrates by antagonizing features important for 5' splice site cleavage, including Prp8, Isy1, U2/U6 helix I and U2 stem IIc. Mutations in these features can suppress a prp16 mutant, relieving stalled optimal substrates, but these same mutations also enhance Prp16-mediated rejection of a suboptimal substrate [22, 23, 25, 26]. Furthermore, biochemical studies have revealed that Prp16 promotes an optimal substrate after 5' splice site cleavage by allowing the release of Cwc25 and Yju2. This study also suggests that Prp16 likely antagonizes a suboptimal substrate before 5' splice site cleavage by allowing the release of Cwc25, which interacts only weakly with mutated branch sites [31].
Finally, Prp22 both rejects suboptimal substrates and promotes optimal substrates by antagonizing components that are important for anchoring exons to the spliceosome and juxtaposing splice sites, such as Prp8 and the U5 loop. Mutations thought to destabilize these interactions suppress a cold-sensitive prp22 mutant by relieving stalled optimal substrates, while also increasing Prp22-mediated rejection of suboptimal substrates [24, 32]. These two Prp22 roles could be mediated by a single mechanism: translocation from the downstream exon in the 3' to 5' direction, a polarity that is characteristic of the DEAH-box family [33–35]. Before exon ligation translocation would dissociate the 3' splice site from the catalytic core of the spliceosome; whereas after exon ligation translocation would dissociate the 5' exon and consequently mRNA [14]. Indeed, a Prp22 ATPase mutant engages the 5' end of the 3' exon, consistent with a pre-translocation state [36]; when exon ligation is impaired, Prp22 crosslinks just upstream of the 3' splice site [37], consistent with a post-translocation rejected state.
Consistent with highly related dual activities, recent biochemical studies establish that the difference in outcome depends simply on the timing of DExD/H-box ATPase activity (Figure 1). Prp16 antagonizes suboptimal substrates just before 5' splice site cleavage and promotes optimal substrates just after this reaction [15, 27, 31]. Equivalently, Prp22 antagonizes suboptimal substrates just before exon ligation and promotes optimal substrates just after exon ligation [14, 29, 30, 36]. Genetic studies similarly imply that Prp5 antagonizes suboptimal substrates just before an undefined transition in branch site recognition and that Prp5 promotes optimal substrates just after this transition [12, 13, 28]. Thus, any DExD/H-box ATPase may proofread a biochemical step by simply acting before, rather than after, the proofread step.
Models for the kinetic basis of substrate specificity
A critical unanswered question is how DExD/H-box ATPases promote substrate specificity. In two non-mutually exclusive models, DExD/H-box ATPases function as timers and/or as sensors of authenticity (Figure 2). In either model, the rate of each ATPase must be tuned to the rate of the proofread step.
Figure 2.
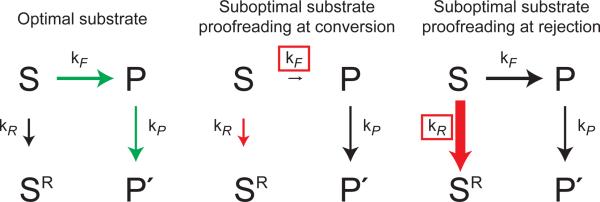
Alternative manifestations of kinetic proofreading in the spliceosome. In the case of an optimal substrate (left panel), the rate of the forward on-pathway conversion of substrate (S) to product (P) (kF) is faster than the rate of DExD/H-box ATPase mediated rejection (kR). Consequently, ATP hydrolysis by the proofreading DExD/H-box ATPase occurs after the conversion and results in transition to a productive conformation (P') at rate kP. In the case of a suboptimal substrate (middle and right panels), the rate of rejection effectively competes with the forward rate of conversion to antagonize splicing, resulting in formation of a rejected conformation (SR). Two non-mutually exclusive models can explain specificity for rejection of suboptimal substrates. In proofreading at the conversion step (middle panel), the rate of rejection may be constant for optimal and suboptimal substrates while the conversion rate (boxed) is sufficiently reduced in the case of a suboptimal substrate such that the rate of rejection is faster than the rate of conversion. In proofreading at the rejection step (right panel), the rate of conversion may be constant for optimal and suboptimal substrates while the rate of rejection (boxed) is enhanced for a suboptimal substrate relative to an optimal substrate, similarly leading to a faster rate of rejection than rate of conversion. Such relative enhancement of the rejection rate for a suboptimal substrate might be achieved by stimulation of rejection by a suboptimal substrate or by repression of rejection by an optimal substrate. The sizes of the arrows reflect the magnitude of their rates. Green arrows represent utilization of the productive pathway; red arrows represent utilization of the rejection pathway; black arrows represent unutilized pathways.
In the timer model, the DExD/H-box ATPase restricts the window of time allotted for a particular splicing event (Figure 2, middle). If a substrate proceeds through the proofread step faster than the DExD/H-box ATPase acts, then the ATPase would function after the step and promote a conformational change that is productive. Conversely, if the substrate proceeds through the proofread step slower than the DExD/H-box ATPase acts, then the ATPase would function before the step and promote a conformational change that is antagonistic; for example, Prp5 would antagonize substrates if they were slow to fully engage U2 snRNA, Prp16 would antagonize substrates if they were slow to undergo 5' splice site cleavage and Prp22 would antagonize substrates if they were slow to undergo exon ligation. This model is attractive because all three of these DExD/H-box ATPases discriminate against substrates having intronic sequences that deviate from the consensus and likely delay assembly and/or catalysis [7, 12–14]. Further, this mode of kinetic proofreading parallels modes observed in other systems, including transcription and translation [2, 3].
Recent results provide evidence for the timer model in splicing. When a U6 metal-ligand interaction that is subject to Prp16-mediated proofreading is disrupted, spliceosomes undergo 5' splice site cleavage an order of magnitude more slowly than when the metal-ligand interaction is intact, as determined in the absence of Prp16-mediated proofreading [15]. However, it remains to be determined whether the rate of Prp16-dependent rejection is tuned appropriately to discriminate between such rates. Additionally, it remains to be proven that suboptimal substrates undergo 5' splice site cleavage more slowly than an optimal substrate.
In the alternative sensor model, a DExD/H-box ATPase rejects a suboptimal substrate more quickly than an optimal substrate (Figure 2, right). Such differential rejection could be established by at least two mechanisms. In one mechanism, differential rejection could reflect differences in the stability of substrate-spliceosome interactions, as in kinetic proofreading in translation [3]. Indeed, several DExD/H-box ATPases destabilize less stable substrates faster than stable ones (Box 2). In a second mechanism, differential rejection is established by regulation of the proofreading DExD/H-box ATPase. In support of this model, several members of this family are positively or negatively regulated (Box 2). Note that DEAH-box protein Prp43 discards optimal and suboptimal substrates with equal probability (see below), so the contribution of Prp43 to fidelity likely depends on differences in forward rates [38].
Spliceosomal proofreading: unique features
In kinetic proofreading, as originally proposed by Hopfield and Ninio, NTP hydrolysis drives an irreversible step that introduces a new, high energy intermediate that must partition between two kinetically competing pathways (Figure 3a–c) [1–3,8,9]. This competition allows additional scrutiny of a substrate beyond the initial stage of enzyme binding. Splicing also employs energy for kinetic proofreading but utilizes energy in a distinct manner (Figure 3d,e). Specifically, DExD/H-box mediated ATP hydrolysis drives the rejection branch to accelerate this route relative to the competing productive pathway [16, 17, 39]. This unique framework for spliceosomal proofreading by DExD/H-box ATPases has significant implications.
Figure 3.

Distinct manifestations of kinetic proofreading across diverse biochemical pathways. (a) Obligatory kinetic proofreading scheme employed during translation and DNA replication [8, 9]. Unbound substrate (SU) can engage the enzyme. The initial enzyme-substrate complex (SB) can dissociate or undergo an irreversible transition to an activated intermediate complex (S*) through the input of energy in the form of NTP hydrolysis. From the activated complex, conversion to a new on-pathway intermediate (P) competes with a branch in the pathway that results in rejection of the substrate (SR). (b) Kinetic proofreading during aminoacyl-tRNA selection on the ribosome [3]. Aminoacid-charged tRNA (green, amino acid; orange, tRNA) binds the ribosome (purple) as part of a ternary complex with EF-Tu and GTP (red). The initial recognition complex then undergoes irreversible GTP hydrolysis by EF-Tu, which then dissociates. In the subsequent inspection, accommodation of the aminoacyl-tRNA into the catalytic core of the ribosome where peptide bond formation occurs (preferred for cognate tRNAs) competes with dissociation of the activated aminoacyl tRNA (preferred for near-cognate tRNAs). (c) Kinetic proofreading during DNA polymerization [1]. An incoming dNTP (blue oval) forms base-paring interactions with the tempate strand (solid blue line) in the active site of DNA polymerase (red oval). The incoming dNTP is incorporated into the nascent strand (dashed blue line) through irreversible phosphodiester bond formation resulting in release of pyrophosphate. Base-pairing at the nascent 3' end of DNA is then inspected a second time as additional productive rounds of dNTP incorporation (preferred for a matched dNMP) compete with cleavage of the nascent phosphodiester bond after relocalization to the polymerase editing site (preferred for a mismatched dNMP), resulting in dissociation of dNMP (blue rectangle). (d) A distinct, non-obligatory kinetic proofreading scheme employed during splicing and protein translocation [8, 9, 16, 17, 39]. Unbound substrate (SU) binds the enzyme, forming an enzyme substrate complex (SB). From the enzyme–substrate complex, conversion to a new on-pathway intermediate (P) competes with an energy-dependent branch that results in substrate rejection (SR). However, if NTP hydrolysis occurs after the productive step then hydrolysis results in a new productive conformation (P'). (e) Kinetic proofreading during exon ligation [14]. At exon ligation, the lariat intermediate 3' splice site binds the spliceosome (blue oval) in a catalytic conformation. In this catalytic conformation, ligation of the exons (yellow rectangles) (preferred for optimal splice sites) competes with ATP hydrolysis by Prp22, resulting in rejection of the 3' splice site (preferred for suboptimal splice sites). ATP hydrolysis by Prp22 after exon ligation results in productive mRNA release [29, 30]. (f) Kinetic proofreading during protein targeting to the translocation machinery [10]. During protein targeting, a nascent signal peptide-ribosome complex is recognized by GTP-bound signal recognition particle (SRP, blue oval), which forms a complex with GTP-bound SRP receptor (SR, pink oval). At the translocon (red ovals), productive loading of the nascent peptide cargo into the translocation machinery (preferred for correct cargoes) competes with GTP hydrolysis by the SRP-SR complex, which results in dissociation of the cargo-SRP-SR complex (preferred for incorrect cargoes). GTP hydrolysis by SRP and SR after engagement of the cargo with translocation apparatus results in productive recycling of SRP and SR.
First, the acceleration of rejection allows for a more rapid proofread step. Second, kinetic proofreading is not obligatory for splicing, in contrast to replication, transcription and translation, for which kinetic proofreading is obligatory due to the on-pathway requirement for NTP hydrolysis (Figure 3). For example, a GTPase mutation in EF-Tu increases the fidelity of translation by allowing more time for dissociation of ternary complexes prior to on-pathway GTP hydrolysis [40]. By contrast, ATPase mutations in PRP5, PRP16 or PRP22 decrease the fidelity of splicing by allowing more time for on-pathway events for suboptimal splicing substrates [7, 12–15]. Moreover, a rejection branch can be entirely eliminated in vitro: ATP depletion eliminates both the Prp16-dependent and Prp22-dependent proofreading branches, thereby allowing 5' splice site cleavage of suboptimal splice sites and exon ligation of suboptimal substrates, respectively [14,15].
An important implication of non-obligatory proofreading is that maximal discrimination requires that the proofreading DExD/H-box protein bind the spliceosome and ATP before the proofread step. In principle, the DExD/H-box ATPase can only bind before the proofread step for slowly-reacting suboptimal substrates, although in that context NTPase activity cannot be regulated to enhance specificity. Alternatively, the binding of the DExD/H-box ATPase can be required for the efficiency of the proofread step. Indeed, Prp22 is ATP-independently required for exon ligation, at least for introns that have a long branchpoint to 3' splice site distance [29]. Interestingly, Prp16 is similarly ATP-independently required for 5' splice site cleavage, at least for introns having a mutation at the branch site [31].
Another important implication of non-obligatory proofreading is the possibility that DExD/H-box ATPases could be regulated locally or globally to relax proofreading, allowing utilization of alternative, suboptimal splice sites. Currently, the strongest candidate for such regulation is telomerase RNA (TER1) from Schizosaccharomyces pombe. Remarkably, TER1 corresponds to a 5' exon and the spliceosome functions as a 3' end processing factor [41]. The intron immediately downstream of TER1 recruits the spliceosome, which cleaves the 5' splice site separating TER1 from the intron. Although the intron includes a consensus 3' splice site, suggesting exon ligation can occur under some conditions, in exponentially growing cells exon ligation is prevented and splicing is aborted, allowing biogenesis of TER1 as a 5' exon. This release of intermediates conspicuously mimics the discard of suboptimal substrates by Prp43, following rejection by Prp22 (discussed later). If Prp22 and Prp43 promote TER1 biogenesis, repression of either factor could lead to upregulation of exon ligation and downregulation of TER1. In the future, it will be important to test the role of proofreading activities in regulating splicing.
A third distinguishing feature of spliceosomal proofreading is that all on-pathway steps in the process are reversible. In replication and transcription, the hydrolysis of NTP that is required for phosphodiester bond formation is irreversible. Similarly, in translation the hydrolysis of GTP by EF-Tu that is required for accommodation of aminoacyl tRNA is irreversible. Thus, errors at these stages cannot be corrected through reversal and instead require nucleic acid hydrolysis or discard of aminoacyl tRNA. However, in splicing, the first studies of single spliceosomes by total internal reflection fluorescence (TIRF) microscopy have revealed that all detected transitions are reversible [42, 43] and a recent biochemical study has verified that each of the two chemical steps of splicing are reversible [44]. Thus, in the future it will be important to determine whether the spliceosome reverses the advancement of suboptimal splice sites to allow the subsequent advancement of alternative, optimal splice sites.
A final distinguishing feature of spliceosomal proofreading is that Prp16, Prp22 and possibly Prp5 reject suboptimal splicing substrates in a reversible manner [14, 15], in contrast to the irreversible hydrolysis of incorrect nucleotides in nucleic acid polymerization or the irreversible dissociation of near-cognate aminoacyl tRNA in translation. In splicing, rejection alone is not sufficient to dissociate a substrate from the spliceosome, so a splice site can re-engage the catalytic core and undergo splicing. This reversibility of rejection suggests that proofreading DExD/H-box ATPases facilitate both the sampling of competing alternative splice sites and the selection of an optimal splice site, and thereby function as RNA chaperones.
Discard of rejected substrates
Although reversibility may allow the selection of an optimal splice site after rejection of a suboptimal site, if an optimal site is not available then the cell degrades the substrate by one of several pathways [45]. First, substrates can undergo turnover by nuclear nucleases that compete with splicing [46–48]. Second, substrates can dissociate from the spliceosome, undergo export to the cytoplasm, and then suffer turnover by cytoplasmic nucleases [49]. Alternatively, after export to the cytoplasm, rejected substrates can engage the ribosome. For instance, rejected lariat intermediates that have been engineered to have an internal ribosome entry site (IRES) can undergo translation [38]. Furthermore, suboptimal pre-mRNAs can undergo translation-dependent nonsense-mediate decay (NMD) [49–53]. Short introns are under strong selective pressure, across eukaryotes, to maintain in-frame pre-mature termination codons, which highlights the significance of NMD in limiting the accumulation of unspliced pre-mRNAs, [54]. While some NMD-targeted pre-mRNAs may fail to ever engage the spliceosome, recent results indicate that suboptimal pre-mRNAs can bind the spliceosome but then suffer rejection [15].
Genetic and biochemical studies have established that suboptimal substrates are dissociated or discarded from the spliceosome by the DEAH-box ATPase Prp43, which is conserved from budding yeast to humans. For optimal substrates, Prp43 promotes release of the excised lariat intron following release of the ligated mRNA [55–58]. Mutations in Prp43 or its cofactor Spp382/Ntr1 suppress mutations in splicing factors that compromise spliceosome assembly, suggesting an additional role in quality control and disassembly of stalled spliceosomes [59]. Furthermore, in vitro studies have shown that Prp43 discards pre-mRNA rejected by Prp16, due to disruption of a critical metal-ligand interaction [15], and it discards splicing intermediates that are stalled due to suboptimal consensus sites [38]. Prp43-dependent discard of intermediates, and possibly pre-mRNA, contributes to fidelity, because maximal repression of a cryptic splice 3' splice site requires Prp43 [38]. However, Prp43 also promotes discard of wild-type lariat intermediates [38, 49], indicating that Prp43-dependent proofreading acts nonspecifically; nevertheless, Prp43 especially limits accumulation of spliceosomes that are stalled due to a suboptimal substrate (Box 4). These data support a model in which Prp16 and Prp22 antagonize forward progression of suboptimal substrates, while Prp43 acts in conjunction to promote dissociation of these stalled substrates.
Because the role of Prp43 in the discard of suboptimal substrates parallels the role of Prp43 in the release of the excised intron, Prp43 functions as a general splicing terminator. Translation activities that function in termination similarly perform parallel roles in quality control (Box 4). Additionally, factors that promote disassembly of the translocon docking apparatus similarly function in a second capacity to promote specificity (discussed later). Thus, common activities function to disassemble both canonical termination complexes and suboptimal complexes broadly across diverse processes.
Implications for proofreading by other NTPases within and beyond the DExD/H-box family
Although kinetic proofreading through direct coupling of ATP hydrolysis to substrate rejection departs significantly from canonical kinetic proofreading, this particular manifestation of kinetic proofreading has been implicated for four different spliceosomal DExD/H-box ATPases, thereby establishing the generality of this mechanism. Thus, a wide range of DExD/H-box ATPases may promote fidelity in disparate RNA-dependent processes. Where a DExD/H-box ATPase functions productively at a particular step in a process, such a DExD/H-box ATPase could also compete kinetically with the preceding step to proofread this step (Figure 3d).
Importantly, this manifestation of kinetic proofreading need not to be restricted to the DExD/H-box class of NTPases. Indeed, proofreading of splicing by DExD/H-box ATPases is functionally indistinguishable from a recently discovered mechanism for proofreading of protein translocation by GTPases (Figure 3f) [10]. In translocation, the signal recognition particle (SRP), which includes a universally conserved GTPase, recognizes the signal sequence of a nascent protein and co-translationally targets the protein for secretion by binding in a GTP-dependent manner to the SRP receptor (SR), which also includes a GTPase. Binding is followed by transfer of the nascent peptide to the translocation machinery, the translocon. Subsequently, GTP hydrolysis triggers disassembly of the SRP-SR complex, much as, subsequent to release of ligated mRNA, ATP hydrolysis triggers Prp43-dependent disassembly of the spliceosome.
In addition to binding optimal nascent peptides, SRP binds suboptimal peptides that do not ultimately translocate, indicating downstream fidelity mechanisms [10]. In one mechanism, the transfer of cargo to the translocon is challenged by a competitive GTP-dependent branch that disassembles the SRP-SR complex. In the case of suboptimal cargo, the SRP-SR complex hydrolyzes GTP and disassembles before releasing cargo into the translocon. Interestingly, the rate of GTP hydrolysis with suboptimal cargo is similar to the rate without cargo, implying that early disassembly occurs by default. Indeed, in the case of optimal cargo, GTP hydrolysis is repressed 6- to 8-fold, thereby delaying disassembly until after transfer of cargo to the translocon. Thus, while the regulation of proofreading DExD/H-box ATPases remains to be investigated, the GTPase activity of the SRP-SR complex functions analogously to the ATPase activity of spliceosomal DExD/H-box ATPase fidelity factors in selectively rejecting suboptimal substrates within a kinetic proofreading framework. This similarity suggests that other GTPases, as well as other ATPases, may proofread in addition to promoting a process, especially when the NTPase normally rearranges or disassembles a complex.
Concluding remarks
Although a general framework for kinetic proofreading was proposed nearly 40 years ago [8, 9], in the past several years the specific manifestation of kinetic proofreading that requires an NTP-dependent branch has emerged as a recurrent and important fidelity mechanism. In splicing, this mechanism utilizes DExD/H-box ATPases to both reject and discard suboptimal substrates [7, 12–15, 22, 23, 38]. In translocation, this mechanism utilizes GTPases to reject illegitimate substrates [10]. In the future, it will be important to consider broadly the role of such NTPases in establishing competitive branches that promote fidelity. Recent advances have also raised a number of intriguing and important, broad and mechanistic questions (Outstanding Questions Box). For example, is the newly-discovered link between splicing and transcriptional pausing [60, 61] mediated by fidelity factors? Answers to these questions will require ingenuity as well as an array of cutting edge strategies, ranging from single-molecule to genome-wide approaches. Without a doubt, exciting times lie ahead, as the field addresses these questions and determines how the spliceosome as well as other processes stay on message.
Box 1. A working definition of splicing fidelity.
The notion of fidelity, which implies the possibility of errors, can be a somewhat muddled concept because errors in one context can be advantageous in another. For example, in DNA replication slipped-strand mispairing, which can reversibly alter promoters or coding regions, underlies regulation of a number of phase-like switches between bacterial phenotypic states [62]. Also, in translation mis-acylation of non-Met tRNAs with Met, which increases ten-fold upon oxidative stress, is thought to promote scavenging of reactive oxygen species [63]. The concept of fidelity in splicing can be especially muddled given splice site degeneracy and alternative splice site utilization, which alters the pathway for the two transesterifcation reactions (Figure I). For the purpose of this review we define splicing fidelity as the mechanisms by which the spliceosome catalyzes intron excision to favor optimal splice sites while discriminating against suboptimal splice sites. We define optimal splice sites as those empirically demonstrated to splice efficiently, and suboptimal splice sites as those observed to splice inefficiently. Frequently, optimal splice sites conform to consensus sequences for the 5' splice site, branch site and 3' splice site, as defined by conservation across introns within a species (Figure I). However, deviation from the splice site consensus sequence does not necessarily render a splice site suboptimal; in some cases, non-consensus splice sites can splice more efficiently than a corresponding consensus splice site [64]. Suboptimal splice sites generally fall into two categories: those in which an optimal splice site is mutated so it is no longer efficiently recognized, and the cryptic splice sites that may resemble optimal splice sites in sequence but only efficiently compete for recognition upon inactivation of an optimal splice site. Importantly, the definition of fidelity used in this review may run counter to favorable gene expression. At least 15% and potentially as much as 50% of disease phenotypes derive from point mutations that convert optimal to suboptimal splice sites or suboptimal to optimal splice sites, resulting in exon skipping or cryptic splicing events, respectively [65, 66]. In such cases, fidelity mechanisms may contribute to the discrimination against suboptimal but favorable splice sites or the selection of optimal but undesirable splice sites.
Box 2. DExD/H-box ATPases remodel RNP complexes by distinct mechanisms.
The eight ATPases conserved in splicing include members of the DEAD-box, DEAH-box, and Ski2-like ATPase families of the SF2 superfamily of helicases [67]. These families are characterized by motifs that confer NTP binding and hydrolysis, RNA binding, and RNA rearrangements [33]. Despite sharing similar architectures, the DEAD-box family remodels RNP complexes by a mechanism that is distinct from the DEAH-box and Ski2-like families. DEAD-box ATPases rearrange RNA duplexes through local strand separation in which the DEAD-box protein loads, upon ATP binding, onto one strand of a duplex, thereby inducing a local opening and allowing the remainder of the duplex to unwind passively [68]. In contrast, DEAH-box and Ski2-like proteins act as translocases capable of disrupting extensively base-paired helices [69]. Recent crystal structures of the spliceosomal DEAH-box ATPase Prp43 and Ski2-like ATPase Brr2 have revealed structural similarities to the DNA helicase Hel308 [35, 70–72]. The Prp43 and Brr2 Hel308-like folds form i) a channel predicted to clamp onto single-stranded RNA and to confer processivity and ii) a helix predicted to contact RNA and through ratcheting, to promote unidirectional movement. Regardless of the specific mechanism, spliceosomal DExD/H-box ATPases utilize cycles of ATP binding and hydrolysis to disrupt RNA structures, to both promote splicing of optimal substrates and to antagonize splicing of suboptimal substrates. DExD/H-box ATPases can be modulated by a variety of factors including positive and negative regulators and the stability of a substrate itself. For example, the nuclear mRNA export factor and DEAD-box ATPase Dbp5 is activated by InsP6-Gle1, which may confer directionality to mRNA export, given the localization of Gle1 to the cytoplasmic fibrils of the nuclear pore complex [73, 74]. Conversely, the ATPase activity of eIF4AIII is repressed by MAGOH-Y14 upon deposition upstream of an exon-exon junction, enabling it to stably clamp an exon-exon junction complex onto spliced mRNA [75]. Furthermore, the DEAD-box ATPase CYT-19, which chaperones folding of group I intron RNA, more rapidly destabilizes non-native conformations than the more stable, native conformation [76]. Thus, through modulation of their activity, spliceosomal DExD/H-box ATPases might enhance specificity by either preferentially stabilizing interactions with optimal substrates and/or preferentially destabilizing interactions with suboptimal substrates. However, it remains to be determined to what extent these different modes of regulation are utilized by spliceosomal DExD/H-box ATPases.
Box 3. Equilibration between distinct spliceosomal conformations promotes fidelity.
Many different spliceosomal mutations suppress defects in splicing suboptimal substrates, thereby compromising fidelity [7, 12–14, 22–24, 38, 59]. Curiously, many suppressors of exon ligation defects compromise 5' splice site cleavage and vice versa, inspiring a model in which the two catalytic spliceosomal conformations compete to promote fidelity [22, 24] (Figure Ia). In this model, splicing intermediates that are defective for exon ligation destabilize the exon ligation conformation and consequently repopulate the 5' splice site cleavage conformation (Figure Ib). Similarly, pre-mRNAs that are defective for 5' splice site cleavage destabilize the 5' splice site cleavage conformation and instead populate an exon ligation-like conformation. In both cases, populating the competing conformation sequesters suboptimal substrates and favors discard rather than splicing of the substrate. In the model, exon ligation defects are suppressed by mutations that effectively destabilize the 5' splice site cleavage conformation, allowing efficient population of the exon ligation conformation (Figure Ic) and vice versa [22], [24].
A distinct class of suppressors of exon ligation defects implicates a third conformation important for fidelity [25, 26]. Such suppressors do not exacerbate 5' splice site cleavage defects and indeed act by weakening snRNA structures absent at the 5' splice site cleavage stage. Thus, these suppressors destabilize a unique conformation that is intermediate between the two catalytic conformations (Figure Id). Like the catalytic conformations, this conformation acts as a sink for suboptimal substrates and may represent an open state of the spliceosome that normally permits substrate rearrangement.
Equilibration of spliceosomal conformations suggests that the spliceosome may repeatedly sample and reject even an optimal 3' splice site before ultimately splicing. Recent structural studies of RNA Polymerase II suggest an analogous model for promoter proofreading during transcription initiation [11]. The initial transcription of a short RNA results in a product that binds the DNA template weakly and frequently dissociates (i.e., suffers rejection). In this model, at a spurious promoter this abortive initiation provides an additional opportunity for polymerase dissociation (i.e., discard) subsequent to initial binding, whereas at a genuine promoter, which is replete with transcription factors and sequence-specific contacts, polymerase remains bound and re-initiation is favored. Thus, equilibration of promoter binding through abortive transcription would ensure polymerase transits to an elongating complex only at genuine promoters. Although the fidelity of splicing appears enhanced by equilibration of a suboptimal substrate to alternative conformations, it remains unknown whether splicing of an optimal substrate is also subject to equilibration.
Box 4. Spliceosomal discard: parallels with translation termination.
Prp43 functions to both discard suboptimal substrates and release the excised intron product [15, 38, 56], suggesting that Prp43 functions in a general splicing termination pathway. Interestingly, the ribosome also appears to use a general termination pathway in both quality control and termination at a stop codon. The translation quality control mechanism no-go decay (NGD) results in dissociation of ribosomal subunits and peptidyl tRNA after ribosomal stalling due to stable mRNA secondary structure, rare codons or ribosome decoding mutations [77, 78]. This pathway requires Dom34-Hbs1, a complex that dissociates stalled ribosomes through a mechanism that mimics peptide release by the eRF1-eRF3 complex. eRF1 specifically recognizes stop codons in the ribosome A site and, stimulated by the GTPase activity of eRF3, promotes hydrolysis of peptidyl-tRNA [79]; eIF3, aided by eIF1, eIF1A, and eIF3j, then stimulates subunit dissociation and release of P-site tRNA and mRNA [80]. The structurally similar Dom34-Hbs1 complex [81, 82] utilizes Dom34 to recognize an empty ribosomal A site in a codon-independent manner and to promote peptide release, as well as subunit dissociation, upon stimulation by the GTPase activity of Hbs1 but without hydrolysis of peptidyl-tRNA [83]. It has been proposed that Dom34-Hbs1 is restricted to stalled ribosomal complexes due to a kinetic competition in which additional elongation cycles, including ternary complex binding (Box 3, Figure Ib), are kinetically favored for functional ribosomes and NGD only becomes kinetically favored when translation is stalled and Dom34-Hbs1 can effectively compete with a ternary complex [84]. Interestingly, Cheng and colleagues have recently shown that productive splicing factors compete with Ntr2, a Prp43 recruitment factor [85], for binding to the spliceosome at different stages in the splicing pathway and further that binding of the productive splicing factors is favored unless splicing is stalled (H.C. Chen et al., unpublished). Thus, the spliceosome and the ribosome rely on common mechanisms to disassemble both canonical termination complexes and stalled complexes, and these mechanisms appear to be regulated by kinetic competition between productive factors and disassembly factors. Prp43 also apparently functions to displace small nucleolar RNAs (snoRNAs) from pre-60S ribosomal subunits during ribosomal RNA (rRNA) biogenesis, suggesting that Prp43 may act as a termination factor for small nucleolar ribonucleoprotein (snoRNP) complexes during ribosome biogenesis as well [86, 87]. In the future, it will be interesting to determine whether Prp43 acts in a quality control capacity to promote dissociation of non-functional or inappropriately assembled snoRNP during rRNA maturation.
Outstanding Questions.
The prevalence of spliceosomal DExD/H-box ATPase-dependent proofreading that has been recently uncovered has introduced a suite of new questions that must be addressed to comprehend how proofreading influences splicing. Chief among these questions is: what is the kinetic basis for the observed specificity of DExD/H-box ATPase-dependent proofreading? As discussed, DExD/H-box ATPases may function as timers tuned to reject suboptimal substrates that progress slowly or as sensors of substrate identity that reject suboptimal substrates at a faster rate. An explicit analysis of the rates of rejection and the forward, proofread step will be required to determine the contributions of the timer and sensor models to the specificity of DExD/H-box ATPase-dependent proofreading.
Another critical question is: what are the molecular mechanisms of DExD/H-box-mediated rejection? In no case has the direct target for rejection been defined. Although Prp22 has been shown to promote an optimal substrate after the proofread step by interacting directly with the substrate to effect mRNA release [36], it is unclear whether the pathway of Prp22-dependent rejection involves a similar mechanism. Direct molecular targets must be identified for Prp22 as well as Prp5, Prp16, and Prp43, to understand how these DExD/H-box ATPases promote splicing fidelity.
We also need to determine whether rejection of suboptimal substrates by DExD/H-box ATPases could represent a specific manifestation of a broader role for these factors in promoting a search for an optimal splice site. For example, because the spliceosome can catalyze exon ligation at multiple competing 3' splice sites, the spliceosome might readily bind a wide range of candidate sites. Prp22 could, through rejection of suboptimal sites, promote sampling of alternative sites and ultimately identification of an optimal 3' splice site. Furthermore, we must determine whether DExD/H-box ATPase-dependent splice site sampling is regulated to influence alternative splicing events as well.
In the cell, splicing is tightly coupled to transcription, raising questions concerning the impact of co-transcriptional splicing on spliceosomal fidelity and vice versa. The presence of a functional intron can induce RNA polymerase II to pause during transcription [60, 61]. This pausing is eliminated by splice site mutations that are subjected to DExD/H-box ATPase-dependent proofreading [60]. Therefore, it will be important to determine whether RNA polymerase II pausing is sensitive to the activity of spliceosomal fidelity factors.
Finally, it will be important to determine the breadth of the NTPase-dependent rejection mode of kinetic proofreading across disparate cellular processes.
Figure I (Box 3).
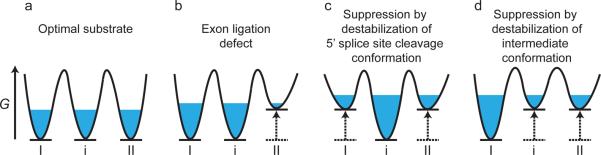
A three-state equilibrium model rationalizes genetic suppressors of substrate mutations. (a) In the model, a population of spliceosomes (blue) assembled on an optimal substrate equilibrates between 5' splice site cleavage (I) and exon ligation (II) conformations as well as an intermediate conformation (i). Note: free energies of each state are arbitrarily set to the same level. (b) The model implies that a mutation that impairs exon ligation destabilizes the exon ligation conformation relative to the 5' splice site cleavage and intermediate conformations, resulting in sequestration of the spliceosomes in these alternative conformations. (c and d) In the model, the exon ligation defect shown in (b) is suppressed by second-site mutations that repopulate the exon ligation conformation by destabilizing either the 5' splice site cleavage conformation (c) or the intermediate conformation (d), relative to the exon ligation conformation. Note that in all panels, destabilization of a particular state is illustrated as underlying changes in equilibria, but stabilization of alternative states would result in similar changes in equilibria. G, Gibbs free energy; in each panel the reaction coordinate proceeds from left to right.
ACKNOWLEDGEMENTS
Due to space constraints, we were unfortunately unable to highlight all relevant stories. We apologize to those whose work was not discussed or cited. We thank P. Koodathingal, D. Qin, A. Wlodaver, Y. Zeng, K. Nielsen and C. Query for comments on the manuscript. Research in our laboratory into splicing fidelity has been funded by the NIH (GM062264).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Loeb LA, Kunkel TA. Fidelity of DNA synthesis. Annu. Rev. Biochem. 1982;51:429–457. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]
- 2.Libby RT, Gallant JA. The role of RNA polymerase in transcriptional fidelity. Mol. Microbiol. 1991;5:999–1004. doi: 10.1111/j.1365-2958.1991.tb01872.x. [DOI] [PubMed] [Google Scholar]
- 3.Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pickrell JK, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox-Walsh KL, Hertel KJ. Splice-site pairing is an intrinsically high fidelity process. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1766–1771. doi: 10.1073/pnas.0813128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitrovich QM, et al. Evolution of yeast noncoding RNAs reveals an alternative mechanism for widespread intron loss. Science. 2010;330:838–841. doi: 10.1126/science.1194554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgess SM, Guthrie C. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell. 1993;73:1377–1391. doi: 10.1016/0092-8674(93)90363-u. [DOI] [PubMed] [Google Scholar]
- 8.Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. U. S. A. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ninio J. Kinetic amplification of enzyme discrimination. Biochimie. 1975;57:587–595. doi: 10.1016/s0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, et al. Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science. 2010;328:757–760. doi: 10.1126/science.1186743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, et al. Initiation complex structure and promoter proofreading. Science. 2011;333:633–637. doi: 10.1126/science.1206629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu YZ, Query CC. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol. Cell. 2007;28:838–849. doi: 10.1016/j.molcel.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perriman R, Ares M., Jr. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol. Cell. 2010;38:416–427. doi: 10.1016/j.molcel.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayas RM, et al. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat. Struct. Mol. Biol. 2006;13:482–490. doi: 10.1038/nsmb1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koodathingal P, et al. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5' splice site cleavage during pre-mRNA splicing. Mol. Cell. 2010;39:385–395. doi: 10.1016/j.molcel.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarus M. Proofreading, NTPases and translation: constraints on accurate biochemistry. Trends Biochem. Sci. 1992;17:130–133. doi: 10.1016/0968-0004(92)90320-9. [DOI] [PubMed] [Google Scholar]
- 17.Yarus M. Proofreading, NTPases and translation: successful increase in specificity. Trends Biochem. Sci. 1992;17:171–174. doi: 10.1016/0968-0004(92)90257-a. [DOI] [PubMed] [Google Scholar]
- 18.Yu Y, et al. Dynamic regulation of alternative splicing by silencers that modulate 5' splice site competition. Cell. 2008;135:1224–1236. doi: 10.1016/j.cell.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgess S, et al. A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell. 1990;60:705–717. doi: 10.1016/0092-8674(90)90086-t. [DOI] [PubMed] [Google Scholar]
- 20.Soares LM, et al. Intron removal requires proofreading of U2AF/3' splice site recognition by DEK. Science. 2006;312:1961–1965. doi: 10.1126/science.1128659. [DOI] [PubMed] [Google Scholar]
- 21.Tavanez JP, et al. hnRNP A1 Proofreads 3' Splice Site Recognition by U2AF. Mol Cell. 2012;45:314–329. doi: 10.1016/j.molcel.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 22.Query CC, Konarska MM. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol. Cell. 2004;14:343–354. doi: 10.1016/s1097-2765(04)00217-5. [DOI] [PubMed] [Google Scholar]
- 23.Villa T, Guthrie C. The Isy1p component of the NineTeen Complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev. 2005;19:1894–1904. doi: 10.1101/gad.1336305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, et al. Opposing classes of prp8 alleles modulate the transition between the catalytic steps of pre-mRNA splicing. Nat. Struct. Mol. Biol. 2007;14:519–526. doi: 10.1038/nsmb1240. [DOI] [PubMed] [Google Scholar]
- 25.Hilliker AK, et al. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev. 2007;21:821–834. doi: 10.1101/gad.1536107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mefford MA, Staley JP. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA. 2009;15:1386–1397. doi: 10.1261/rna.1582609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwer B, Guthrie C. A conformational rearrangement in the spliceosome is dependent on Prp16 and ATP hydrolysis. EMBO J. 1992;11:5033–5039. doi: 10.1002/j.1460-2075.1992.tb05610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruby SW, et al. Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev. 1993;7:1909–1925. doi: 10.1101/gad.7.10.1909. [DOI] [PubMed] [Google Scholar]
- 29.Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre- mRNA splicing. EMBO J. 1998;17:2086–2094. doi: 10.1093/emboj/17.7.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner JD, et al. The DEAH-box protein Prp22 is an ATPase that mediates ATP-dependent mRNA release from the spliceosome and unwinds RNA duplexes. EMBO J. 1998;17:2926–2937. doi: 10.1093/emboj/17.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng CK, et al. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA. 2011;17:145–154. doi: 10.1261/rna.2459611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aronova A, et al. Functional interactions between Prp8, Prp18, Slu7, and U5 snRNA during the second step of pre-mRNA splicing. RNA. 2007;13:1437–1444. doi: 10.1261/rna.572807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem. Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider S, Schwer B. Functional domains of the yeast splicing factor Prp22p. J. Biol. Chem. 2001;276:21184–21191. doi: 10.1074/jbc.M101964200. [DOI] [PubMed] [Google Scholar]
- 35.He Y, et al. Structural basis for the function of DEAH helicases. EMBO Rep. 2010;11:180–186. doi: 10.1038/embor.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwer B. A conformational rearrangement in the spliceosome sets the stage for Prp22-dependent mRNA release. Mol. Cell. 2008;30:743–754. doi: 10.1016/j.molcel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McPheeters DS, et al. Interaction of the yeast DExH-box RNA helicase Prp22p with the 3' splice site during the second step of nuclear pre-mRNA splicing. Nucleic Acids Res. 2000;28:1313–1321. doi: 10.1093/nar/28.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayas RM, et al. Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10020–10025. doi: 10.1073/pnas.0906022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgess SM, Guthrie C. Beat the clock: paradigms for NTPases in the maintenance of biological fidelity. Trends Biochem. Sci. 1993;18:381–384. doi: 10.1016/0968-0004(93)90094-4. [DOI] [PubMed] [Google Scholar]
- 40.Zeidler W, et al. Site-directed mutagenesis of Thermus thermophilus elongation factor Tu. Replacement of His85, Asp81 and Arg300. Eur. J. Biochem. 1995;229:596–604. doi: 10.1111/j.1432-1033.1995.tb20503.x. [DOI] [PubMed] [Google Scholar]
- 41.Box JA, et al. Spliceosomal cleavage generates the 3' end of telomerase RNA. Nature. 2008;456:910–914. doi: 10.1038/nature07584. [DOI] [PubMed] [Google Scholar]
- 42.Abelson J, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat. Struct. Mol. Biol. 2010;17:504–512. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoskins AA, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–1295. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tseng CK, Cheng SC. Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science. 2008;320:1782–1784. doi: 10.1126/science.1158993. [DOI] [PubMed] [Google Scholar]
- 45.Egecioglu DE, Chanfreau G. Proofreading and spellchecking: a two-tier strategy for pre-mRNA splicing quality control. RNA. 2011;17:383–389. doi: 10.1261/rna.2454711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bousquet-Antonelli C, et al. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000;102:765–775. doi: 10.1016/s0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 47.Lemieux C, et al. A pre-mRNA degradation pathway that selectively targets intron-containing genes requires the nuclear poly(A)-binding protein. Mol. Cell. 2011;44:108–119. doi: 10.1016/j.molcel.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 48.Egecioglu DE, et al. Quality control of MATa1 splicing and exon skipping by nuclear RNA degradation. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr864. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilleren PJ, Parker R. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol. Cell. 2003;12:1453–1465. doi: 10.1016/s1097-2765(03)00488-x. [DOI] [PubMed] [Google Scholar]
- 50.Chang YF, et al. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 51.Sayani S, et al. Widespread impact of nonsense-mediated mRNA decay on the yeast intronome. Mol. Cell. 2008;31:360–370. doi: 10.1016/j.molcel.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Legrain P, Rosbash M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell. 1989;57:573–583. doi: 10.1016/0092-8674(89)90127-x. [DOI] [PubMed] [Google Scholar]
- 53.He F, et al. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1- mutant. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7034–7038. doi: 10.1073/pnas.90.15.7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaillon O, et al. Translational control of intron splicing in eukaryotes. Nature. 2008;451:359–362. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- 55.Arenas JE, Abelson JN. Prp43: An RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11798–11802. doi: 10.1073/pnas.94.22.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin A, et al. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J. Biol. Chem. 2002;277:17743–17750. doi: 10.1074/jbc.M200762200. [DOI] [PubMed] [Google Scholar]
- 57.Tsai RT, et al. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev. 2005;19:2991–3003. doi: 10.1101/gad.1377405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Small EC, et al. The EF-G-like GTPase Snu114p regulates spliceosome dynamics mediated by Brr2p, a DExD/H box ATPase. Mol. Cell. 2006;23:389–399. doi: 10.1016/j.molcel.2006.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pandit S, et al. Inhibition of a spliceosome turnover pathway suppresses splicing defects. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13700–13705. doi: 10.1073/pnas.0603188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexander RD, et al. Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell. 2010;40:582–593. doi: 10.1016/j.molcel.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carrillo Oesterreich F, et al. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell. 2010;40:571–581. doi: 10.1016/j.molcel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 62.Henderson IR, et al. Molecular switches — the ON and OFF of bacterial phase variation. Mol. Microbiol. 1999;33:919–932. doi: 10.1046/j.1365-2958.1999.01555.x. [DOI] [PubMed] [Google Scholar]
- 63.Netzer N, et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009;462:522–526. doi: 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konarska MM, et al. Repositioning of the reaction intermediate within the catalytic center of the spliceosome. Mol. Cell. 2006;21:543–553. doi: 10.1016/j.molcel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 65.Krawczak M, et al. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 66.Lopez-Bigas N, et al. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–1903. doi: 10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 67.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 68.Yang Q, et al. DEAD-box proteins unwind duplexes by local strand separation. Mol. Cell. 2007;28:253–263. doi: 10.1016/j.molcel.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 69.Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008;37:317–336. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, et al. Structural evidence for consecutive Hel308-like modules in the spliceosomal ATPase Brr2. Nat. Struct. Mol. Biol. 2009;16:731–739. doi: 10.1038/nsmb.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pena V, et al. Common design principles in the spliceosomal RNA helicase Brr2 and in the Hel308 DNA helicase. Mol. Cell. 2009;35:454–466. doi: 10.1016/j.molcel.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 72.Walbott H, et al. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J. 2010;29:2194–2204. doi: 10.1038/emboj.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alcazar-Roman AR, et al. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat. Cell Biol. 2006;8:711–716. doi: 10.1038/ncb1427. [DOI] [PubMed] [Google Scholar]
- 74.Weirich CS, et al. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat. Cell Biol. 2006;8:668–676. doi: 10.1038/ncb1424. [DOI] [PubMed] [Google Scholar]
- 75.Ballut L, et al. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat. Struct. Mol. Biol. 2005;12:861–869. doi: 10.1038/nsmb990. [DOI] [PubMed] [Google Scholar]
- 76.Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD-box chaperone. Nature. 2007;449:1014–1018. doi: 10.1038/nature06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cole SE, et al. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol. Cell. 2009;34:440–450. doi: 10.1016/j.molcel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alkalaeva EZ, et al. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 80.Pisarev AV, et al. Recycling of eukaryotic posttermination ribosomal complexes. Cell. 2007;131:286–299. doi: 10.1016/j.cell.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song H, et al. The crystal structure of human eukaryotic release factor eRF1--mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 82.Chen L, et al. Structure of the Dom34-Hbs1 complex and implications for no go decay. Nat. Struct. Mol. Biol. 2010;17:1233–1240. doi: 10.1038/nsmb.1922. [DOI] [PubMed] [Google Scholar]
- 83.Shoemaker CJ, et al. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Passos DO, et al. Analysis of Dom34 and its function in no-go decay. Mol. Biol. Cell. 2009;20:3025–3032. doi: 10.1091/mbc.E09-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai RT, et al. Dynamic interactions of Ntr1-Ntr2 with Prp43 and with U5 govern the recruitment of Prp43 to mediate spliceosome disassembly. Mol. Cell Biol. 2007;27:8027–8037. doi: 10.1128/MCB.01213-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leeds NB, et al. The splicing factor Prp43p, a DEAH box ATPase, functions in ribosome biogenesis. Mol. Cell. Biol. 2006;26:513–522. doi: 10.1128/MCB.26.2.513-522.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bohnsack MT, et al. Prp43 bound at different sites on the pre-rRNA performs distinct functions in ribosome synthesis. Mol. Cell. 2009;36:583–592. doi: 10.1016/j.molcel.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wahl MC, et al. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]