

ABSTRACT
Staphylococcus aureus is a significant cause of infections worldwide and is able to utilize aerobic respiration, anaerobic respiration, or fermentation as the means by which it generates the energy needed for proliferation. Aerobic respiration is supported by heme-dependent terminal oxidases that catalyze the final step of aerobic respiration, the reduction of O2 to H2O. An inability to respire forces bacteria to generate energy via fermentation, resulting in reduced growth. Elucidating the roles of these energy-generating pathways during colonization of the host could uncover attractive therapeutic targets. Consistent with this idea, we report that inhibiting aerobic respiration by inactivating heme biosynthesis significantly impairs the ability of S. aureus to colonize the host. Two heme-dependent terminal oxidases support aerobic respiration of S. aureus, implying that the staphylococcal respiratory chain is branched. Systemic infection with S. aureus mutants limited to a single terminal oxidase results in an organ-specific colonization defect, resulting in reduced bacterial burdens in either the liver or the heart. Finally, inhibition of aerobic respiration can be achieved by exposing S. aureus to noniron heme analogues. These data provide evidence that aerobic respiration plays a major role in S. aureus colonization of the host and that this energy-generating process is a viable therapeutic target.
IMPORTANCE
Staphylococcus aureus poses a significant threat to public health as antibiotic-resistant isolates of this pathogen continue to emerge. Our understanding of the energy-generating processes that allow S. aureus to proliferate within the host is incomplete. Host-derived heme is the preferred source of nutrient iron during infection; however, S. aureus can synthesize heme de novo and use it to facilitate aerobic respiration. We demonstrate that S. aureus heme biosynthesis powers a branched aerobic respiratory chain composed of two terminal oxidases. The importance of having two terminal oxidases is demonstrated by the finding that each plays an essential role in colonizing distinct organs during systemic infection. Additionally, this process can be targeted by small-molecule heme analogues called noniron protoporphyrins. This study serves to demonstrate that heme biosynthesis supports two terminal oxidases that are required for aerobic respiration and are also essential for S. aureus pathogenesis.
Introduction
The Gram-positive bacterium Staphylococcus aureus is a significant cause of morbidity and mortality despite innocuously colonizing the anterior nares of approximately 30% of the human population (1). When protective barriers are compromised, this organism can cause a variety of distinct infections, including cellulitis, endocarditis, osteomyelitis, bacteremia, and septic shock (2, 3). The ability to colonize multiple niches within a single vertebrate host is likely due to the ability of S. aureus to fine-tune its cellular physiology to meet challenges presented by these diverse environments.
Uncovering the metabolic pathways that sustain bacterial growth within the host is paramount to the identification of novel targets for therapeutic intervention (4, 5). Three distinct energy-generating pathways support bacterial proliferation. First, in the presence of oxygen, bacteria respire aerobically. Second, bacteria can anaerobically respire in environments devoid of oxygen when an alternative terminal electron acceptor is present. Third, in the absence of oxygen and alternative terminal electron acceptors, bacteria are limited to fermentation for energy production (6, 7). The energy output of aerobic or anaerobic respiration is greater than that of fermentation, as both forms of respiration harness the energy provided by the generation of a membrane potential and a chemical gradient referred to collectively as the proton motive force (PMF). The final step of aerobic respiration, the reduction of O2 to H2O, is mediated by heme-dependent terminal oxidases (6–10). Bacteria often utilize multiple terminal oxidases in order to support aerobic respiration, and in these cases the respiratory pathway is said to be branched (6–12). Branched-chain respiratory pathways enable bacteria to cope with rapid changes in the aerobic capacity of the environment (13).
Given the diverse host niches colonized by bacteria, it is likely that metabolic plasticity is required to generate the energy needed for survival and proliferation. For pathogens, these metabolic pathways must be active in the presence of a robust innate immune response that, in the case of S. aureus, is composed primarily of neutrophil influx to the site of infection (14). Each organ presents distinct respiratory challenges due to variations in (i) the amount of available oxygen, (ii) the availability of alternative electron acceptors, (iii) the concentration of respiratory inhibitors, such as nitric oxide produced by neutrophils, and (iv) the availability of cofactors required for energy production. S. aureus is particularly adept at overcoming respiratory challenges given its inherent resistance to nitric oxide and ability to persist within the host as respiratory-deficient small-colony variants (SCVs) (15, 16).
SCVs are characterized by reduced growth and are an etiological agent of persistent staphylococcal infections. The slow growth of S. aureus SCVs is due to genetic mutations that inactivate the biosynthesis of cofactors required for respiration, such as menaquinone or heme (15, 17). The respiration defect in SCVs is thought to lead to an increased ability to resist antibiotic treatment due to loss of the PMF, preventing antibiotic transport across the bacterial membrane. Therefore, antibiotic treatment selects for the formation of SCVs (15, 18, 19). The finding that heme biosynthesis mutants are often isolated from patients experiencing persistent infection suggests that de novo heme biosynthesis plays a significant role in staphylococcal infection. However, the importance of endogenous heme biosynthesis and aerobic respiration to systemic infection has not been defined.
We hypothesize that a heme-dependent branched respiratory chain allows S. aureus to overcome respiratory challenges it encounters during infection. In keeping with this, previous biochemical and bioinformatic analyses suggest that S. aureus encodes two terminal oxidases, one encoded by the cydAB locus and another encoded by the qoxABCD locus (8–10, 16, 20–22). However, our understanding of the terminal oxidases that comprise the electron transport chain of S. aureus and their role during systemic infection is incomplete. In this report, we demonstrate that endogenous heme biosynthesis is required for initiating systemic infection. Furthermore, we establish a requirement for terminal oxidases and aerobic respiration during infection as well as identify the energy-generating pathways that are required for colonization of specific vertebrate organs. Inhibiting respiration with noniron metalloporphyrin heme analogues decreases growth by inducing fermentation, providing evidence that this critical energy-generating process can be targeted by small molecules.
RESULTS
Endogenous heme biosynthesis is required for S. aureus systemic infection.
HemA catalyzes the first committed step of the C5 heme biosynthesis pathway (23, 24). An S. aureus hemA mutant was constructed in order to determine the contribution of heme biosynthesis to systemic infection (Table 1). hemA mutants are SCVs that can be chemically complemented by supplementation of solid growth medium with the universal heme precursor δ-aminolevulinic acid (ALA) or heme (25, 26) (data not shown). Consistent with this, growth of the hemA mutant in liquid medium is reduced compared to that of the wild type (WT), but growth can be restored to WT levels when ALA or heme is added to the medium (Fig. 1A). The hemA growth defect can also be complemented when hemA is expressed in trans from a plasmid (data not shown). Chemical complementation of the hemA growth defect by supplementing the medium with heme supports the idea that the hemA mutant can utilize heme it scavenges from the environment. This finding implies that exogenous heme acquired during infection will support WT-like growth of the hemA mutant within the host. The restoration of the hemA growth defect upon ALA supplementation allows for the comparison of virulence between the hemA and WT strains without the results being confounded by differential in vitro growth kinetics prior to infection. To ascertain the colonization capacity of the hemA mutant in comparison to that of the WT, a murine model of systemic infection was employed. In these experiments, bacteria were rarely recovered from hearts and livers of mice infected with the hemA mutant, compared to considerable colonization in each of these organs by the WT strain (Fig. 1B). In contrast, bacterial burdens were not significantly reduced in the kidney; however, a biphasic distribution of enumerated bacteria suggests that the ability of hemA to establish colonization in this organ is impaired (Fig. 1B). Importantly, bacteria recovered from mice with high burdens of the hemA mutant retained the ALA-responsive SCV phenotype. These data support the conclusion that de novo heme biosynthesis is required for S. aureus to establish colonization of murine hearts and livers.
TABLE 1 .
Strains used in this study
FIG 1 .
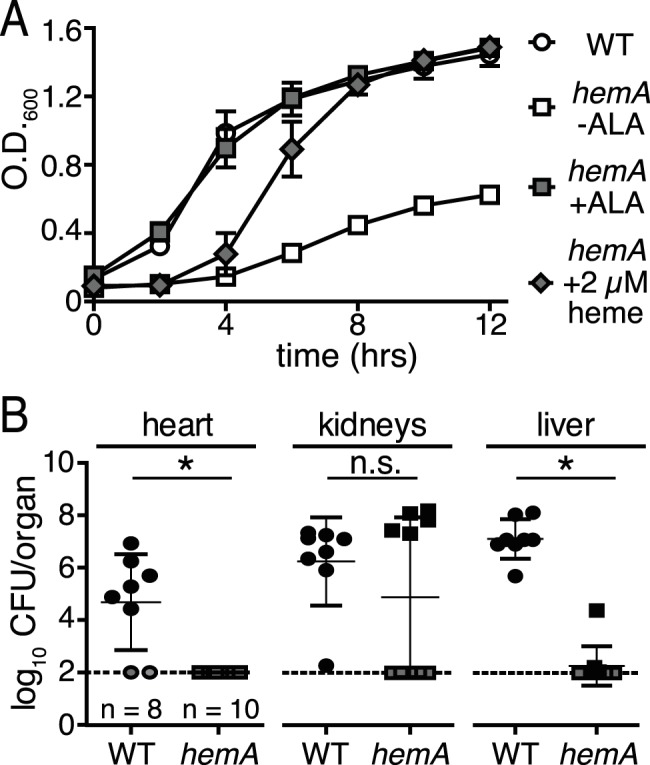
Endogenous heme biosynthesis is required for full S. aureus pathogenesis. (A) The growth of the wild type (WT; circles) or an hemA mutant (open squares) of S. aureus was monitored over time by optical density at 600 nm. The final concentration of aminolevulinic acid (ALA) and heme added to the growth medium was 75 µg ml−1 (gray squares) and 2 µM (gray diamonds), respectively. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (B) Female BALB/c mice were systemically infected with 107 CFU of the wild type (WT; circles) or an hemA mutant (squares) of S. aureus, and bacterial burdens were determined 96 h postinfection. The dotted line represents the limit of detection. Error bars represent one standard deviation from the mean. The mean is represented by a horizontal bar. *, P < 0.0001, via an unpaired Student t test. Gray symbols represent organs where no bacteria were recovered. None of the mice infected with the hemA mutant had recoverable CFU in the heart. Five mice had recoverable CFU in the kidneys and the liver when infected with the hemA mutant.
S. aureus encodes two terminal oxidases that support aerobic growth.
The SCV phenotype results from the inability of S. aureus to aerobically respire (15). Two well-defined S. aureus SCVs, the hemB and menB mutants, are unable to synthesize heme and menaquinone, respectively. These two mutants have significant aerobic growth defects in both liquid and solid media (18, 27) (Fig. 2A; see also Fig. S1 in the supplemental material). Heme is an essential cofactor for terminal oxidases, and menaquinone is the electron carrier utilized by S. aureus (15, 17, 28). Therefore, the reduced growth of the hemB and menB mutants represents a phenotype that should be mimicked by loss of terminal oxidases. Previous biochemical analyses and a genomic search for terminal oxidase homologues revealed that S. aureus contains two terminal oxidases, an annotated cytochrome bd encoded by cydBA and an annotated cytochrome aa3 encoded by qoxABCD (9, 10, 21). qoxB and cydB mutants were created and used to assess the contribution of these cytochromes to staphylococcal growth (Table 1). The cydB mutant exhibits no growth defect when cultured in liquid medium (Fig. 2A). In contrast, the qoxB mutant is slightly reduced for aerobic growth, suggesting that it is the dominant terminal oxidase utilized under these conditions. When grown on solid medium, the colony morphologies of both mutant strains are similar to that of the WT (see Fig. S1) (20).
FIG 2 .
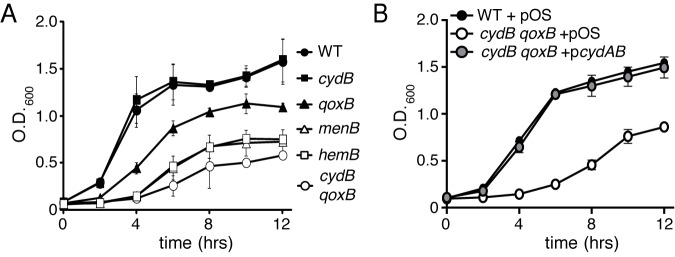
Inactivation of cydB and qoxB reduces the growth of S. aureus. (A) Wild-type (WT) and respective mutant strains of S. aureus were grown overnight, subcultured into fresh medium, and incubated at 37°C. The growth of the strains was monitored over time by measuring optical density at 600 nm. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (B) Wild-type (WT) S. aureus and the qoxB cydB mutant harboring a plasmid control (pOS) or a plasmid containing the cydAB operon (pcydAB) were grown overnight, subcultured into fresh medium, and incubated at 37°C. The growth of the strains was monitored over time by measuring optical density at 600 nm. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean.
A cydB qoxB double mutant was constructed in order to determine the importance of both terminal oxidases to staphylococcal aerobic growth. This mutant is significantly reduced for growth in liquid medium compared to that of either of the representative single mutants (Fig. 2A). The growth reduction of the cydB qoxB mutant is comparable to that of the respiratory-deficient menB and hemB mutants (Fig. 2A). When grown on solid medium, the cydB qoxB mutant exhibits an SCV phenotype, suggesting that this mutant is unable to aerobically respire (see Fig. S1A).
In order to determine if a single terminal oxidase is sufficient for WT growth, a cydB qoxB strain harboring a plasmid providing the constitutive expression of the cydAB operon was constructed (pcydAB). The cydB qoxB mutant containing pcydAB had growth comparable to that of the WT, but the cydB qoxB mutant harboring a vector control was significantly reduced for growth (Fig. 2B; see also Fig. S1B). These data demonstrate that the activity of either cytochrome is sufficient to support aerobic growth of S. aureus.
The terminal oxidases of S. aureus are required for aerobic respiration.
The reduced growth phenotype of the cydB qoxB mutant suggests that aerobic respiration is inhibited in these cells. In this case, the capacity of cydB qoxB to generate a PMF should be impaired, and this strain should be limited to fermentation to produce energy and maintain redox balance. The ability of these strains to generate a component of the PMF, the membrane potential, was assessed using the fluorescent dye 3′3′-diethyloxacarbocyanine iodide (DiOC2). When bacteria generate a membrane potential, DiOC2 accumulates intracellularly and self-associates, resulting in a shift from emitting green fluorescence to emitting red fluorescence. Cells unable to generate a membrane potential accumulate less dye and emit green fluorescence (29). A shift to the red spectrum, indicative of the presence of a membrane potential, was observed when the WT and cydB and qoxB mutant strains were grown to mid-log phase and incubated in the presence of DiOC2 (Fig. 3A). No shift was observed when the menB, hemB, or cydB qoxB mutant strains were grown in similar conditions, indicating that these strains are significantly impaired for the ability to generate a membrane potential (Fig. 3A). To test the hypothesis that the terminal oxidase cydB qoxB double mutant is fermenting in order to support growth, the amount of l- and d-lactate that accumulated in the supernatant when S. aureus was grown to stationary phase was measured. A significant increase in the amount of both isomers of lactate was observed in the supernatant of the cydB qoxB double mutant strain compared to that of the WT and cydB or qoxB single-mutant strain (Fig. 3B). The amounts of l- and d-lactate produced by cydB qoxB were comparable to either the menB or hemB respiratory-deficient mutants. Expression of the cydAB operon from a plasmid is sufficient to reverse the loss of a membrane potential and reduce the amount of l- and d-lactate produced by the cydB qoxB double mutant (Fig. 3C and D). These results support the conclusion that cytochromes encoded by the qox and cyd operons are the exclusive terminal oxidases utilized by S. aureus to generate a membrane potential and facilitate aerobic respiration.
FIG 3 .

Inactivation of both cydB and qoxB limits the metabolic flexibility of S. aureus. (A) The membrane potential was measured as the mean ratio of red/green fluorescence of wild-type (WT) and mutant strains of S. aureus grown to mid-exponential phase and incubated with 30 µM of the dye 3′3′-diethyloxacarbocyanine iodide (DiOC2). The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (B) l-Lactate and d-lactate production in aerobically respiring and fermenting strains of S. aureus after 15 h of growth. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (C) The membrane potential of wild-type (WT) S. aureus and the qoxB cydB mutant harboring a plasmid control (pOS) or a plasmid containing the cydAB operon (pcydAB) was measured as the mean ratio of red/green fluorescence when the strains were grown to mid-exponential phase and incubated with 30 µM of the dye 3′3′-diethyloxacarbocyanine iodide (DiOC2). The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (D) l-Lactate and d-lactate produced in the supernatants of wild-type (WT) S. aureus and the qoxB cydB mutant harboring a plasmid control (pOS) or a plasmid containing the cydAB operon (pcydAB) grown after 12 h of growth. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean.
Branched-chain electron transport is required for full staphylococcal virulence.
Each vertebrate organ presents a unique metabolic challenge to bacterial colonization, and given the reduced colonization of the hemA mutant, a heme-dependent branched respiratory chain may allow S. aureus to meet and overcome these challenges. Therefore, the ability of the cydB or qoxB strains to colonize the heart, kidneys, and liver was evaluated using a murine model of systemic infection. Mice were infected with WT S. aureus or each of the single terminal oxidase mutants, cydB or qoxB. Bacterial burdens were decreased in the heart by approximately 2 logs following challenge with cydB, whereas bacterial burdens in the liver were decreased by approximately 4 logs when qoxB was inactivated (Fig. 4). The liver colonization defect of qoxB is consistent with previously published studies (20). The in vitro growth defect of qoxB does not explain the liver colonization defect, as this mutant colonizes the heart and kidneys to levels comparable to those of the WT (Fig. 4). The differential colonization of cydB and qoxB is also consistent with the hemA infection data, which demonstrate reduced bacterial loads in both the heart and liver (Fig. 1B). These data support the idea that the cytochromes have distinct roles during pathogenesis and are not functionally redundant. These findings also suggest that targeting terminal oxidases might be a viable antimicrobial strategy.
FIG 4 .
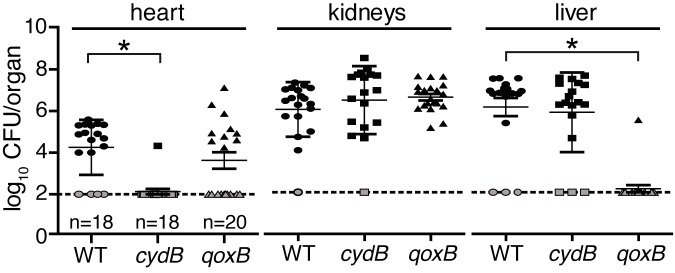
Two terminal oxidases power organ-specific colonization of the host. Female BALB/c mice were systemically infected with 107 CFU of the wild type (WT; circles) or cydB (squares) or qoxB (triangles) mutants of S. aureus, and bacterial burdens were determined 96 h postinfection. The dotted line represents the limit of detection. Error bars represent one standard deviation from the mean. The mean is represented by a horizontal bar. *, P < 0.0001, via an unpaired Student t test. Gray symbols represent organs where no bacteria were recovered. One mouse had recoverable CFU in the heart when infected with the cydB mutant, and one mouse had recoverable CFU in the liver when infected with the qoxB mutant.
Noniron metalloporphyrins restrict the metabolic flexibility of S. aureus by inhibiting aerobic respiration.
Previous reports have demonstrated that noniron metalloporphyrins inhibit the growth of many species of bacteria in iron-limited conditions (30). It has been proposed that the mechanism by which these molecules exert their antimicrobial activity is through incorporation into cytochromes, resulting in respiration arrest (30). In iron-replete conditions, S. aureus traffics heme (iron-protoporphyrin IX) to the plasma membrane (31). In similar growth conditions, increasing concentrations of either gallium protoporphyrin (GaPPIX) or zinc protoporphyrin (ZnPPIX) significantly reduces S. aureus growth and induces the SCV phenotype (Fig. 5A; see also Fig. S2). Analogous to observations in Gram-negative bacteria, GaPPIX and ZnPPIX toxicity in S. aureus is more potent in aerobic growth conditions (Fig. 5B) and when iron is depleted from the growth medium (Fig. 5C) (30). A mutant inactivated for heme transport through the iron-regulated surface determinant system (isdC) does not abrogate the toxicity of GaPPIX or ZnPPIX in iron-replete or iron-deplete growth conditions (data not shown).
FIG 5 .

Noniron metalloporphyrins inhibit staphylococcal growth. (A) Growth of S. aureus in the presence of increasing concentrations of GaPPIX or ZnPPIX. The growth of the strains was monitored over time by measuring optical density at 600 nm. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (B) Aerobic (+O2) or anaerobic (−O2) growth of S. aureus in the presence of increasing concentrations of GaPPIX or ZnPPIX. Growth was monitored as the optical density at 12 h and compared to that of cells grown in the absence of the noniron PPIXs. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (C) S. aureus was grown in the absence or presence of 1 mM 2,2′-dipyridyl (±DIP) and 0.1 µM GaPPIX or ZnPPIX. Growth was monitored as the optical density at 12 h and compared to that of cells grown in the absence of the noniron PPIXs. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean.
To determine the localization of GaPPIX within the cell, inductively coupled plasma mass spectrometry was employed, and the localization of the gallium atom in cell fractions of S. aureus was measured when the bacteria were exposed to either GaPPIX or Ga(NO3)3. Greater than 50% of total gallium is found in the plasma membrane fraction when S. aureus is grown in the presence of GaPPIX (Fig. 6A). However, the majority of the gallium localizes to the cell wall when S. aureus is grown in the presence of Ga(NO3)3 (Fig. 6A). These results demonstrate that when gallium is complexed within a protoporphyrin ring, it is preferentially trafficked to the plasma membrane.
FIG 6 .

GaPPIX and ZnPPIX inhibit S. aureus aerobic respiration and are trafficked to the plasmid membrane. (A) ICP-MS tracking of GaPPIX in S. aureus. Percentage of total gallium atoms measured in the cell wall (W), membrane (M), and cytoplasmic (C) fractions. Error bars represent the standard deviation from three independent samples. Asterisks represent statistically significant differences between GaPPIX and Ga(NO3)3 localization, as determined by a Student t test (*, P < 0.02; **, P < 0.05). (B) Mean ratio of red/green fluorescence of S. aureus grown to mid-exponential phase and incubated with 30 µM of the dye 3′3′-diethyloxacarbocyanine iodide (DiOC2). The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (C) Measurement of d- and l-lactate in the supernatants of S. aureus grown in the presence of GaPPIX or ZnPPIX after 15 h of growth. The amount of d- and l-lactate accumulated in the supernatants of anaerobically grown S. aureus is also presented for comparison. The average from three independent experiments is shown. Error bars represent one standard deviation from the mean. (D) ICP-MS tracking of GaPPIX in S. aureus (WT) and the isogenic cydB qoxB mutant. Percentage of total gallium atoms measured in the cell wall (W), membrane (M), and cytoplasmic (C) fractions. Error bars represent the standard deviation from three independent samples. Asterisks represent statistically significant differences between GaPPIX and Ga(NO3)3 localization, as determined by a Student t test (*, P < 0.003).
The growth reduction in the presence of GaPPIX and ZnPPIX combined with the plasma membrane localization of GaPPIX suggests that these noniron metalloporphyrins are inhibiting aerobic respiration. Consistent with this, the ability of S. aureus to generate a membrane potential is reduced when the bacteria are exposed to GaPPIX or ZnPPIX (Fig. 6B). This collapse in membrane potential was associated with a concomitant increase in the amount of both l- and d-lactate measured in the supernatant of bacteria exposed to increasing concentrations of both GaPPIX and ZnPPIX (Fig. 6C). These results support the hypothesis that the noniron metalloporphyrins incorporate into the cytochromes, whereby they inhibit aerobic respiration and force S. aureus to ferment in order to support growth. Consistent with this hypothesis, we observed an increase in the percentage of GaPPIX found within the cell wall fraction of the cydB qoxB mutant, implying that trafficking of noniron metalloporphyrins to the membrane is impaired in this strain (Fig. 6D). Given the attenuation of the cydB and qoxB mutants in the systemic model of infection, these findings establish small-molecule targeting of the terminal oxidases as a valid strategy to develop antimicrobials to treat S. aureus infections.
DISCUSSION
In this report, we demonstrate that heme-dependent aerobic growth of S. aureus is critical for pathogenesis. Endogenous heme powers a branched respiratory chain that utilizes two terminal oxidases. Limiting the staphylococcal respiratory chain to a single terminal oxidase impairs the infectious capacity of this pathogen. Inactivation of heme biosynthesis further exacerbates S. aureus infection of the host, as measured by a decrease in liver and heart colonization. Branched respiratory chains ensure efficient energy production in diverse aerobic environments. This feature of branched respiratory chains is particularly important for bacterial pathogens that experience rapid changes in the host environment during infection. For example, Mycobacterium tuberculosis differentially regulates components of a branched respiratory chain in order to adapt to the host immune response. In the transition from acute to chronic infection, M. tuberculosis concomitantly downregulates cytochrome aa3 and upregulates cytochrome bd (4). The shift to cytochrome bd-mediated respiration facilitates energy generation in the presence of increased nitric oxide and decreased oxygen levels. Consistent with these findings, M. tuberculosis cytochrome bd mutants are not able to establish persistent lung infections (4). Inactivation of cytochrome bd also has deleterious effects in an Escherichia coli murine intestine colonization model and leads to attenuation of Shigella flexneri virulence (32, 33). These results highlight the importance of cytochrome bd to bacterial colonization of the host and provide further evidence that targeting the flexibility of aerobic respiration is a potent strategy for combating a variety of bacterial infections.
It should be noted that the cytochromes encoded by cydAB and qoxABCD are annotated as a cytochrome bd and a cytochrome aa3, respectively. The name given to a cytochrome is determined by its spectral signature, which is dependent upon the heme cofactor bound to the cytochrome. Therefore, a cytochrome bd is predicted to utilize a heme b and heme d, whereas a cytochrome aa3 is predicted to use two heme a cofactors. However, elegant biochemical experiments have demonstrated that a cytochrome bo and a cytochrome ba3 are the predominant terminal oxidases in S. aureus, suggesting that the cydAB and qoxABCD operons are incorrectly annotated (9, 10, 21, 22). In support of this, the S. aureus CydA and CydB share limited homology with CydA and CydB from other bacterial species (21). Our work demonstrates that the structural components encoded by cydAB and qoxABCD are the exclusive terminal oxidases in S. aureus but highlights the need for biochemical analysis of the cydB and qoxB mutants in order to determine the heme moieties that are found within these cytochromes.
The differential colonization of host organs by S. aureus single terminal oxidase mutants underscores the importance of a branched respiratory chain to staphylococcal pathogenesis. Despite the fact that both the cydB mutant and the qoxB mutant are capable of aerobic respiration, these two strains differ significantly in their capacity to colonize the host. Several factors likely represent barriers to colonization in each organ. One of these factors is the ability to generate energy in the presence of a robust innate immune response. Neutrophil recruitment to the site of infection leads to a burst in the respiration inhibitor nitric oxide. Upon exposure to nitric oxide, S. aureus increases the expression of cydAB (16). In Escherichia coli, a cytochrome bd facilitates respiration in low-oxygen environments containing nitric oxide due to its high affinity for oxygen and rapid nitric oxide dissociation rate (34). The biophysical properties of the staphylococcal terminal oxidases and the heme moieties that complex with each cytochrome have yet to be elucidated; however, it is tempting to speculate that one mechanism by which S. aureus resists neutrophil-mediated killing is by rerouting electrons to a terminal oxidase that is functional in the presence of nitric oxide.
The lack of a functional terminal oxidase leads to an inability to respire and a severe growth defect in vitro known as the SCV phenotype. These strains are clinically relevant given their ability to cause persistent infections and to resist to antimicrobial therapy (15). The virulence capacity of the hemB and menB SCVs has previously been tested in a rabbit endocarditis model of infection. Results from this study demonstrated that both menaquinone and heme biosynthesis are dispensable for colonizing the heart valves in rabbits. Interestingly, the menB mutant strain persistently colonized the kidney in this model (35). In our study, the kidney is the only organ where the respiratory-arrested hemA mutant and both the cydB and qoxB mutants colonize to appreciable levels. Consistent with this, mutants of S. aureus that are unable to reduce pyruvate to l-lactate are reduced in their ability to form renal abscesses and are outcompeted by WT S. aureus in this organ (36). These findings support a model in which aerobic-independent metabolic pathways facilitate colonization of the kidney. Given the ability of S. aureus to fine-tune its aerobic respiration to overcome challenges presented by the host, limiting the metabolic flexibility of S. aureus has the potential to promote host clearance of this microbial threat in an organ-specific manner.
GaPPIX and ZnPPIX are examples of small molecules that inhibit staphylococcal aerobic respiration and restrict the energy-generating pathways available to this pathogen. GaPPIX and ZnPPIX inhibit the generation of a membrane potential and force S. aureus to generate energy via fermentation. The ability of these noniron metalloporphyrins to inhibit aerobic respiration suggests that these molecules exert their effect by incorporating into the terminal oxidases (30). Interestingly, chemical inhibition of aerobic respiration via the noniron metalloporphyrins results in an increased ratio of l-lactate to d-lactate compared to the genetic inactivation of aerobic respiration (Fig. 6C). In SCVs, the amounts of l-lactate and d-lactate produced are comparable (Fig. 3B). A similar result is observed when S. aureus is exposed to nitric oxide, suggesting that chemical inhibition of aerobic respiration alters the fermentative activity of this pathogen (36).
Previous work has demonstrated that the acquisition of host heme is essential for S. aureus pathogenesis and is the preferred source of nutrient iron (31). Under iron-replete conditions, exogenous heme is trafficked to the membrane (31). Given the finding that the heme analogue GaPPIX is also trafficked to the membrane, we propose a model in which exogenously acquired heme is transported to the terminal oxidases in order to support aerobic respiration during infection of host tissues when an alternative iron source is present.
Due to the clinical significance of S. aureus and the high prevalence of antibiotic-resistant strains, new therapeutics are required to combat this pathogen. The metabolic pathways that support staphylococcal growth within the host are beginning to be elucidated, and these pathways may prove to be candidate targets for therapeutic intervention. Heme-dependent aerobic respiration is one pathway that is required for full S. aureus pathogenesis, and it is inhibited by the noniron metalloporphyrins GaPPIX and ZnPPIX. However, respiration-impaired SCVs are etiological agents of persistent infections, suggesting that therapeutic intervention that induces SCVs may result in a more insidious disease state (27, 37). Therefore, small molecules that are toxic to SCVs could provide novel dual-treatment therapies that would also be efficacious for people suffering from persistent infection (38). SCVs and small molecules that induce the SCV phenotype provide a discovery platform for these potential alternative strategies (37). Given the ability of many microbial pathogens to develop drug-resistant persistent subpopulations, the elucidation of the metabolic pathways that sustain the adaptation to the persistent phenotype will aid in the development of treatments for persistent infections (39).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in this study are described in Table 1. All strains are derivatives of the human clinical isolate, S. aureus Newman (40). The hemA, menB, and qoxB isogenic in-frame deletion mutants were constructed using previously described methods (41). The cydB::erm mutant was isolated from a Tn917 transposon insertion library of S. aureus Newman. PCR revealed that the Tn917 inverted repeat sequence mapped adjacent to the 1,058,914 bp of the S. aureus Newman genome in the cydB mutant. This is 65 bp downstream from the putative cydB start codon. A backcross of the erythromycin cassette-disrupted cydB allele into WT S. aureus Newman was produced via phage Φ85-mediated transduction (42). The backcrossed cydB::erm strain was used in the studies described here. Phage Φ85-mediated transduction of the cydB erythromycin cassette-disrupted allele was also used to construct the cydB qoxB double terminal oxidase mutant. The cydAB operon was PCR amplified and cloned into the pOS1 Plgt plasmid (43) using primers 5′ GCGCTCGAGATGGATACAGTTGAAATCAGTC 3′ and 5′ GCGGGATCCTTATGATTTCTTTCCTTCAACATA 3′. An S. aureus 8325-4 hemB::erm mutant was kindly provided by the Friedrich Götz laboratory (18). Transduction of the hemB::erm mutant allele into wild-type S. aureus Newman was mediated by phage Φ85 (42). Antibiotic selection of erythromycin cassette-containing resistant recipient cells was achieved with 10 µg ml−1 erythromycin. Antibiotic selection of strains containing the pOS plasmid was achieved by using 10 µg ml−1 of chloramphenicol. Bacteria were routinely grown in Trypticase soy broth (TSB) at 37°C. All strains were diluted 1:100 from overnight cultures into fresh TSB-containing 96-well round-bottom plates, and growth was measured by optical density at 600 nm. Stock solutions of porcine hemin (Sigma), gallium protoporphyrin (GaPPIX), and zinc protoporphyrin (ZnPPIX) (Frontier Scientific) were resuspended to a final concentration of 10 mM in dimethyl sulfoxide (DMSO) and used at the indicated concentrations. Growth experiments were conducted in the absence of light to reduce the exposure of the cells to the protoporphyrin-induced photodynamic effect. ALA was supplemented to the growth medium at a final concentration of 75 µg ml−1. Anaerobiosis was achieved by growing the bacteria in 15-ml conical tubes containing an abundance of degassed TSB. The tubes were then placed within a growth receptacle containing an AnaeroGen compact anaerobic atmosphere-generating pouch (Oxoid Ltd., England) and were incubated at 37°C. Anaerobic indicator strips (Becton, Dickson and Company) were used to ensure that anaerobiosis was achieved.
Measurement of the membrane potential.
The BacLight bacterial membrane potential kit (Invitrogen) was used to monitor the membrane potential of S. aureus mutants. Mid-exponential bacterial suspensions were added to a 1-ml phosphate-buffered saline (PBS) solution containing 30 µM DiOC2. After a 30-minute incubation at 37°C, samples were analyzed by the Vanderbilt Flow Core. For each sample, the membrane decoupler, CCCP (5 µM), was used as a positive control for loss of the membrane potential.
Lactate production.
Bacterial strains were subcultured 1:100 from cultures grown overnight into fresh TSB. These cultures were grown for 15 h at 37°C. The cells were pelleted and the supernatant was collected. The amount of l- and d-lactate in the culture supernatants was analyzed using the commercially available Boehringer Mannheim enzymatic UV method (Roche).
Systemic mouse infections.
Eight-week-old female BALB/c mice were retro-orbitally infected with 107 CFU of bacteria resuspended in PBS. After 96 h of infection, animals were sacrificed and the heart, kidneys, and liver were harvested and homogenized in 1 ml PBS. Bacterial burdens within each organ were quantified by counting CFU obtained from serial dilutions of the organ homogenate. Infections were performed at the Vanderbilt University Medical Center under the principles and guidelines described in the Guide for the Care and Use of Laboratory Animals. Vanderbilt University Medical Center is an American Association for Laboratory Animal Science (AALAS)-accredited facility using Institutional Animal Care and Use Committee (IACUC)-approved (7 September 2012) protocol M10165. The Vanderbilt University Medical Center is registered with the Office of Laboratory Animal Welfare (OLAW), assurance number A-3227-01.
Inductively coupled plasma mass spectrometry (ICP-MS) tracking.
Teflon sample collection tubes were washed in 6 N HCl for 2 weeks and then washed at least 5 times with deionized water to remove all metals. Overnight cultures of S. aureus grown in TSB medium were washed and normalized by optical density. Bacteria were then subcultured into medium containing 2 µM GaPPIX or 2 µM GaNO3 and were incubated at 37°C for 4 h. The cultures were then normalized and pelleted. The supernatants were removed and concentrated at room temperature by vacuum. The pellets were then washed three times in Tris-buffered saline (50 mM Tris base, 150 mM NaCl, pH 7.5) and transferred to flat-bottom Teflon tubes. The cell wall was removed by lysostaphin (0.1 mg) treatment in Tris sucrose magnesium buffer (50 mM Tris base, 500 mM sucrose, and 10 mM MgCl2, pH 7.5), followed by pelleting. The cell wall fraction (supernatant) was transferred to Teflon tubes, while the pellets were resuspended in Tris-buffered saline and sonicated, followed by ultracentrifugation (100,000 × g) for 1 h. The cytoplasmic fractions (supernatant) were transferred to clean Teflon tubes, and the membrane fractions (pellets) were resuspended in Tris-buffered saline. Nitric acid (1 ml; Optima) and hydrogen peroxide (100 µl) were added to each Teflon tube, and samples were boiled at 130°C until dry. Dried samples were sent for analysis by ICP-MS at Applied Speciation and Consulting (Bothell, WA).
Statistical analyses.
Statistical significance was determined with the Student t test (GraphPad Prism). Results were considered significant if the P value was less than 0.05.
SUPPLEMENTAL MATERIAL
The cydB qoxB mutant is a small-colony variant. (A) Overnight cultures of the respective strains were grown in TSB and streaked for isolated colonies. (B) Overnight cultures of wild-type (WT) S. aureus and the qoxB cydB mutant harboring a plasmid control (pOS) or a plasmid containing the cydAB operon (pcydAB) were grown in TSB containing chloramphenicol and streaked for isolated colonies on agar plates also containing chloramphenicol. Pictures were recorded after 48 h of growth at 37°C. Download
The noniron protoporphyrins, GaPPIX and ZnPPIX, induce the SCV phenotype. Overnight cultures of the respective strains were grown in TSB and streaked for isolated colonies on TSA plates or TSA plates supplemented with 10 µM ZnPPIX or GaPPIX. Growth was recorded after 15 h at 37°C. Download
ACKNOWLEDGMENTS
We thank members of the Skaar laboratory for critically evaluating the manuscript.
Work in the Skaar laboratory is supported by NIH grants AI0169233, AI073843, and AI057157. E.P.S. is a Burroughs Wellcome Fund Investigator in the pathogenesis of Infectious Diseases. N.D.H. was supported by National Institutes of Health Ruth L. Kirschstein fellowship F32-AI091244-01 and is currently a Cystic Fibrosis Foundation Ann Weinberg Memorial Research Fellow (HAMMER13QI0). Flow cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485).
Footnotes
Citation Hammer ND, Reniere ML, Cassat JE, Zhang Y, Hirsch AO, Indriati Hood M, Skaar EP. 2013. Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. mBio 4(4):e00241-13. doi:10.1128/mBio.00241-13.
REFERENCES
- 1. Kuehnert MJ, Kruszon-Moran D, Hill HA, McQuillan G, McAllister SK, Fosheim G, McDougal LK, Chaitram J, Jensen B, Fridkin SK, Killgore G, Tenover FC. 2006. Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis. 193:172–179 [DOI] [PubMed] [Google Scholar]
- 2. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763
- 3. Kuehnert MJ, Hill HA, Kupronis BA, Tokars JI, Solomon SL, Jernigan DB. 2005. Methicillin-resistant-Staphylococcus aureus hospitalizations, United States. Emerg. Infect. Dis. 11:868–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. U. S. A. 102:15629–15634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Bäumler AJ. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neidhardt FC, Curtiss R. 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 7. White D, Drummond JT, Fuqua C. 2012. The physiology and biochemistry of prokaryotes, 4th ed. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 8. Frerman FE, White DC. 1967. Membrane lipid changes during formation of a functional electron transport system in Staphylococcus aureus. J. Bacteriol. 94:1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taber HW, Morrison M. 1964. Electron transport in staphylococci. Properties of a particle preparation from exponential phase Staphylococcus aureus. Arch. Biochem. Biophys. 105:367–379 [DOI] [PubMed] [Google Scholar]
- 10. Tynecka Z, Szcześniak Z, Malm A, Los R. 1999. Energy conservation in aerobically grown Staphylococcus aureus. Res. Microbiol. 150:555–566 [DOI] [PubMed] [Google Scholar]
- 11. Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. 2011. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807:1398–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iuchi S, Weiner L. 1996. Cellular and molecular physiology of Escherichia coli in the adaptation to aerobic environments. J. Biochem. 120:1055–1063 [DOI] [PubMed] [Google Scholar]
- 13. Poole RK, Cook GM. 2000. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 43:165–224 [DOI] [PubMed] [Google Scholar]
- 14. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 15. Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4:295–305 [DOI] [PubMed] [Google Scholar]
- 16. Richardson AR, Dunman PM, Fang FC. 2006. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol. Microbiol. 61:927–939 [DOI] [PubMed] [Google Scholar]
- 17. Hammond RK, White DC. 1969. Formation of vitamin K2 isoprenologues by Staphylococcus aureus. J. Bacteriol. 100:573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. von Eiff C, Heilmann C, Proctor RA, Woltz C, Peters G, Götz F. 1997. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J. Bacteriol. 179:4706–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Eiff C, McNamara P, Becker K, Bates D, Lei XH, Ziman M, Bochner BR, Peters G, Proctor RA. 2006. Phenotype microarray profiling of Staphylococcus aureus menD and hemB mutants with the small-colony-variant phenotype. J. Bacteriol. 188:687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lan L, Cheng A, Dunman PM, Missiakas D, He C. 2010. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J. Bacteriol. 192:3068–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voggu L, Schlag S, Biswas R, Rosenstein R, Rausch C, Götz F. 2006. Microevolution of cytochrome bd oxidase in staphylococci and its implication in resistance to respiratory toxins released by Pseudomonas. J. Bacteriol. 188:8079–8086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clements MO, Watson SP, Poole RK, Foster SJ. 1999. CtaA of Staphylococcus aureus is required for starvation survival, recovery, and cytochrome biosynthesis. J. Bacteriol. 181:501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anzaldi LL, Skaar EP. 2010. Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect. Immun. 78:4977–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panek H, O’Brian MR. 2002. A whole genome view of prokaryotic haem biosynthesis. Microbiology 148:2273–2282 [DOI] [PubMed] [Google Scholar]
- 25. Petricek M, Rutberg L, Schröder I, Hederstedt L. 1990. Cloning and characterization of the hemA region of the Bacillus subtilis chromosome. J. Bacteriol. 172:2250–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wulff DL. 1967. Delta-aminolevulinic acid-requiring mutant from Escherichia coli. J. Bacteriol. 93:1473–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Proctor RA. 2000. Respiration and small colony variants of Staphylococcus aureus, p 345–350 In Vincent A, Fischetti RPN, Ferretti JJ, Portnoy DA, Rood JI, Gram-positive pathogens: staphylococci. American Society for Microbiology Press, Washington, DC [Google Scholar]
- 28. Wakeman CA, Hammer ND, Stauff DL, Attia AS, Anzaldi LL, Dikalov SI, Calcutt MW, Skaar EP. 2012. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol. Microbiol. 86:1376–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sims PJ, Waggoner AS, Wang CH, Hoffman JF. 1974. Studies on the mechanism by which cyanine dyes measure membrane potential in red blood cells and phosphatidylcholine vesicles. Biochemistry 13:3315–3330 [DOI] [PubMed] [Google Scholar]
- 30. Stojiljkovic I, Kumar V, Srinivasan N. 1999. Non-iron metalloporphyrins: potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 31:429–442 [DOI] [PubMed] [Google Scholar]
- 31. Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. 2004. Iron-source preference of Staphylococcus aureus infections. Science 305:1626–1628 [DOI] [PubMed] [Google Scholar]
- 32. Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration of Escherichia coli in the mouse intestine. Infect. Immun. 75:4891–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Way SS, Sallustio S, Magliozzo RS, Goldberg MB. 1999. Impact of either elevated or decreased levels of cytochrome bd expression on Shigella flexneri virulence. J. Bacteriol. 181:1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mason MG, Shepherd M, Nicholls P, Dobbin PS, Dodsworth KS, Poole RK, Cooper CE. 2009. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat. Chem. Biol. 5:94–96 [DOI] [PubMed] [Google Scholar]
- 35. Bates DM, von Eiff C, McNamara PJ, Peters G, Yeaman MR, Bayer AS, Proctor RA. 2003. Staphylococcus aureus menD and hemB Mutants are as infective as the parent strains, but the menadione biosynthetic mutant persists within the kidney. J. Infect. Dis. 187:1654–1661 [DOI] [PubMed] [Google Scholar]
- 36. Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676 [DOI] [PubMed] [Google Scholar]
- 37. Biswas L, Biswas R, Schlag M, Bertram R, Götz F. 2009. Small-colony variant selection as a survival strategy for Staphylococcus aureus in the presence of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 75:6910–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mike LA, Dutter BF, Stauff DL, Moore JL, Vitko NP, Aranmolate O, Kehl-Fie TE, Sullivan S, Reid PR, Dubois JL, Richardson AR, Caprioli RM, Sulikowski GA, Skaar EP. 2013. Activation of heme biosynthesis by a small molecule that is toxic to fermenting Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 110:8206–8211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372 [DOI] [PubMed] [Google Scholar]
- 40. Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6:95–107 [DOI] [PubMed] [Google Scholar]
- 41. Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63 [DOI] [PubMed] [Google Scholar]
- 42. Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. 2003. Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299:906–909 [DOI] [PubMed] [Google Scholar]
- 43. Schneewind O, Model P, Fischetti VA. 1992. Sorting of protein A to the staphylococcal cell wall. Cell 70:267–281 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cydB qoxB mutant is a small-colony variant. (A) Overnight cultures of the respective strains were grown in TSB and streaked for isolated colonies. (B) Overnight cultures of wild-type (WT) S. aureus and the qoxB cydB mutant harboring a plasmid control (pOS) or a plasmid containing the cydAB operon (pcydAB) were grown in TSB containing chloramphenicol and streaked for isolated colonies on agar plates also containing chloramphenicol. Pictures were recorded after 48 h of growth at 37°C. Download
The noniron protoporphyrins, GaPPIX and ZnPPIX, induce the SCV phenotype. Overnight cultures of the respective strains were grown in TSB and streaked for isolated colonies on TSA plates or TSA plates supplemented with 10 µM ZnPPIX or GaPPIX. Growth was recorded after 15 h at 37°C. Download