

ABSTRACT
Acinetobacter baumannii is a Gram-negative, opportunistic pathogen. Recently, multiple A. baumannii genomes have been sequenced; these data have led to the identification of many genes predicted to encode proteins required for the biogenesis of type IV pili (TFP). However, there is no experimental evidence demonstrating that A. baumannii strains actually produce functional TFP. Here, we demonstrated that A. baumannii strain M2 is naturally transformable and capable of twitching motility, two classical TFP-associated phenotypes. Strains were constructed with mutations in pilA, pilD, and pilT, genes whose products have been well characterized in other systems. These mutants were no longer naturally transformable and did not exhibit twitching motility. These TFP-associated phenotypes were restored when these mutations were complemented. More PilA was detected on the surface of the pilT mutant than the parental strain, and TFP were visualized on the pilT mutant by transmission electron microscopy. Thus, A. baumannii produces functional TFP and utilizes TFP for both natural transformation and twitching motility. Several investigators have hypothesized that TFP might be responsible, in part, for the flagellum-independent surface-associated motility exhibited by many A. baumannii clinical isolates. We demonstrated that surface-associated motility was not dependent on the products of the pilA, pilD, and pilT genes and, by correlation, TFP. The identification of functional TFP in A. baumannii lays the foundation for future work determining the role of TFP in models of virulence that partially recapitulate human disease.
IMPORTANCE Several investigators have documented the presence of genes predicted to encode proteins required for the biogenesis of TFP in many A. baumannii genomes. Furthermore, some have speculated that TFP may play a role in the unique surface-associated motility phenotype exhibited by many A. baumannii clinical isolates, yet there has been no experimental evidence to prove this. Unfortunately, progress in understanding the biology and virulence of A. baumannii has been slowed by the difficulty of constructing and complementing mutations in this species. Strain M2, a recently characterized clinical isolate, is amenable to genetic manipulation. We have established a reproducible system for the generation of marked and/or unmarked mutations using a modified recombineering strategy as well as a genetic complementation system utilizing a modified mini-Tn7 element in strain M2. Using this strategy, we demonstrated that strain M2 produces TFP and that TFP are not required for surface-associated motility exhibited by strain M2.
IMPORTANCE
Several investigators have documented the presence of genes predicted to encode proteins required for the biogenesis of TFP in many A. baumannii genomes. Furthermore, some have speculated that TFP may play a role in the unique surface-associated motility phenotype exhibited by many A. baumannii clinical isolates, yet there has been no experimental evidence to prove this. Unfortunately, progress in understanding the biology and virulence of A. baumannii has been slowed by the difficulty of constructing and complementing mutations in this species. Strain M2, a recently characterized clinical isolate, is amenable to genetic manipulation. We have established a reproducible system for the generation of marked and/or unmarked mutations using a modified recombineering strategy as well as a genetic complementation system utilizing a modified mini-Tn7 element in strain M2. Using this strategy, we demonstrated that strain M2 produces TFP and that TFP are not required for surface-associated motility exhibited by strain M2.
Introduction
Acinetobacter baumannii is an aerobic Gram-negative, nonflagellated opportunistic pathogen; of late, some strains have developed resistance to most antimicrobial therapies (1). Hospital-acquired pneumonia is the most common clinical manifestation of A. baumannii infections; moreover, these infections frequently occur in mechanically ventilated patients, suggesting that environmental exposure to A. baumannii followed by subsequent accidental inoculation associated with the endotracheal tube may lead to infection (2). Interestingly, we now know that A. baumannii is not ubiquitous in nature but actually is isolated primarily within hospital settings on medical equipment, hospital workers, and patients and should not be considered just an environmental contaminant (3). Key to A. baumannii’s persistence in hospital environments is its ability to resist desiccation, as it survives on surfaces, including bed rails, bedside tables, surfaces of ventilators, and even mattresses (4, 5). From 1993 to 2004, multidrug-resistant (MDR) Acinetobacter infections increased 23% in intensive care units, more than double the rate of any other Gram-negative bacillus (6). A. baumannii has gained attention as an “ESKAPE” pathogen, one of a cohort of microorganisms that cause the majority of MDR nosocomial infections within the United States, aptly named for their ability to escape the effects of modern antimicrobial therapies (7). Furthermore, the competency of A. baumannii to acquire antibiotic resistance genes has now resulted in strains that are characterized as extensively drug resistant (XDR) and pan-drug resistant (PDR), prompting the suggestion that we may be nearing the end of the antibiotic era for this important Gram-negative pathogen (8, 9). In addition, the prevalence of MDR A. baumannii, an increase of infection incidence, and its recalcitrance to desiccation signifies the clinical importance of this opportunistic pathogen.
Although A. baumannii has earned global recognition for its ability to infect the immunocompromised patient population, little is actually known about how A. baumannii causes disease. Recent characterization of A. baumannii indicates that a few molecular factors are required for virulence in models that partially recapitulate human disease, including but not limited to outer membrane protein A (OmpA), phospholipase D, biofilm-associated protein (Bap), an O-glycosylation system, the Acinetobacter trimeric autotransporter (Ata), the Csu chaperone-usher type pilus, the acinetobactin-mediated iron acquisition system, and a secreted serine protease (10–18). Despite these significant findings, there is still a clear need to better define the bacterial determinants that facilitate colonization and subsequent disease.
Type IV pili (TFP) are multiprotein bacterial surface appendages assembled by many Gram-negative bacteria (19, 20). Due to their dynamic nature, TFP are able to be rapidly assembled and disassembled, participating in processes such as natural transformation, twitching motility, and adherence to abiotic and biotic surfaces. Natural transformation, or the ability of an organism to acquire exogenous DNA in a horizontal fashion, is a multistep process involving the uptake of DNA, processing of DNA, and ensuing homologous recombination (21). TFP-associated genes and their products are linked to DNA uptake; however, TFP alone are not sufficient for natural transformation. TFP also mediate a unique form of flagellum-independent motility termed twitching motility. Twitching motility involves the assembly of TFP, attachment of the pilus, and subsequent retraction of the pilus, facilitating the translocation of the cell body toward the point of attachment (22). Interestingly, the term “twitching motility” was coined in 1965 by Lautrop to describe the jerky locomotion exhibited by Acinetobacter calcoaceticus, thereby laying the foundation early for a link between Acinetobacter spp. and TFP (23).
Recently, Smith et al. sequenced the genome of A. baumannii strain ATCC 17978 and described several genes whose products might play a role in transformation; some of these were predicted to encode components of a TFP-like system (24). Furthermore, a recent analysis of the published A. baumannii genomes revealed many genes whose products have homology to proteins required for the biogenesis of TFP found in other Gram-negative bacteria; thus, it appears that some A. baumannii strains contain the coding potential required to assemble TFP (25). In addition to this analysis, Antunes et al. demonstrated that A. baumannii strain AYE exhibits twitching motility. A complete list of TFP biogenesis gene homologs in three A. baumannii reference strains can be found in the study by Antunes et al. (25). Eijkelkamp and coworkers simultaneously reported that several genes in A. baumannii strain ATCC 17978, which likely encode proteins required for TFP biogenesis, were slightly down-regulated under low-iron growth conditions and that surface-associated motility was lost (26). In other organisms, TFP are associated with numerous phenotypes, including natural transformation and/or twitching motility (27, 28). Ramirez et al. identified a clinical isolate of A. baumannii isolated from blood that was naturally transformable; however, the molecular basis for this phenotype was not explored (29). Also, some A. baumannii clinical isolates are capable of twitching motility (reviewed in reference 30). Recently, Eijkelkamp et al. reported that all of the international clone I A. baumannii clinical isolates tested in their study as well as some clinical strains that did not belong to a currently characterized clonal lineage were capable of twitching motility, indicating that these strains may produce functional TFP (31). It has been speculated that TFP might play a role in the unique, flagellum-independent surface-associated motility displayed by many A. baumannii clinical isolates, yet there is no experimental evidence demonstrating that A. baumannii strains produce functional TFP (26, 32).
In this study, we demonstrated that A. baumannii strain M2, a clinical isolate, is naturally transformable and that natural transformation is dependent on genes which encode orthologs of proteins required for the biogenesis of TFP found in other Gram-negative organisms. These gene products were also required for twitching motility, another TFP-associated phenotype. Similar to observations made in other organisms (Neisseria gonorrhoeae, Pseudomonas aeruginosa, and Dichelobacter nodosus), the M2∆pilT mutant exhibited more surface-exposed PilA than the wild-type strain (33–35). TFP were readily visualized by transmission electron microscopy on pilT mutant cells but not on pilA or pilA pilT mutant cells. Collectively, these data led us to conclude that A. baumannii strain M2 produces functional TFP and that these structures are required for natural transformation and twitching motility. Lastly, we demonstrated that the unique surface-associated motility exhibited by many clinical isolates is not dependent on the production of functional TFP in strain M2.
RESULTS
Identification and arrangement of TFP biosynthesis gene clusters in A. baumannii strain M2.
Roche 454 and Ion Torrent instruments were employed to obtain raw genomic sequence for strain M2. These data were assembled, and the resulting contig sets (R. S. Munson, Jr., and P. N. Rather, unpublished data) were queried for genes whose products have homology to proteins required for the biogenesis of TFP found in other Gram-negative organisms as well as in completed A. baumannii genomes. Antunes et al., in Table S2 of their article, published a list of putative TFP-associated genes in three A. baumannii genomes (25). Eijkelkamp and coworkers examined 11 fully sequenced A. baumannii genomes and reported that all contained genes likely to encode proteins involved in TFP biogenesis (31). In the strain M2 genome, we identified homologs of genes whose products are known to be critical in TFP biogenesis in other organisms. We chose three of these genes for further investigation, as our primary goal was to determine whether functional TFP are produced by strain M2. The TFP biogenesis-related gene clusters were arranged similarly to the gene clusters identified in other A. baumannii strains (25). One gene cluster was identified that contains genes that encode a putative traffic ATPase (PilB), a putative inner-membrane platform protein (PilC), and a putative prepilin peptidase (PilD) (Fig. 1). A second gene cluster contains genes that encode homologs of the TFP retraction ATPases PilT and PilU. A third locus contains a gene that encodes the putative major pilin subunit, PilA. PilA has a predicted six-amino-acid leader peptide, an N-terminal hydrophobic region, and a processed length of 138 amino acids (aa) (31). The protein has two cysteines, which are predicted to form one disulfide bridge. Together, these data indicate that strain M2 has genes encoding a type IVa pilus system (36). The gene directly downstream of the pilA gene in strain M2 is predicted to encode a 436-aa hypothetical protein that contains a conserved Wzy_C domain found in the O-antigen ligase-like protein family. A gene encoding a member of the O-antigen ligase-like protein family is found immediately downstream of the pilA gene in some strains of P. aeruginosa. This Wzy_C domain-containing protein in P. aeruginosa is a pilin glycosylase (37). However, in the absence of functional data, we have designated the gene encoding this Wzy_C domain-containing protein in strain M2 pgyA, for “putative glycosylase A.”
FIG 1 .
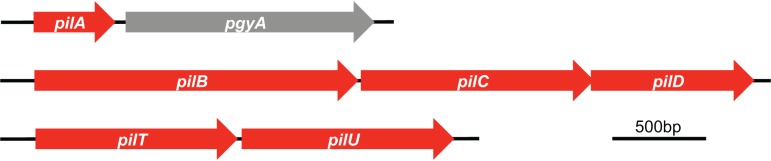
The pil gene loci in A. baumannii strain M2 employed in this study. Genes predicted to encode subunits of a TFP system in A. baumannii strain M2 are shown. We identified a pilA gene predicted to encode the major pilin subunit followed by pgyA, possibly coding for a type IV pilus O-glycosylase. Two other pil gene clusters were identified, (i) a pilBCD gene cluster, encoding a putative traffic ATPase (pilB), a putative inner membrane platform protein (pilC), and a putative prepilin peptidase (pilD), and (ii) a pilTU gene cluster, encoding two putative retraction ATPases.
The pil gene products were required for natural transformation.
In addition to the identification of TFP-encoding genes, we observed that strain M2 was naturally transformable, a phenotype associated with the production of TFP in other naturally transformable Gram-negative organisms. We constructed strains containing mutations in the pilA, pilD, or pilT gene and then determined the transformation efficiency of each strain. As shown in Fig. 2, the transformation efficiency for all mutant strains was below the level of detection in our assay. Each mutation was complemented by cloning the parental gene under the control of its predicted native promoter into a mini-Tn7 element, which was then transposed into the attTn7 site downstream of the glmS2 gene in the strain M2 chromosome. Transformation levels similar to parental levels were observed in the complemented pilA and pilD mutants. In contrast, we were unable to complement the pilT mutation with the parental pilT allele. In A. baumannii, pilT and pilU are contiguous on the chromosome, as they are in P. aeruginosa (Fig. 1). However, in P. aeruginosa, pilT and pilU are independently transcribed, indicating that the promoter for pilU may be within the pilT open reading frame (34). Thus, it is likely that our inability to complement the pilT mutation was due to polar effects on pilU. We therefore reintroduced a complete pilTU gene cluster, using the mini-Tn7 system, into the pilT strain; the natural transformation phenotype was restored, suggesting that we may have affected transcription of pilU in the pilT mutant. In addition, we introduced an empty mini-Tn7 element into the pilA mutant and demonstrated that this strain did not have detectable levels of transformation, indicating that mini-Tn7 alone does not restore the transformation phenotype (Munson, unpublished data). We also generated a strain containing a deletion of the pgyA gene. This mutant was naturally transformable.
FIG 2 .
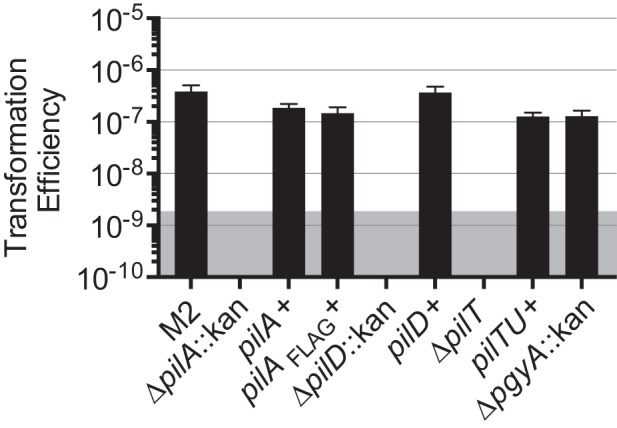
Natural transformation of strain M2 was reliant upon pil gene products. Strain M2, the pilA, pilD, and pilT isogenic mutants, and their respective complemented strains, including a ∆pilA strain complemented with the pilA gene fused to a FLAG tag, were tested for their transformation efficiencies. The pilA, pilD, and pilT mutants had transformation efficiencies below our level of detection, but the complemented strains, including the strain expressing PilA-FLAG, regained the natural transformation phenotype. The pgyA mutant also retained parental levels of transformation. The shaded area represents the level of detection of the assay. Transformation efficiency was calculated as the number of transformants/ml divided by the total CFU/ml for a given reaction. Bars show the means from three independent experiments with two technical replicates each, and error bars represent the standard errors of the means.
TFP-like structures were observed on the surface of strain M2.
Type IV pili are long, narrow fibers, generally <8 nm in diameter, that can be readily visualized on the surface of many Gram-negative bacteria, including Neisseria species and P. aeruginosa (36). We failed to observe TFP-like structures when whole cells of strain M2 were examined by transmission electron microscopy (TEM). However, the inability to detect TFP by EM is similar to what we observed with TFP in H. influenzae; that is, unless the TFP regulon was overexpressed, TFP were not observed by EM (27). Others have demonstrated that strains containing mutations in the pilT gene of N. gonorrhoeae, P. aeruginosa, and D. nodosus are hyperpiliated compared to the parental strain (33–35). PilT is an ATPase that is required for disassembly of TFP; thus, bacteria with mutations in pilT are frequently observed to be hyperpiliated, as the pili cannot retract. Since pilus retraction is required for twitching motility and transformation, the pili observed in the pilT mutant are not functional. We readily observed TFP-like structures on the pilT mutant by TEM (Fig. 3). To verify that these structures were in fact TFP, we constructed a pilT pilA mutant strain and examined it for the loss of TFP-like structures. This strain was devoid of TFP-like structures, except for 1 cell out of the >1,000 viewed which displayed a structure with dimensions similar to those of TFP. These data are consistent with the observed transformation phenotype in that both data sets affirm the conclusion that TFP are produced by strain M2.
FIG 3 .
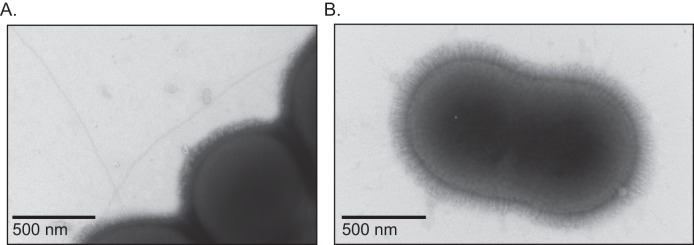
Observation of TFP on the ∆pilT mutant. (A) Type IV pilus-like appendages were readily observed on the surface of the ∆pilT mutant. (B) Type IV pilus-like appendages were not observed on the surface of a ∆pilT pilA::strAB mutant.
The major pilin subunit, PilA, was surface exposed.
Type IV pili are polymers composed predominately of the major pilin subunit, PilA, and minor pilins, so named for their relative abundance within the pilus fiber. By shearing pili from the surface of bacteria, the fiber can be separated from whole cells (38, 39), disassembled into its components, and separated by SDS-PAGE analysis, and protein composition can be determined via mass spectrometry. Cells of the parental M2 strain as well as the isogenic pilA and pilT mutant strains were vortexed to shear off surface structures. Whole cells were separated from the sheared protein fraction by centrifugation. The sheared protein fractions from each strain were then analyzed by SDS-PAGE. Selected bands were excised, and the proteins were identified by mass spectrometry. As shown in Fig. 4, when the sheared protein fractions were examined on a silver-stained SDS-PAGE gel, a band was present in the parental fraction slightly above the predicted mass of PilA (13.9 kDa). This band was absent in the fraction prepared from the pilA mutant. When preparations from the pilT mutant were characterized, a band which had the same mobility as the band in the parental fraction was observed. In addition, a band with a lower apparent molecular weight was present. The regions where the bands were present were excised from a Coomassie-stained SDS-PAGE gel, trypsinized, and subjected to MALDI-TOF (matrix-assisted laser desorption ionization–time of flight) mass spectrometry. As expected, the upper band present in the both the parental and pilT mutant fraction was identified as PilA. The PilA protein was not found in this region of the preparation from the pilA mutant. Interestingly, the lowest band in the pilT mutant fraction was also identified as PilA protein. These data taken together with the TEM images provide additional evidence that strain M2 produced TFP and that PilA is the major pilin subunit.
FIG 4 .
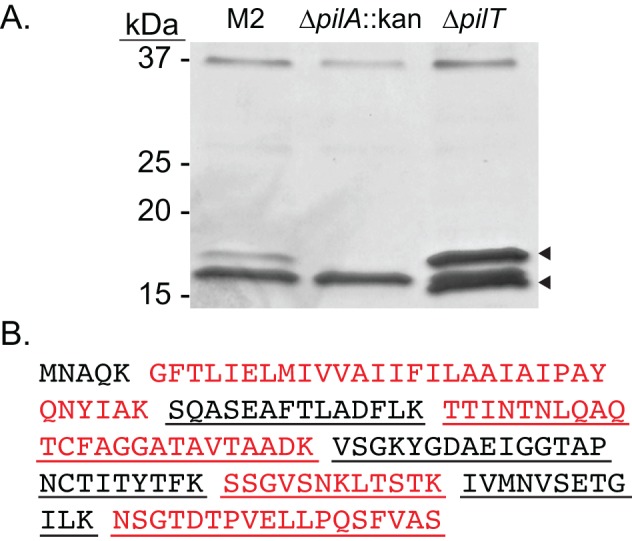
PilA, the major pilin subunit, is surface exposed. (A) The parental strain and the pilA and pilT mutant strains were resuspended in DPBS from L agar plates and vortexed to remove surface appendages. The sheared proteins were separated by SDS-PAGE, excised, and examined by MALDI-TOF mass spectrometry. The upper band in both the parental and pilT mutant fraction was identified as PilA. Interestingly, the lowest band present in the pilT mutant fraction was also identified as PilA. PilA was not identified in the pilA mutant fraction. (B) The primary amino acid sequence of the unprocessed prepilin, PilA, is shown. Predicted tryptic peptides are separated by spaces and shown in alternating colors for clarity. Underlined peptides were identified in both samples analyzed from the pilT mutant. The predominant band in the sample from the pilA mutant strain (also seen in the other preparations) is a predicted pilin produced by a chaperone/usher system. This is 87% identical to a putative biofilm synthesis protein (YP_001713377) in A. baumannii strain AYE.
Lastly, a band running at an intermediate molecular weight between the upper and lower bands of the pilT mutant fraction was observed in all three samples. This band was identified as a protein containing a spore coat domain by Blast; however, structural prediction analysis (PHYRE2) (40) of the primary amino acid sequence revealed that this protein may be a pilin of a chaperone usher pilus system that is not yet characterized in A. baumannii. This protein is highly homologous to a putative biofilm synthesis protein (NCBI accession number YP_001713377) found in A. baumannii strain AYE. We are currently investigating the role of this putative type I pilin in the biology of A. baumannii.
The strain M2 PilD homolog was required for processing of PilA.
Based on homology, we predicted that pilD encodes the prepilin peptidase, a protein required in other TFP systems that acts by cleaving an N-terminal leader peptide from immature PilA. To test this prediction, we compared the apparent molecular weights of PilA proteins produced in pilD+ and pilD backgrounds. As we do not currently have an antiserum that recognizes strain M2’s PilA, we constructed a mini-Tn7 element carrying sequence that encoded PilA with a C-terminal FLAG tag. Strains expressing the pilA-FLAG gene in a pilA and pilA pilD mutant background were constructed. We then compared whole-cell lysates of the two strains by Western blot analysis, probing with an anti-FLAG antibody. In Fig. 5, we show that PilA-FLAG was detected in both strains; however, PilA-FLAG from the pilA pilD mutant strain migrated as a higher-molecular-mass protein than PilA-FLAG produced in the pilA mutant background. These results are consistent with our hypothesis that PilD is the prepilin peptidase involved in processing of strain M2’s major pilin, PilA.
FIG 5 .
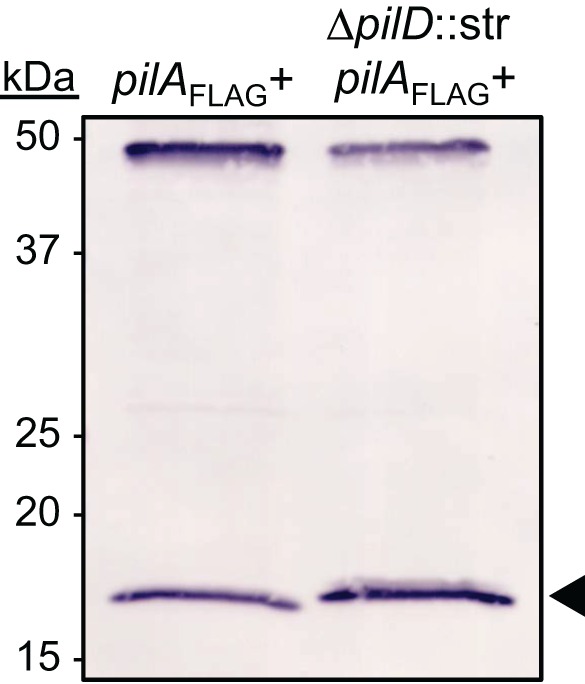
The strain M2 PilD homolog acted as a prepilin peptidase. Whole-cell lysates of the M2∆pilA::kan(pilA-FLAG+) and M2∆pilA::kan∆pilD::strAB(pilAFLAG+) strains were examined by Western blot analysis for processed and unprocessed PilA-FLAG. The nonspecific band around 50 kDa was included to demonstrate equal migration of proteins. PilA-FLAG from the M2∆pilA::kan(pilA-FLAG+) migrated to a slightly lower position than PilA-FLAG from M2∆pilA::kan∆pilD::strAB(pilAFLAG+). The leader peptide of PilA is predicted to be six amino acids.
The pilA, pilD, and pilTU gene products were required for twitching motility but not surface-associated motility.
Twitching motility, a TFP-dependent phenotype, is generally observed at the interphase of the petri dish and an agar or agarose surface. Given that transformation of strain M2 was dependent on TFP, we hypothesized that strain M2 would be capable of twitching motility. Classically, twitching motility assays have been conducted with medium containing 1% agar; however, agarose may be substituted. Media containing 1% agarose were used in these experiments. As shown in Fig. 6, strain M2 exhibited twitching motility. In contrast, the pilA, pilD, and pilT mutants did not exhibit twitching motility. Parental levels of twitching motility were observed when the complemented mutants were tested. The pilA mutant containing an empty mini-Tn7 element did not exhibit twitching motility (Munson, unpublished). The pgyA mutant strain retained parental levels of twitching motility.
FIG 6 .
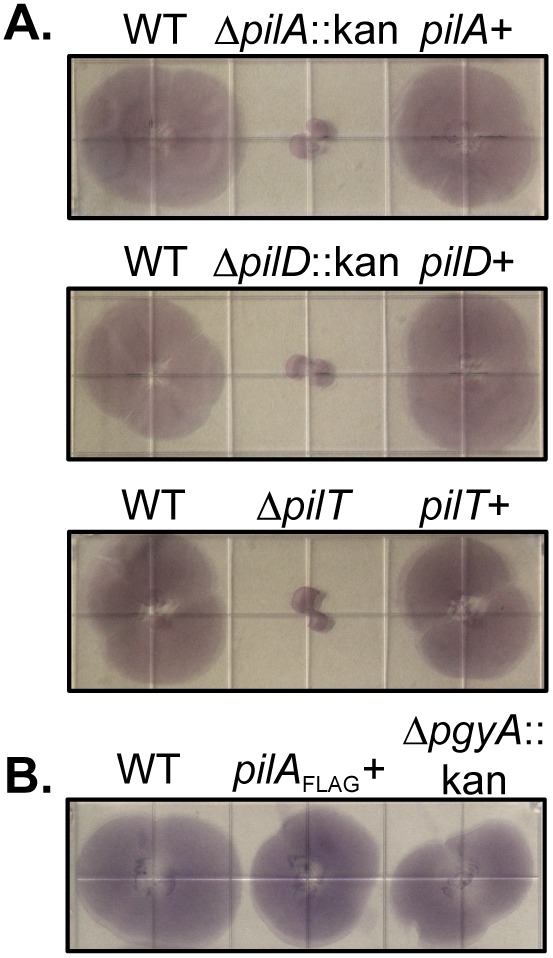
Twitching motility is reliant upon the pil gene products. (A) Twitching motility was observed at the agarose/plastic interface for M2 and the complemented mutants but not for the pilA, pilD, and pilT mutants. Each strain was inoculated by stabbing through the agarose to the surface of a plastic petri dish followed by incubation at 37°C for 18 h. The agarose was removed, and the nonadherent bacteria were removed by washing with PBS. The adherent bacteria were visualized by staining with 0.1% crystal violet. Each square in the grid on the plate is 13 mm wide. (B) The C-terminal FLAG tag on PilA did not impede twitching motility. The pgyA mutant retained the twitching motility phenotype.
Some A. baumannii isolates are also capable of a flagellum-independent surface-associated motility exhibited on semisolid media (41, 42). Clemmer et al. demonstrated that strain M2 was capable of surface-associated motility (32). In their study, surface-associated motility was impaired but not absent in a pilT mutant. To further explore the role of TFP in A. baumannii strain M2 surface-associated motility, we assessed the isogenic TFP mutant strains for their ability to translocate across the surface of semisolid media. Interestingly, the surface-associated motilities of the pilA, pilD, and pilT mutants were similar to that of the parental M2 strain (Fig. 7).
FIG 7 .

The pil gene products were not required for surface-associated motility. Strain M2 and the pilA, pilD, and pilT mutants were inoculated on the surface of a semisolid agarose plate (0.5%) and incubated for 18 h at 37°C. The pilA, pilD, and pilT mutant strains demonstrated no defect in surface-associated motility compared to the parental strain. Grids measure 13 mm2.
DISCUSSION
Several investigators have noted that A. baumannii genomes contain genes that likely encode proteins required for the biogenesis of TFP, and it has been suggested that TFP might be involved in the surface-associated motility phenotype demonstrated by some A. baumannii isolates (32). We sought to definitively demonstrate the production of TFP in an A. baumannii clinical isolate and determine whether the classically defined TFP-associated phenotypes, transformation and twitching motility, required A. baumannii TFP.
In A. baumannii strain M2, we identified several genes whose products were known to be involved in TFP biogenesis in other organisms (27, 43). We demonstrated that strain M2 was naturally transformable and exhibited twitching motility; both phenotypes were absent in the pilA, pilD, and pilT mutants. The pilA, pilD, and pilT mutations were then complemented, restoring the TFP-dependent phenotypes.
Major and minor pilins are processed prior to being polymerized into a fiber by the removal of a leader peptide by a dedicated prepilin peptidase, PilD (44). We tested for the predicted PilD activity by constructing strains that expressed C-terminal FLAG-tagged PilA in a pilA mutant and a pilA pilD mutant background. When examined by Western blot, PilA-FLAG produced in the pilA pilD mutant ran with a slightly higher apparent molecular weight than the protein produced in the pilD+ strain, consistent with cleavage of the predicted six-amino-acid leader peptide.
Downstream of the pilA gene is a gene encoding a hypothetical protein containing an O-antigen ligase (Wzy_C) domain. This gene is found in a subset of genomes of the clinical isolates that have been sequenced. These include A. baumannii strain ACICU (accession no. CP000863), TYTH-1 (accession no. CP-003856), MDR-TJ (accession no. CP003500), MDR-ZJ06 (accession no. CP001937), TCDC-AB0715 (accession no. CP002522), and 1656-2 (accession no. CP001921). It is possible that PgyA is a pilin glycosylase, as PilO produced by P. aeruginosa 1244 also contains a Wzy_C domain, and catalyzes the addition of a trisaccharide to serine 148 of PilA (45). Eleven serine residues are present in the M2 PilA; however, the location of serines within the strain M2 protein are not the same as those identified as glycosylation targets in the P. aeruginosa 1244 pilin. Additional studies are required to determine the activity of PgyA.
The pgyA gene is not to be confused with the recently characterized pglL gene identified in A. baumannii ATCC 17978, although the pglL gene is present downstream of the pilA gene in strain ATCC 17978. In strains whose genomes contain the pgyA gene, the pglL gene is found immediately downstream of pgyA, as is the case in strain M2. In strain ATCC 17978, the pglL gene product is required for the O-glycosylation of several proteins of unknown function (14).
Type IV pili are surface appendages that have diameters ranging from 5 to 8 nm and can measure several micrometers in length. TFP have been visualized on, and purified from, many Gram-negative organisms (36). We employed numerous strategies to visualize TFP on the surface of strain M2, including using immunogold labeling and TEM to view pili on M2 strains expressing PilA-FLAG. These attempts were unsuccessful. However, when we examined the pilT mutant strain by negative-stain transmission electron microscopy, we were able to visualize structures emanating from the surface of the outer membrane that resembled TFP. These structures were present on a subset of the cells in the pilT mutant population and found free on the grid surface. In addition, these structures were rarely on the parent and absent in a pilT pilA mutant, indicating that these structures are most likely TFP. These observations are expected, given the increase in relative abundance of surface exposed PilA on the pilT mutant compared to the parental M2 strain.
Enrichment for bacterial extracellular appendages by shearing bacteria in suspension without excessive cell lysis has been demonstrated for many organisms, including P. aeruginosa and nontypeable H. influenzae (27, 46). Therefore, sheared cell supernatants from the parental M2 as well as the pilA and pilT mutant strains were examined by Coomassie blue-stained SDS-PAGE. An additional band was present in the fraction from strain M2 when compared to the fraction from the pilA mutant. This same band was present in the fraction from the pilT mutant along with another lower molecular weight band which was absent in both the fraction from the strain M2 and the pilA mutant. Our observation was concordant with those obtained by Han et al., where a pilT mutant in D. nodosus demonstrated an increase in surface-exposed PilA compared to the parental strain (35). The upper band present in the fraction from the parent and the pilT mutant contained PilA when analyzed by MALDI-TOF mass spectrometry, confirming that strain M2 is able to export PilA to the surface of the cell and, by inference, assemble TFP. Interestingly, the lowest band in the fraction from the pilT mutant also contained PilA. We hypothesized that PilA in the lower band could simply be truncated or a proteolysis product; however, when we analyzed the mass spectrometry data for differential peptide identification, we found that the same peptides were present in both fractions. All peptides, with the exception of the hydrophobic amino terminal peptide, were present.
Genes predicted to encode TFP biogenesis proteins have been identified in all of the fully sequenced genomes of A. baumannii clinical isolates; however, most of the A. baumannii clinical isolates do not exhibit twitching motility (31), a classic phenotype associated with functional TFP. Additional work will be required to determine if the TFP-associated genes in these genomes are transcribed and under what conditions.
Recently, Eijkelkamp and coworkers reported that surface-associated motility and twitching motility were not always observed together and concluded that there are different mechanisms accounting for the different motility phenotypes displayed by A. baumannii clinical isolates (31). Twitching motility is a form of bacterial motility independent of flagella that requires the TFP to assemble, bind to a substratum, and then disassemble, resulting in movement of the cell body toward the point of attachment (22). Twitching motility was observed with strain M2, and this phenotype was dependent on the pilA, pilD, and pilT gene products, as twitching motility was absent in the mutant strains but restored to parental levels in the complemented strains. Eijkelkamp et al. demonstrated that there is sequence conservation between the PilA protein of A. baumannii clinical isolates within the same clonal lineage and that strains within the same clonal lineage exhibited similar motility patterns. Thus, their data suggested that specific PilA sequences might partially influence the ability of a strain to exhibit twitching motility. However, when we compared the sequence of strain M2’s PilA to those identified in the study by Eijkelkamp et al., we found that M2’s PilA sequence had 92% identity to that of A. baumannii strain 19606, a clinical isolate that did not exhibit twitching motility. Thus, it appears that PilA sequence does not correlate with ability of a particular strain to exhibit twitching motility.
Many strains of A. baumannii have also been shown to exhibit a unique surface-associated motility on semisolid media that resembles swarming motility of P. aeruginosa; however, swarming motility, as defined by Henrichsen, is flagellum-coordinated movement of groups of cells across a solid surface (47). Acinetobacter baumannii surface-associated motility is a flagellum-independent, multifactorial, complex process that is partially dependent on 1,3-diaminopropane synthesis, quorum sensing, iron concentration, the composition of lipopolysaccharide and is sensitive to blue light when cells are grown at 24°C (30, 32, 41, 42). It has been speculated that TFP might be involved in the surface-associated motility of this bacterium. Recently, Clemmer et al. demonstrated, in strain M2, that a mutant deficient in expression of the PilT protein was partially impaired with respect to surface-associated motility (32). In contrast, the pilT mutant we characterized as well as the pilA and pilD mutants exhibited parental levels of surface-associated motility. Occasionally, the pilD mutant exhibited a variable motility compared to the parental strain as well as the other mutant strains. Rather and coworkers (unpublished data) have observed two subtle but distinct colony morphologies, opaque and translucent, when M2 is streaked on 1% L agar. These colony morphologies are independent of the TFP phenotype. When a translucent clone containing the pilT mutation was tested, surface-associated motility of the pilT mutant was impaired. In contrast, an opaque clone containing a pilT mutation retained parental levels of surface-associated motility. When streaked on a plate, colonies of the pilT mutant described by Clemmer et al. are primarily translucent (32). Colonies of the pilT mutant described herein were primarily opaque, which partially explains the discrepant results. Overall, however, it is clear that surface-associated motility is not strictly dependent on the expression of TFP in the opaque form. A complete understanding of the basis for the surface-associated motility will, in part, depend on the identification of the molecular basis for the translucent and opaque colony types.
Herein, we have provided genetic evidence that twitching motility and surface-associated motility are distinct phenotypes confirming and extending the observations of Eijkelkamp and coworkers (31). We also have demonstrated that PilA sequence does not correlate with the ability to exhibit twitching motility. Importantly, we have provided the first visual evidence of TFP production by an A. baumannii strain through the observation of TFP on the surface of the strain M2 pilT mutant, a phenotype that is dependent upon the pilA gene product. In addition, we demonstrated that PilA is a major surface-exposed protein, and taking these data together with the TEM images, we conclude that PilA is the major pilin subunit. Natural transformation and twitching motility were dependent on expression of TFP, while the mechanisms responsible for surface-associated motility remain to be determined; however, our data emphatically illustrate that TFP are not required for surface-associated motility of strain M2. Importantly, our study demonstrated that TFP are produced by and have a role in the biology of an A. baumannii clinical isolate. Future studies will be directed at defining the conditions that promote TFP expression and determining the role of TFP in the virulence of this emerging pathogen.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
Bacterial strains and plasmids used for this study are listed in Table S1 in the supplemental material. All bacterial strains were grown on L agar or in LB broth. When appropriate, antibiotics were added to the A. baumannii cultures at the following concentrations: 750 µg ampicillin/ml, 20 µg kanamycin/ml, 50 µg streptomycin/ml, or 12.5 µg chloramphenicol/ml. When appropriate, Escherichia coli cultures were supplemented with antibiotics at the following concentrations: 50 µg ampicillin/ml for strains containing plasmids other than pGEM derivatives, 100 µg ampicillin/ml for strains containing pGEM derivatives, 25 µg streptomycin/ml, or 20 µg kanamycin/ml.
Construction of the pilA, pilD, and pilT mutants.
Primers for this study were obtained from Integrated DNA Technologies (Coralville, IA) and are listed in Table S2 in the supplemental material. We constructed a mutant deficient in expression of the pilA gene as follows. The pilA gene plus approximately 1 kb of DNA 5′ and 3′ of pilA was amplified by PCR using primer set 1. The amplicon was then ligated into pGEM-T-Easy (Promega, Madison, WI), and the ligation products were transformed into E. coli DH5α. A plasmid with the correct insert was saved as pGEM-pilA, and the sequence of the insert was verified by sequencing. The pilA gene in pGEM-pilA was then replaced with a kanamycin resistance cassette flanked by FRT sites from pKD13 using a lambda recombinase-based strategy (48, 49). An amplicon was generated that contained 47 nucleotides (nt) of DNA 5′ of pilA along with the translational start site, the cassette, the last 21 nt of the pilA gene and 29 nt downstream of pilA using primer set 2 and pKD13 as the template. The amplicon and pGEM-pilA were electroporated into E. coli strain DY380 that had been heat shocked at 42°C for 15 min prior to electroporation to induce expression of the lambda recombinase genes. Clones containing a plasmid with the pilA gene replaced by the cassette were identified on L agar supplemented with kanamycin. Plasmids were isolated and characterized; a plasmid containing FRT-kan-FRT (Flp recombination target) in place of the pilA gene was saved as pGEM-∆pilA::kan.
Natural transformation was then used to move the ∆pilA::kan fragment into M2. The plasmid pGEM-∆pilA::kan and primer set 1 were used to generate an amplicon containing ∆pilA::kan and flanked by 1 kb of DNA up- and downstream of pilA. An overnight culture of strain M2 was diluted 1:10 in 500 µl of LB broth and incubated at 37°C with shaking at 180 rpm for 2 h. Following the 2 h incubation, 1 µg of the amplicon was added to the bacterial culture. Cells were then transferred to an L agar plate and incubated at 37°C for 4 h. Cells were collected from the plate and resuspended in 500 µl of LB broth, and dilutions were plated on L agar supplemented with kanamycin at 37°C. Kanamycin-resistant clones were characterized by PCR analysis and sequencing to verify allele exchange. A clone was saved as M2∆pilA::kan. The identical strategy was used to construct M2∆ pilD::kan using primer sets 3 and 4.
Since a mutation in pilT might be polar on pilU, we modified our strategy to remove the cassette, leaving a short open reading frame containing the translational start site of pilT, a small scar left by the excision of the cassette, and the last 21 nt of pilT. For this construction, pilTU were amplified from M2 genomic DNA with primer set 5 and cloned as described above. A clone was saved as pGEM-pilTU.
To construct an unmarked mutation in pilT, a derivative of pKD13 containing FRT-kan-sacB-FRT, pRSM3542, was used as the template in a PCR with primer set 6 (50). The amplicon, along with pGEM-pilTU, was electroporated into heat-shocked E. coli DY380 as described above. Restriction digest analysis and sequencing were performed on plasmid purified from kanamycin-resistant clones. A correct clone was saved as pGEM-∆pilT::[kan-sacB] pilU. To introduce this mutation into strain M2, a PCR was performed using pGEM-∆pilT::[kan-sacB] pilU as the template and primer set 5. This amplicon was introduced into strain M2 by natural transformation as described above. A kanamycin-resistant clone with the correct sequence was saved as M2∆pilT::kan-sacB.
A triparental mating strategy was employed to transiently introduce the pFLP2 plasmid, which expresses the FLP recombinase, into M2∆pilT::kan-sacB as described by Carruthers et al. (50). Briefly, 100 µl of stationary cultures normalized to an optical density at 600 nm (OD600) of 2.0 of each of DH5α(pFLP2), HB101(pRK2013), and M2∆pilT::kan-sacB were added to 700 µl of warm LB broth. The bacterial suspension was washed twice by centrifugation at 7,000 × g followed by resuspension of the bacterial pellet in 1 ml of 32°C LB broth. On the final wash, the bacterial pellet was resuspended in 25 µl of LB broth, and the suspension was spotted on a prewarmed L agar plate and incubated for 16 h at 32°C. The plates were then transitioned to 37°C for 2 h to transiently induce the FLP recombinase. Bacteria were then scraped from the plate and resuspended in 1 ml of LB broth. Serial dilutions were plated on L agar containing 10% sucrose and 12.5 µg of chloramphenicol/ml and incubated at 26°C to select for transconjugants that had lost sacB and to select against E. coli strains. Strain M2 is naturally resistant to chloramphenicol at the concentrations used in this study. Clones demonstrating sucrose resistance were analyzed by PCR to verify loss of the kan-sacB cassette. A correct clone was sequenced and saved as M2∆pilT.
Construction of the pgyA mutant strain.
The pGEM-pilA vector was constructed so that it contained the pilA gene as well as flanking DNA, which included the pgyA gene. Thus, to construct a pgyA mutant strain, the following strategy was employed. EcoRV, a unique restriction site 64 bp from the 5′ end of pgyA, was used to digest pGEM-pilA. The kanamycin cassette from pUC4K was excised with HincII and ligated to the EcoRV-linearized pGEM-pilA and transformed into DH5α, and clones were selected for on L agar supplemented with kanamycin. A clone demonstrating the correct restriction pattern was used to generate a PCR amplicon containing the interrupted pgyA gene along with approximately 1 kb of flanking DNA using primer set 1. The resulting amplicon was transformed into strain M2 via natural transformation as described above. Transformants were selected for on L agar supplemented with kanamycin. A clone with the correct insertion was verified with colony PCR and sequencing to confirm the mutation.
Construction of complemented mutant strains.
A mini-Tn7 transposon system was used to complement mutations made in strain M2 in single copy, under the control of each gene’s predicted promoter. Construction of the mini-Tn7-containing plasmid, pRSM3510, employed for this study has been described (50). The pilA gene along with 430 bp upstream of the start codon was amplified with primer set 7. The forward primer contained a BamHI site on the 5′ end, while the reverse primer of each primer set contained KpnI on the 3′ end. The amplicon was digested with BamHI and KpnI and ligated to pRSM3510 that had been digested with the same enzymes. The ligation mixture was electroporated into E. coli EC100D, and clones were selected on L agar containing ampicillin. Plasmids containing the pilA gene were identified by restriction digestion and verified by sequencing. A clone was saved as pRSM3510-pilA. Similarly pRSM3510-pilTU, which contains 312 bp upstream of the start codon of pilT, was constructed using primer set 8 with the following exceptions. The reverse primer did not have a KpnI site on the 3′ end and was instead phosphorylated for use in a blunt cloning strategy. pRSM3510 was digested with BamHI and StuI, the pilTU amplicon was digested with BamHI, and the two were subsequently ligated together.
Since pilD is the last gene in a cluster of TFP-related genes (Fig. 1), we assumed that pilD transcription would be driven from a promoter 5′ of pilB. Thus, we constructed a clone containing the putative pilB promoter driving expression of pilD. Plasmid pGEM-pilBCD was generated using primer set 3 and M2 genomic DNA as the template as described for pGEM-pilA. Inverse PCR using pGEM-pilBCD as the template and primer set 9 was utilized to create an in-frame deletion of pilBC, which left the putative promoter, the start codon of pilB, and the last 21 bp of pilC immediately 5′ to the pilD gene. This amplicon was self-ligated and electroporated into DH5α. Clones were selected for on L agar supplemented with ampicillin, and a plasmid containing the pilBC deletion was saved as pGEM-pilD. pGEM-pilD was used as a template in a PCR with primer set 10 to produce an amplicon that was cloned into pRSM3510 as described above for pRSM3510-pilA. Ligations were electroporated into EC100D, and clones were selected for on L agar supplemented with ampicillin. A correct clone was saved as pRSM3510-pilD.
Primer set 11 was used to amplify the template, pRSM3510-pilA, with a FLAG tag immediately upstream of the stop codon of the pilA gene. The amplicon was self-ligated and electroporated into EC100D. Transformants were selected on L agar supplemented with ampicillin and a correct clone was sequence verified and saved as pRSM3510-pilA-FLAG.
Insertion of the mini-Tn7 constructs into the A. baumannii mutants.
The pRSM3510 derivatives expressing the pilA, pilD, or pilTU genes were introduced into the respective A. baumannii M2 mutants via a four-parent conjugal strategy modified from that described by Kumar et al. (51). Briefly, 100 µl of stationary cultures normalized to an OD600 of 2.0 of the recipient strain (A. baumannii mutant), HB101(pRK2013), EC100D(pTNS2), and EC100D containing the appropriate pRSM3510 derivative were added to 600 µl of warm LB broth. Each suspension was washed twice by centrifugation at 7,000 × g followed by resuspension of the bacterial pellet in 1 ml of warm LB broth. On the final wash, the bacterial pellet was resuspended in 25 µl of LB broth, and the suspension was spotted on a prewarmed L agar plate and incubated overnight at 37°C. The bacteria were scraped from the plate and resuspended in 1 ml of LB broth, vortexed for 8 s, and serial dilutions were plated on L agar plates supplemented with chloramphenicol to select against E. coli strains and ampicillin to select for A. baumannii strain M2 exconjugants that had received the mini-Tn7 constructs. To verify that mini-Tn7 had successfully transposed downstream of the glmS2 gene, primer set 12 (Tn7L forward and downstream glmS2 reverse) were used to amplify a 400-bp fragment that would produce a product only if mini-Tn7 had been inserted at the predicted attTn7 in the correct orientation. The four strains were saved as M2∆pilA::kan(pilA+), M2∆ pilD::kan(pilD+), M2∆pilT(pilTU+), and M2∆pilA::kan(pilA-FLAG+).
Construction of pGEM-blsA::strAB. The blsA gene along with 1 kb of upstream and downstream DNA was amplified by PCR with the primer set 13 using A. baumannii strain M2 genomic DNA as the template. The amplicon was A tailed, TA cloned into pGEM-T Easy, and electroporated into DH5α. Transformants were selected on L agar supplemented with ampicillin, and correct clones were verified by restriction digestion and sequencing. The strA and strB genes along with approximately 400 bp of flanking DNA were amplified from A. baumannii strain RUH134 with the phosphorylated primer set 14. This PCR product was ligated to an EcoRV-digested pGEM-blsA and electroporated into DH5α. Clones were selected for on L agar supplemented with streptomycin. A clone with the correct insertion was saved as pGEM-blsA::strAB. The interruption in the blsA gene is 72 bp downstream of the ATG start site.
Construction of the pilT pilA mutant.
To construct a pilT pilA mutant, an insertionally inactivated pilA gene was introduced into the pilT mutant strain. Briefly, pGEM-pilA was used as the template to construct pGEM-∆pilA, in which inverse PCR was performed using primer set 16 to create a deletion of pilA. A PCR product containing the streptomycin resistance genes strAB was amplified from RUH134 using primer set 14. The pGEM-∆pilA- and strAB-containing PCR products were ligated together, and this ligation product was transformed into E. coli DH5α. This plasmid was saved as pGEM-∆pilA::strAB. The fragment containing ∆pilA::strAB was cut from pGEM-∆pilA::strAB and cloned into pKNOCK-Km. This plasmid was saved as pKNOCK-Km-∆pilA::strAB. E. coli EC100D cells harboring pKNOCK-∆pilA::strAB, E. coli HB101 harboring pRK2013, and A. baumannii strain M2∆pilT were used as donor, helper, and recipient strains, respectively, in a triparental conjugation. Exconjugants containing ∆pilA::strAB were selected for on L agar containing chloramphenicol and streptomycin. Exconjugants were verified by PCR to confirm the pilA mutation. A verified clone was saved as M2∆pilT∆ pilA::strAB.
Construction of the pilA pilD mutant.
To construct a pilA pilD mutant, we introduced a marked deletion of the pilD gene into the M2∆pilA::kan(pilA-FLAG+) strain via natural transformation. Briefly, pGEM-pilBCD was amplified by inverse PCR using primers set 15 to create an in-frame deletion of the pilD gene, in which the ATG start site as well as the last 21 nt of the pilD gene was left. The streptomycin cassette from A. baumannii RUH134 was amplified by PCR using primer set 14 which was 5′ phosphorylated. The two amplicons were ligated and electroporated into DH5α, and clones were selected for on L agar plates containing streptomycin. A clone demonstrating the correct sequence was used to generate an amplicon encoding the pilD deletion interrupted with the streptomycin cassette as well as 1 kb of flanking DNA. The PCR product was transformed via natural transformation into the pilA-FLAG complemented pilA mutant as described above. A correct clone was verified by restriction digest and sequencing as M2∆pilA::kan ∆ pilD::strAB(pilA-FLAG+).
Transformation efficiency assays.
A single colony, from plates incubated overnight at 37°C, was used to inoculate 2 ml of LB broth. After overnight growth at 37°C and 180 rpm, 50 µl of culture was diluted with 450 µl of fresh LB broth and grown for 2 h at 37°C and 180 rpm. One microgram of pGEM-blsA::strAB, linearized with PstI, was added to cultures. After 2 h, the bacterium-DNA suspension was spotted on an L agar plate and incubated for 4 h at 37°C. Bacteria were removed from the plate, resuspended in 500 µl LB broth, and normalized to an OD600 of 50, and serial dilutions were plated on L agar to enumerate total CFU. In addition, serial dilutions were plated on L agar supplemented with streptomycin to enumerate CFU of transformants. Transformation efficiency was calculated by dividing the CFU of transformants by the total CFU. Experiments were conducted on at least three separate occasions.
Electron microscopy.
M2 and the pilA, pilT, and pilT pilA mutant strains were streaked onto L agar and incubated overnight at 37°C. One hundred microliters of Dulbecco’s phosphate-buffered saline (DPBS) was added to isolated colonies to create a slightly turbid bacterial suspension. Twenty microliters of the bacterial suspension was pipetted onto a piece of Parafilm, and a Formvar carbon film on 300 square mesh nickel grids (Electron Microscopy Sciences) was placed in each droplet for 5 min. The grids were blotted on filter paper and subsequently incubated in 20 µl of 2% paraformaldehyde in DPBS for 1 h. The grids were washed three times in DPBS, blotted on filter paper, and then stained in a 2.0% (wt/vol) ammonium molybdate, 2.0% (wt/vol) ammonium acetate solution diluted 1:1 with deionized water for 5 min. The grids were blotted dry on filter paper and dried overnight. Images were captured on an FEI Tecnai G2 Spirit transmission electron microscope at the Campus Microscopy and Imaging Facility at The Ohio State University.
Pilus shear preparations.
M2 and the pilA and pilT mutant strains were each streaked onto six L agar plates and incubated overnight at 37°C. In order to purify enough surface-exposed protein to visualize by Coomassie staining on a polyacrylamide gel, bacteria were collected from agar plates and resuspended in 5 ml of phosphate-buffered saline containing protease inhibitor cocktail (Roche), yielding a concentrated cell suspension. Appropriate dilutions were made, and cell suspensions were normalized via absorbance at 600 nm to an optical density of 50. Cells were vortexed at high speed for 1 min, followed by incubation on ice for 1 min. After centrifugation at 16,000 × g for 1 min at 4°C, the resultant supernatants were removed, and ammonium sulfate was added to the supernatants to a final concentration of 30%. The preparations were incubated overnight at 4°C. The precipitated protein was then collected by centrifugation at 20,000 × g and resuspended in 100 µl of SDS-PAGE loading buffer. Twenty microliters of each sample was run in duplicate on a 4 to 20% TGX gel (Bio-Rad) for analysis by silver staining and Coomassie staining. Protein bands were excised and sent off for MALDI-TOF mass spectrometry analysis at the Campus Chemical Instrument Center Mass Spectrometry and Proteomics Facility at The Ohio State University.
Processed and unprocessed PilA-FLAG Western blots.
To determine whether PilD was the leader peptidase, M2∆pilA::kan(pilA-FLAG+) and M2∆pilA::kan ∆pilD::strAB pilA(pilA-FLAG+) were streaked onto L agar and incubated for 16 to 18 h at 37°C. The cells were scraped from the plate into DPBS and normalized to an OD600 of 5.0. Samples were resuspended in 1× loading buffer, boiled for 10 min, run on a 4 to 20% TGX gel, and transferred to nitrocellulose for Western blot analysis with an anti-FLAG antibody (Agilent) following the manufacturer’s protocol.
Twitching motility.
Twitching motility plates, comprised of 10 g tryptone/liter, 5 g NaCl/liter, and 10 g agarose/liter (BP164-100; Fisher), were prepared fresh for each experiment. Each plate was made by pouring 30 ml medium into a 150-mm petri dish and allowing the medium to air-dry with the lid off for 20 min in a laminar flow hood. Each twitching motility plate was stab inoculated with a colony of bacteria at the agarose/petri plate interface and placed in a 37°C, humidified incubator for 18 h. To visualize the bacteria at the interface, agarose was removed from each plate, and the plates were washed with phosphate-buffered saline (PBS) and stained with 0.1% crystal violet (wt/vol) in water for 5 min. To remove excess crystal violet, each plate was gently washed with PBS and allowed to dry. Twitching motility assays were conducted on at least three separate occasions.
Surface-associated motility.
Surface motility plates, comprised of 5 g tryptone/liter, 2.5 g NaCl/liter, and 5 g agarose/liter (0.3%) as previously described (30), were prepared fresh for each surface motility experiment. In order to reduce variation between plates, each plate was poured with 30 ml of surface motility medium in a laminar flow hood with the lids off. The plates were allowed to dry for 30 min and then promptly used for motility assays. Motility plates were inoculated on the surface with 2 µl of a bacterial suspension normalized to an OD600 of 2.0. Each bacterial culture was started in the morning from an isolated colony and incubated to stationary phase at 37°C with shaking at 180 rpm. Motility plates were incubated for 18 h at 37°C. Surface-associated motility assays were conducted on at least three separate occasions.
SUPPLEMENTAL MATERIAL
Bacterial strains and plasmids included in this study.
Primers included in this study.
ACKNOWLEDGMENTS
This work was supported by funds from the Research Institute at Nationwide Children’s Hospital. C.M.H. was funded by the Howard Hughes Medical Institute Med into Grad Initiative.
Footnotes
Citation Harding CM, Tracy EN, Carruthers MD, Rather PN, Actis LA, Munson RS, Jr. 2013. Acinetobacter baumannii strain M2 produces type IV pili which play a role in natural transformation and twitching motility but not surface-associated motility. mBio 4(4):e00360-13. doi:10.1128/mBio.00360-13.
REFERENCES
- 1. Maragakis LL, Perl TM. 2008. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin. Infect. Dis. 46:1254–1263 [DOI] [PubMed] [Google Scholar]
- 2. Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5:939–951 [DOI] [PubMed] [Google Scholar]
- 3. Towner KJ. 2009. Acinetobacter: an old friend, but a new enemy. J. Hosp. Infect. 73:355–363 [DOI] [PubMed] [Google Scholar]
- 4. Weber DJ, Rutala WA, Miller MB, Huslage K, Sickbert-Bennett E. 2010. Role of hospital surfaces in the transmission of emerging health care-associated pathogens: norovirus, Clostridium difficile, and Acinetobacter species. Am. J. Infect. Control 38:S25–S33 [DOI] [PubMed] [Google Scholar]
- 5. Houang ET, Sormunen RT, Lai L, Chan CY, Leong AS. 1998. Effect of desiccation on the ultrastructural appearances of Acinetobacter baumannii and Acinetobacter lwoffii. J. Clin. Pathol. 51:786–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. 2007. Antimicrobial resistance among gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J. Clin. Microbiol. 45:3352–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081 [DOI] [PubMed] [Google Scholar]
- 8. Hsueh PR, Teng LJ, Chen CY, Chen WH, Yu CJ, Ho SW, Luh KT. 2002. Pandrug-resistant Acinetobacter baumannii causing nosocomial infections in a university hospital, Taiwan. Emerg. Infect. Dis. 8:827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giamarellou H, Antoniadou A, Kanellakopoulou K. 2008. Acinetobacter baumannii: a universal threat to public health? Int. J. Antimicrob. Agents 32:106–119 [DOI] [PubMed] [Google Scholar]
- 10. Choi CH, Lee JS, Lee YC, Park TI, Lee JC. 2008. Acinetobacter baumannii invades epithelial cells and outer membrane protein A mediates interactions with epithelial cells. BMC Microbiol. 8:216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee JS, Choi CH, Kim JW, Lee JC. 2010. Acinetobacter baumannii outer membrane protein A induces dendritic cell death through mitochondrial targeting. J. Microbiol. 48:387–392 [DOI] [PubMed] [Google Scholar]
- 12. Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. 2010. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 78:1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brossard KA, Campagnari AA. 2012. The Acinetobacter baumannii biofilm-associated protein plays a role in adherence to human epithelial cells. Infect. Immun. 80:228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwashkiw JA, Seper A, Weber BS, Scott NE, Vinogradov E, Stratilo C, Reiz B, Cordwell SJ, Whittal R, Schild S, Feldman MF. 2012. Identification of a general O-linked protein glycosylation system in Acinetobacter baumannii and its role in virulence and biofilm formation. PLoS Pathog. 8:e1002758. 10.1371/journal.ppat.1002758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bentancor LV, Camacho-Peiro A, Bozkurt-Guzel C, Pier GB, Maira-Litrán T. 2012. Identification of Ata, a multifunctional trimeric autotransporter of Acinetobacter baumannii. J. Bacteriol. 194:3950–3960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tomaras AP, Dorsey CW, Edelmann RE, Actis LA. 2003. Attachment to and biofilm formation on abiotic surfaces by Acinetobacter baumannii: involvement of a novel chaperone-usher pili assembly system. Microbiology 149:3473–3484 [DOI] [PubMed] [Google Scholar]
- 17. Gaddy JA, Arivett BA, McConnell MJ, López-Rojas R, Pachón J, Actis LA. 2012. Role of acinetobactin-mediated iron acquisition functions in the interaction of Acinetobacter baumannii strain ATCC 19606T with human lung epithelial cells, Galleria mellonella caterpillars, and mice. Infect. Immun. 80:1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. King LB, Pangburn MK, McDaniel LS. 2013. Serine protease PKF of Acinetobacter baumannii results in serum resistance and suppression of biofilm formation. J. Infect. Dis. 207:1128–1134 [DOI] [PubMed] [Google Scholar]
- 19. Ayers M, Howell PL, Burrows LL. 2010. Architecture of the type II secretion and type IV pilus machineries. Future Microbiol. 5:1203–1218 [DOI] [PubMed] [Google Scholar]
- 20. Mattick JS. 2002. Type IV pili and twitching motility. Annu. Rev. Microbiol. 56:289–314 [DOI] [PubMed] [Google Scholar]
- 21. Chen I, Dubnau D. 2004. DNA uptake during bacterial transformation. Nat. Rev. Microbiol. 2:241–249 [DOI] [PubMed] [Google Scholar]
- 22. Burrows LL. 2012. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol. 66:493–520 [DOI] [PubMed] [Google Scholar]
- 23. Henrichsen J. 1983. Twitching motility. Annu. Rev. Microbiol. 37:81–93 [DOI] [PubMed] [Google Scholar]
- 24. Smith MG, Gianoulis TA, Pukatzki S, Mekalanos JJ, Ornston LN, Gerstein M, Snyder M. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21:601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Antunes LC, Imperi F, Carattoli A, Visca P. 2011. Deciphering the multifactorial nature of Acinetobacter baumannii pathogenicity. PLoS One 6:e22674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eijkelkamp BA, Hassan KA, Paulsen IT, Brown MH. 2011. Investigation of the human pathogen Acinetobacter baumannii under iron limiting conditions. BMC Genomics 12:126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carruthers MD, Tracy EN, Dickson AC, Ganser KB, Munson RS, Jr, Bakaletz LO. 2012. Biological roles of nontypeable Haemophilus. influenzae type IV pilus proteins encoded by the pil and com operons. J. Bacteriol. 194:1927–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Skerker JM, Berg HC. 2001. Direct observation of extension and retraction of type IV pili. Proc. Natl. Acad. Sci. U. S. A. 98:6901–6904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramirez MS, Don M, Merkier AK, Bistué AJ, Zorreguieta A, Centrón D, Tolmasky ME. 2010. Naturally competent Acinetobacter baumannii clinical isolate as a convenient model for genetic studies. J. Clin. Microbiol. 48:1488–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Skiebe E, de Berardinis V, Morczinek P, Kerrinnes T, Faber F, Lepka D, Hammer B, Zimmermann O, Ziesing S, Wichelhaus TA, Hunfeld KP, Borgmann S, Gröbner S, Higgins PG, Seifert H, Busse HJ, Witte W, Pfeifer Y, Wilharm G. 2012. Surface-associated motility, a common trait of clinical isolates of Acinetobacter baumannii, depends on 1,3-diaminopropane. Int. J. Med. Microbiol. 302:117–128 [DOI] [PubMed] [Google Scholar]
- 31. Eijkelkamp BA, Stroeher UH, Hassan KA, Papadimitrious MS, Paulsen IT, Brown MH. 2011. Adherence and motility characteristics of clinical Acinetobacter baumannii isolates. FEMS Microbiol. Lett. 323:44–51 [DOI] [PubMed] [Google Scholar]
- 32. Clemmer KM, Bonomo RA, Rather PN. 2011. Genetic analysis of surface motility in Acinetobacter baumannii. Microbiology 157:2534–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wolfgang M, Park HS, Hayes SF, van Putten JP, Koomey M. 1998. Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 95:14973–14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Whitchurch CB, Mattick JS. 1994. Characterization of a gene, pilU, required for twitching motility but not phage sensitivity in Pseudomonas aeruginosa. Mol. Microbiol. 13:1079–1091 [DOI] [PubMed] [Google Scholar]
- 35. Han X, Kennan RM, Davies JK, Reddacliff LA, Dhungyel OP, Whittington RJ, Turnbull L, Whitchurch CB, Rood JI. 2008. Twitching motility is essential for virulence in Dichelobacter nodosus. J. Bacteriol. 190:3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pelicic V. 2008. Type IV pili: e pluribus unum? Mol. Microbiol. 68:827–837 [DOI] [PubMed] [Google Scholar]
- 37. Castric P. 1995. pilO, a gene required for glycosylation of Pseudomonas aeruginosa 1244 pilin. Microbiology 141:1247–1254 [DOI] [PubMed] [Google Scholar]
- 38. Tramont EC, Sadoff JC, Boslego JW, Ciak J, McChesney D, Brinton CC, Wood S, Takafuji E. 1981. Gonococcal pilus vaccine. Studies of antigenicity and inhibition of attachment. J. Clin. Invest. 68:881–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schoolnik GK. 1994. Purification of somatic pili. Methods Enzymol. 236:271–282 [DOI] [PubMed] [Google Scholar]
- 40. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 41. Mussi MA, Gaddy JA, Cabruja M, Arivett BA, Viale AM, Rasia R, Actis LA. 2010. The opportunistic human pathogen Acinetobacter baumannii senses and responds to light. J. Bacteriol. 192:6336–6345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McQueary CN, Kirkup BC, Si Y, Barlow M, Actis LA, Craft DW, Zurawski DV. 2012. Extracellular stress and lipopolysaccharide modulate Acinetobacter baumannii surface-associated motility. J. Microbiol. 50:434–443 [DOI] [PubMed] [Google Scholar]
- 43. Rudel T, Facius D, Barten R, Scheuerpflug I, Nonnenmacher E, Meyer TF. 1995. Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 92:7986–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Strom MS, Nunn DN, Lory S. 1993. A single bifunctional enzyme, PilD, catalyzes cleavage and N-methylation of proteins belonging to the type IV pilin family. Proc. Natl. Acad. Sci. U. S. A. 90:2404–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Comer JE, Marshall MA, Blanch VJ, Deal CD, Castric P. 2002. Identification of the Pseudomonas aeruginosa 1244 pilin glycosylation site. Infect. Immun. 70:2837–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baynham PJ, Ramsey DM, Gvozdyev BV, Cordonnier EM, Wozniak DJ. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188:132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henrichsen J. 1972. Bacterial surface translocation: a survey and a classification. Bacteriol. Rev. 36:478–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tracy E, Ye F, Baker BD, Munson RS., Jr. 2008. Construction of non-polar mutants in Haemophilus influenzae using FLP recombinase technology. BMC Mol. Biol. 9:101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carruthers MD, Nicholson PA, Tracy EN, Munson RS., Jr. 2013. Acinetobacter baumannii utilizes a type VI secretion system for inter-bacterial competition. PLoS One 8:e59388. 10.1371/journal.pone.0059388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. 2010. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J. Microbiol. Methods 82:296–300 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial strains and plasmids included in this study.
Primers included in this study.