

Abstract
Prostate cancer (PCa) preferentially metastasizes to the bone marrow stroma of the axial skeleton. This activity is the principal cause of PCa morbidity and mortality. The exact mechanism of PCa metastasis is currently unknown, although considerable progress has been made in determining the key players in this process. In this review, we present the current understanding of the molecular processes driving PCa metastasis to the bone.
Keywords: bone, bone marrow stroma, metastasis, prostate cancer
Metastatic mechanisms in the primary tumour
The principal problem arising from prostate cancer (PCa) is its propensity to metastasize. This tendency arises from specific molecular mechanisms and interactions that together lead to local invasion, extravasation and distal migration from the primary site, followed by endothelial attachment, transmigration and site-specific establishment of metastases at secondary sites. Basic knowledge related to this structured process has improved recently, but many of the key elements are still poorly understood.
Local invasion is one of the fundamental early steps in metastasis, as without it tumour spread cannot occur. To develop invasive potential, the malignant cell must down-regulate its cell–cell and cell–matrix adhesive characteristics, become motile and acquire the ability to break down the extracellular matrix (ECM) using degradative enzymes1. Once the malignant cell has reached the interstitium, it must enter the vascular or lymphatic circulation by breaching the endothelial barriers. From there, the cell must migrate via the blood or lymphatic circulation and arrest at a secondary endothelial site before binding to the endothelium, extravasating and transmigrating through the endothelial layer to reach the interstitium, where it proliferates and/or coalesces with other metastasized cells to form a micro-metastasis (Figure 1)2. It will do this only if the environment at the secondary site is favourable.
Figure 1.
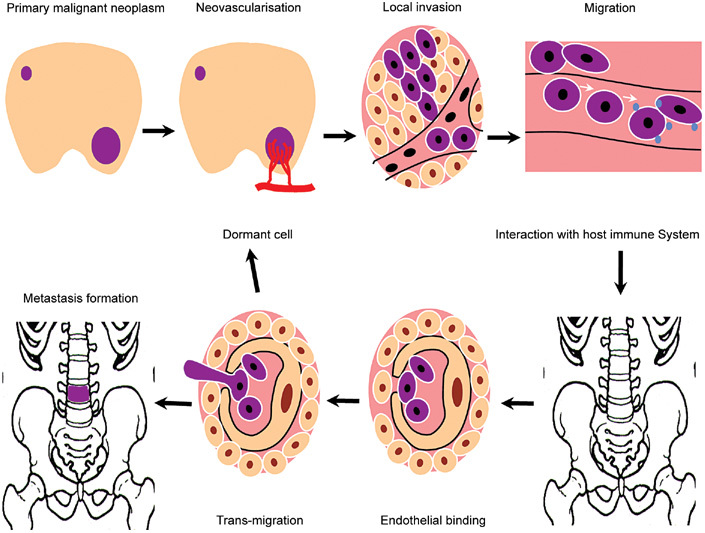
Metastasis is characterized by proliferation, neovascularization and extravasation at the primary site. In the circulation, malignant cells interact with the host immune system, typically resulting in cancer cell destruction or apoptosis. Surviving cells arrest at secondary endothelial sites by a process of lectin binding consolidated by integrin-based stabilization of the epithelial–endothelial binding. The cell then undergoes active transmigration. The binding process is complete within 30–60 min and transmigration within 24 h. Once the cell reaches the interstitium, it may remain dormant for an undefined period or it may coalesce with other cells and proliferate to form a metastatic colony. This will then disturb local physiological function, leading to physiological dysfunction and anatomical disruption. Any metastatic site may produce further metastases (Reproduced with permission from2).
Primary site cell–cell adhesion
Maintenance of organic architecture depends on cell-cell and cell-matrix binding. In the prostate and other structures, a key cell–cell binding regulator is the cadherin–catenin complex, whereas cell–matrix binding is largely mediated by integrins, dimeric binding proteins comprising α-and β-chain subunits.
Cadherins are transmembrane glycoproteins, of which E-cadherin is the best characterized in PCa. It serves critical functions during embryogenesis and organogenesis through intercellular adhesion and signaling3. The locus coding for E-cadherin (16q22.1) is considered to be a tumour-suppressor gene; loss of function enables cell detachment and induces an invasive phenotype4, whereas transfection of E-cadherin complementary DNA (cDNA) into invasive adenocarcinoma cells renders them non-invasive5, 6.
E-cadherin is attached intracellularly to the actin cytoskeleton via intracellular catenin. Once anchored, the transmembrane cadherins bind through their external domains to the binding sites of other cadherins on adjacent cells. The cadherin–catenin complex is essential for both morphogenesis7 and subsequent structural and functional organization of epithelia8, and the disruption of either of the interactive components produces significant alterations in cellular behaviour. E-cadherin has been extensively studied in human cancers, resulting in its nomination as a marker for metastatic biopotential in many tumours9. In primary PCa, reduced E-cadherin expression has been correlated with increased tumour grade or stage, and with bone metastasis and poor prognosis10, 11, 12. Further data have confirmed the correlation with tumour grade, but one study found no relationship between E-cadherin and tumour progression or PCa death13. In animal models, low E-cadherin expression has also been described in both metastasizing and non-metastasizing PCa tumour sublines5. An archival study of this issue14 in paired primary prostate tissue and prostatic bone metastases from the same patients showed decreased expression of E-cadherin messenger RNA (mRNA) in metastases in nine of the total number of cases. The results suggest that E-cadherin down-regulation, although important, is not the foremost step in the metastatic cascade, but this protein is a clinically relevant invasion–metastasis suppressor. Indeed, it is a critical component in the general process of epithelial to mesenchymal transition (EMT). For epithelial cancer to progress and metastasize, cells must undergo this transition, whereby cell polarity and cell–cell binding are lost. These cells assume a mesenchymal phenotype, which gives them the ability to invade the ECM and migrate to distant sites. This process involves the disruption of stable E-cadherin binding, a primary event governing EMT, and it is accompanied by increased expression of mesenchymal N-cadherin. The EMT process may be an 'on–off' phenomenon, and it is possible that transient functional down-regulation of E-cadherin may be a feature of the metastatic process in PCa, with differences in expression at the primary and secondary sites14.
Integrity of the cadherin–catenin complex and its anchorage to the actin cytoskeleton are required for E-cadherin-mediated intercellular adhesion. Absence or dysfunction of the catenin component of this complex may lead to impaired cellular adhesion, despite apparently normal E-cadherin levels14, 15. Clinical studies of PCa confirm the correlation between catenin subtype expression with tumour de-differentiation and local stage16, although aberrant expression of α-catenin is rare in the presence of normal E-cadherin expression. A study of 28 prostatic tumours found consistent abnormalities of E-cadherin and down-regulation of α-catenin17, and although Umbas et al.18 detected these effects in only four out of 52 cases, the combination occurred in patients with advanced disease.
β-Catenin has dual functions in prostatic and other tissues. In addition to its role in the cadherin–catenin complex, it also regulates signal transduction by binding to DNA and activating gene transcription. Few reports describe abnormal β-catenin signalling as a master regulator of PCa (< 4% of primary prostate tumours have β-catenin mutations19), but aberrant β-catenin expression seems to affect the function of the cadherin–catenin complex. This notion is supported in a paired primary or bone metastasis study14, in which 13 out of 14 primary tumours had high β-catenin expression, whereas 12 out of 14 metastases showed down-regulated β-catenin mRNA levels compared with their primary tumours. Therefore, there is a striking contrast in the levels of β-catenin mRNA between primary tumours and metastases, suggesting a major dysfunction of the cadherin–catenin complex. This factor may be an important early step in the metastatic process. There are, however, unexplained observations that run counter to this hypothesis, for example, β-catenin expression in the primary tumour does not appear to reflect the metastatic potential of tumours in some patients, and E-cadherin is not lost from metastatic cells, although it may be re-expressed in the secondary site once it is lost in the primary site. Better understanding of this process is required, but, overall, these observations suggest that the essential E-cadherin–β-catenin complex is often impaired during metastasis.
Cell–matrix adhesion and matrix degradation
Integrins are essential for cell–matrix attachment. Integrin expression varies between tumours, but over-expression of α6 and β3 integrins have been associated with increased invasion20, 21. This may suggest the anchorage of the malignant cell to the basement membrane (BM) or the involvement of signalling pathways related to cell motility. Whatever the actual mechanism, integrins are fundamentally important in the binding and migration processes at metastatic sites, where they work together with enzymes that degrade the ECM and BM. These structures are composed mainly of type IV collagen, laminin, fibronectin, entactin and tenascin22. Leucocytes and malignant cells are thought to be the only cells that are able to breach the BM, a process facilitated by the production of matrix metalloproteinases (MMPs). Twenty-four MMPs have been described to date23 and they act to degrade the ECM. MMP activity is regulated by tissue inhibitors of metalloproteinases (TIMPs), and imbalances in the MMP:TIMP ratio due to either TIMP down-regulation or increased MMP production by tumour cells can induce an invasive phenotype24. In metastasis, this balance is vitally important both in endothelial barrier degradation25 and in the establishment of metastases within bone marrow stroma (BMS)26, 27 (Figure 2). The proteolytic enzyme, urokinase-type plasminogen activator, is also important in the MMP cascade. It has direct lytic activity on fibronectin, and through plasmin it activates procollagenases. It works to initiate MMP action and is particularly critical in the development of PCa metastases27.
Figure 2.
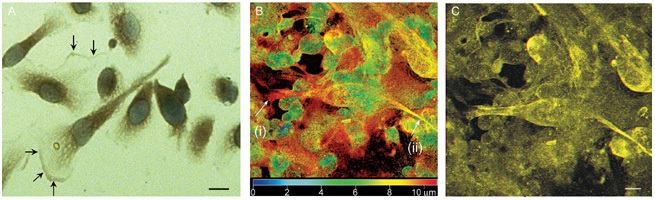
(A): Photomicrograph showing matrix metalloproteinase (MMP)-7 staining of prostate cancer (PCa) cells in culture. High MMP-7 staining is seen at the leading edge of the cell relative to the ruffling border at the margin of the pseudopodial extension (arrows); original magnification (× 400), scale bar = 10 μm. (B): Confocal 3D imaging of PCa cells in bone marrow co-culture showing PCa invasion of bone narrow stroma (BMS). False colour image of a PC-3 cell within the BMS: Blue being 0 μm, closest to the viewer (top of BMS) through to red 10 μm, furthest away from the viewer (bottom of the BMS layer). Using morphology for identification, the arrow (i) shows the leading pseudopodia of a PC-3 cell, with a second arrow (ii) showing the trailing end. (C): 3D image of the same picture showing the PC-3 cell underneath the BMS. MMP concentrations are highest at the leading edge of the cell. The cells also have the extended mesenchymal morphology typical of motile cells. Scale bar = 10 μm (Reproduced with permission from2).
Cell migration and motility: the GTPase axis
Cell motility and migration in prostate and other cancers are linked integrally to Ras and other GTP-binding proteins, for example, Rho and Rac. These proteins are important for general cellular functions, including cytoskeletal assembly, intracellular signalling and physical movement of cell membranes and whole cells28. Ras is a transmembrane glycosylated protein that regulates downstream cellular activities such as cell proliferation, nuclear transcription, apoptosis and invasion29 (Figure 3). It acts as a membrane transducer, as extracellular signals bind to receptor tyrosine kinases, which in turn activate Ras and initiate downstream events30, 31. The Ras family, which has a major influence on cell signalling, comprises h-ras, k-ras, n-ras, r-ras and m-ras, and although Ras mutations are rare in PCa (3%), they are associated with 30% of solid tumours31.
Figure 3.
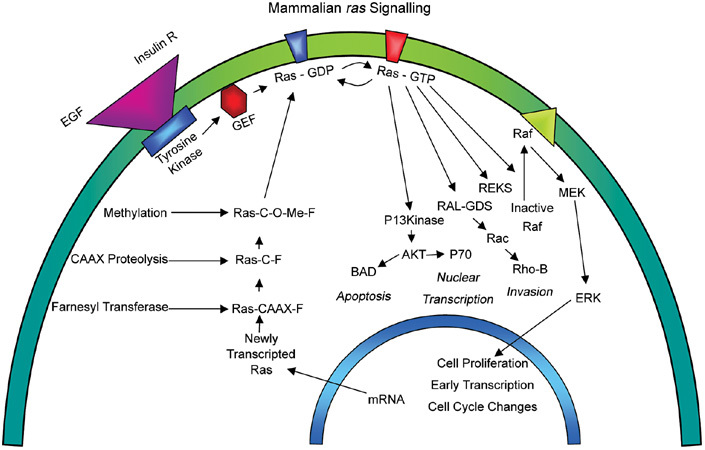
Schematic diagram showing the Ras signalling pathway and its linkage to the GTPase cellular motility axis (Reproduced with permission from2).
The Rho GTPases are similar to Ras in their structure and synthesis; their activation lies downstream of Ras and is therefore Ras-dependent. This family currently comprises RhoA, B, C, E and G; Rac1, Rac2, cdc42-H5 and TC10, all proteins involved in cell motility. It has been suggested32 that Rho GTPases act through actin dynamics, guiding morphological changes, including cell growth and movement. Cell movement may occur via a 'molecular clutch', which involves the extension of filopodia bound to the cortical actin network and a fixed extracellular ligand, resulting in net movement of the whole cell (Figure 4). Prevention of Rho synthesis or activity should result in reduced cell motility, with a corresponding reduction in invasion across endothelial barriers. This Ras–Rho-mediated activity is thought to be important in cellular migration and metastasis in prostate and other cancers. A study using bisphosphonates to inhibit the mevalonate pathway (and thus RhoA) in PCa25 showed that cell motility and transmigration of PCa cells across human bone marrow endothelial (BME) barriers and human BMS were inhibited in the presence of zoledronic acid. A further study that examined the effects of inhibiting the farnesyl and geranyl–geranyl prenylation pathways33 showed that migration and motility of PCa cells were reduced dramatically by inhibition of Ras prenylation (and therefore also inhibition of Rho activation). It is likely that the Ras–Rho axis is activated in PCa metastasis and that this underpins the acquisition of cell motility that is fundamental for successful metastasis.
Figure 4.
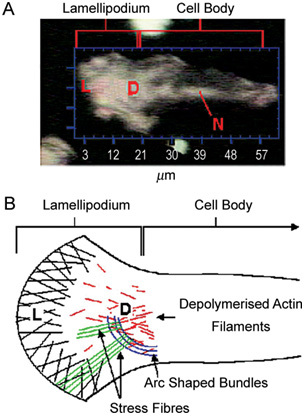
(A): Optical image of a motile prostate cancer (PCa) cell with a lamellipodial extension (L) projecting out in the direction of cellular travel. N, Nucleus; D, de-polymerized actin filaments. (B): Schematic showing the internal architecture of the lamellipodium (L), with actin filaments linking the internal cellular structure to the external lipid cellular membrane. These filaments undergo a constant process of polymerization and destruction (D), resulting in movement of the lamellipodium and forward movement of the cell (Reproduced with permission from2).
Prostate tumour cell clearance from peripheral blood
In solid tumours, malignant cells enter the circulation increasingly as the tumour load grows. Iatrogenic cellular shedding into the circulation occurs in clinical situations, for example, during transurethral resection of the prostate (TURP)34, 35, radical prostatectomy36, prostate biopsy37 and brachytherapy38. This cellular dissemination is unexpectedly not associated with a perceptible increase in metastasis development, perhaps because of the inability of individual cells to propagate or because of other unknown factors. Whatever the reality, tumour growth is accompanied by an ongoing process of cellular clearance from the circulation. Some authors suggest that this cell clearance takes up to 4 weeks37, but this proposed time scale is far too long and the speed of cell clearance from the circulation is almost certainly more rapid. Chambers et al.26 proposed that clearance is mainly attributable to the arrest of relatively large epithelial cells (or cellular clumps) in the first capillary bed they encounter. However, this cannot be the sole explanation; if it were, the incidence of pulmonary and hepatic metastases would be much higher than it actually is in PCa and other cancers. There must be additional relevant factors, predicated on the differential binding of PCa cells and differences in chemo-attraction, that activate cellular motility. In vitro models of prostate epithelial cell (PEC) binding to the human BME have shown that the process of epithelial–endothelial binding is virtually complete within an hour. Once this has occurred, epithelial cell migration through the endothelial barrier occurs within 24 h39 (Figure 5). These laboratory findings are supported by reverse transcriptase–polymerase chain reaction-based measurements in men undergoing TURP, showing that PECs appear in the circulation upon commencement of surgery, but are undetectable within 2 h of the procedure's conclusion40. It is clear, therefore, that once a cell enters the circulation, it is rapidly taken out, probably by endothelial surface binding at a secondary site.
Figure 5.
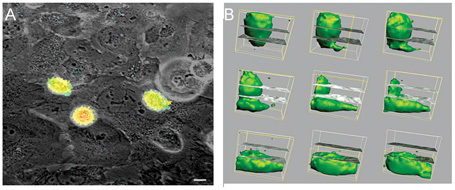
(A): Photomicrograph of a bone marrow endothelial (BME) monolayer (phase contrast) seeded with prostate cancer (PCa) PC-3 cells transfected with green fluorescent protein (GFP) (green). Cells bind to the junctional endothelial areas within 30–60 min. Thereafter they induce BME retraction and migrate into the interstitium. Scale bar = 10 μm. (B): This process involves active cellular movement and cellular expansion as shown by the time-lapse volumetric reconstructions of GFP-marked cells as they extravasate through the endothelial monolayer (Reproduced with permission from2).
Distal attachment and tumour cell transmigration through the endothelium
Tumour cells arrest on endothelial surfaces within the circulatory system and subsequently undergo transendothelial migration; this is a key event in cancer metastasis. Tumour cell–endothelial interactions involve multiple adhesive interactions (docking and locking) at the molecular level21. The initial step is thought to involve selectins, followed by stabilization through integrin binding41. These are not the only binding steps, because antibodies to CD11a, CD18, LFA-1 and CD31 have been shown to interfere with the binding process42.
Site-specific adhesion determinants play a role in preferential metastasis to individual organs. Molecules postulated to be involved in tumour–endothelial adhesion include platelet–endothelial cell adhesion molecule-1 (PECAM-1 or CD31)43, α4β1 integrin and sialyl Lewis X, which bind to the endothelial cells through E-selectin, vascular cell adhesion molecule 1 (VCAM-1)44 and others.
Tumour cells penetrate endothelial junctions after adhering to the surface of endothelial cells (Figures 5 and 6). Endothelial cells appear to be actively involved in transmigration, as dynamic changes occur in the expression and localization of adhesion molecules, including N-cadherin, VE-cadherin and PECAM-145, inducing endothelial cell retraction once the tumour cell adheres to the underlying ECM. Binding to laminin, type IV and type V collagens is mediated by β1 and β4 integrins, whereas binding to hyaluronan, fibronectin, type I collagen and cellular migration is mediated by β1 integrins and CD4444, 46, 47, 48, 49. Understanding the process of secondary site binding and endothelial transmigration in PCa has been facilitated by the development of co-culture models using human BMS and primary PECs and PEC lines27, 39 and the establishment of prostate epithelial colonies in human BMS50, 51. These models have demonstrated that cell–matrix binding depends fundamentally on the β1 integrin component of the integrin-binding mechanism. Studies using various endothelial types as well as benign and malignant PECs have shown that PECs bind more avidly to bone marrow endothelial cells (BMECs) than to other endothelia and that benign and malignant cells have the same binding capacity for those endothelial surfaces. Why, then, do metastases not develop from the PECs known to be present in the circulation during prostatic resection for benign prostatic hyperplasia (BPH)? The answer to this question lies in the differential ability of PECs to migrate across the endothelial barrier. In vitro studies using green fluorescent protein (GFP)-transfected PCa cells in conjunction with time-lapse confocal microscopy have enabled cellular tracking measurements of benign and malignant PECs in epithelial–endothelial co-culture39 (Figures 5 and 6) and have shown that only malignant cells will transmigrate through the endothelial layer. Benign cells will bind in the same way as malignant cells, but they do not cross the endothelial barrier into the interstitium39.
Figure 6.

Reconstructed confocal microscopic image of prostate cancer (PCa) cells transfected with green fluorescent protein (GFP) (green) seeded onto a bone marrow endothelial (BME) monolayer (grey) and photographed sequentially with time-lapse confocal microscopy. (A): PCa cells binding to the junctional areas of the endothelium. (B): The endothelial cells have started to retract, leaving gaps in the endothelial barrier. (C): The epithelial cells then migrate through into the underlying interstitium (Reproduced with permission from2)
Explication of this mechanism is critical to the understanding and potential treatment of metastases in PCa. Once bound, PECs induce rapid endothelial cell retraction (Figure 6), but the precise mechanism inducing this reaction is at present unclear. A major component of the signalling cascade modulating endothelial permeability is intracellular Ca2+52. Studies by Lewalle et al.53 showed that binding of breast epithelial cells to human umbilical vein endothelium cells (HUVECs) induced a transitory rise in the HUVEC intracellular Ca2+ concentration, resulting in endothelial retraction and epithelial migration. This Ca2+ rise and the process of endothelial retraction are entirely dependent on cell–cell contact, and inhibition of the Ca2+elevation inhibited breast epithelial cell transendothelial migration. Binding of PECs and melanoma cells also induces increases in intracellular Ca2+ levels54, correlating with increased binding of the epithelial cells. Further studies of calcium-binding agents support this notion: treatment of BMEC lines with zoledronic acid, a potent calcium-chelating agent, tightens endothelial–endothelial cell binding and limits transmigration25.
The effect of Ras–Rho inhibition of reducing the propensity of PECs to invade across endothelial barriers suggests that a major component affecting cancer cell migration is inhibition of transduction pathways related to the Rho axis. This inhibitory effect has been demonstrated in vitro in PCa using zoledronic acid25 and the prenylation inhibitor AZD340933, 55. The inhibitory effects of these compounds are known to be related to Rho through its interaction with Ras56. Inhibition of this pathway affects downstream prenylation of small GTPases (Rho–Rac), which are known to have an integral involvement in cell motility. Therefore, an early event following integrin β1 binding in PCa cells may be the induction of specific pathways that relate to Ras and subsequently Rho–Rac. These in turn are associated with the epithelial-to-mesenchymal transition, a phenomenon known to be important in the cellular migration process.
Chemo-attraction at the secondary site: chemokines and lipids
The 'seed–soil' hypothesis of Paget is exemplified by PCa, with its predilection for the red bone marrow of the axial skeleton57. Once there, the malignant cells disturb integrated and balanced skeletal functions and displace the red bone marrow, inducing marrow dysfunction and bone marrow failure. Two factors contribute to this homing phenomenon: the presence of chemokines and the affinity for energy-rich sources, such as specific lipids that are freely available within red marrow adipocytes.
The chemokine axis is important in the homing of haematological and immunological cells to specific targets. Cells from various epithelial tumours share many of the trafficking characteristics of this haematopoietic stem cell (HSC) homing system58. Homing of the HSCs to the bone marrow during foetal life and after bone marrow transplantation has been well characterized. The key molecular axis for this process was identified as the CXC chemokine stromal-derived factor-1 (SDF-1 or CXCL12) and its receptor CXCR4 (CD186). This model is supported by the knowledge that both BMECs and osteoblasts express SDF-159, 60, 61, the observation that CXCR4 knockouts do not show haematopoietic engraftment of the bone marrow62 and the recognition that the level of CXCR4 expression in HSCs determines their ability to engraft the bone marrow63. The CXCR4–SDF-1 axis is also known to play an important role in targeting solid tumour metastases to the bone marrow. This is important in various primary tumours, including the breast58, kidney64, lung65, pancreas66 and prostate57, 67. In vitro, CXCR4 and SDF-1 are involved in the motility process: interactions alongside CCR7 or CCL21 trigger pseudopodial invasion by malignant breast epithelial cells through actin polymerization58. These results have led to the hypothesis that CXCR4 is the key component of metastatic implantation in the bone marrow and that it represents an important therapeutic target for metastatic bone disease in PCa and other cancers. Indeed, blockage of CXCR4 signalling in breast cancer by neutralizing antibodies58 or peptide antagonists such as T14068 has been shown to inhibit metastasis in vivo. Sun et al.67 also showed that CXCR4 expression increased with increasing prostatic malignancy; the greatest expression was observed in aggressively metastatic PC-3 cells and in human bone metastasis specimens. This gradient of expression suggests that CXCR4–SDF-1 signalling may be a key signalling pathway for metastatic spread to the bone. It has also been demonstrated57, using a matrigel BM invasion assay, that SDF-1 signalling induced both DU145 and PC-3 cells to invade. However, Hart et al.39 used recombinant SDF-1 and T140 inhibitors to show that the CXCR4–SDF-1 signalling pathway is not the sole chemo-attractant important for the spread of PECs to the bone, confirming that, although SDF-1 is a potent stimulus for invasion, the level of invasion it induces is significantly less than that seen using either BMECs or BMS alone. This phenomenon was reinforced in these experiments by the observation that the use of a specific CXCR4 antagonist peptide (T140), at a concentration that blocked PEC invasion in response to maximum levels of SDF-1 signalling, did not completely block invasion towards either BME or BMS. Thus, although the CXCR4/SDF-1 signalling pathway is important in PCa metastasis, it is not the only chemokine signalling pathway involved39.
Another important stimulus is the requirement for the metastasizing PCa cells to seek a lipid source. Cancer cells are in a state of rapid metabolism and have a fundamental requirement for lipids, to be used either as an energy source or in the processes involved in tumour cell maintenance, proliferation and migration. An in vitro study69 showed that PC-3 cells grew rapidly in the vicinity of lipid cells in bone marrow, and further studies70 showed that PCa cells take up lipids rapidly as soon as they are seeded onto the human bone marrow (Figure 7). Specific lipids also act as strong chemo-attractants for PCa cells. Treatment with arachidonic acid, an omega-6 lipid, results in rapid migration of PC-3 cells towards bone marrow stroma, an effect that is blocked competitively using omega-3 lipids. Further experiments of lipid depletion in the bone marrow confirm this effect: the attractiveness of the human bone marrow to PCa cells decreased dramatically once the BMS was depleted of lipid cells prior to epithelial seeding70, confirming that specific lipids are critical to metastasis and that they may be an important determinant of the site specificity of the bone marrow in PCa.
Figure 7.
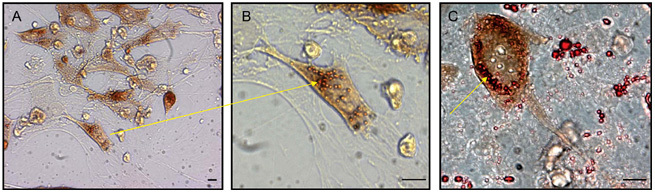
(A): Photomicrograph showing lipid uptake by a prostate cancer (PCa) cell in co-culture. (B): The lipid droplets (Oil Red O-stained vesicles) are taken up rapidly by the PCa cell and have been shown to be intracellular using confocal microscopy. (C): At higher magnification, the lipid droplets are more clearly demonstrated to be inside the PCa cells after co-culture with human BMS. The PCa cells have an affinity for the adipocytes and co-localize with them in the bone marrow. Scale bars = 10 μm (Reproduced with permission from2).
Molecular mechanisms of metastatic prostate cancer in the bone or bone marrow
Once established at the secondary site, prostatic micro-metastases develop in the bone marrow space, often in close association with the bone surface, where the osteoblast–fibroblast microenvironment is disturbed locally. It is postulated that the first event in this metastatic developmental process is osteoclast-mediated bone resorption, leading to the release of stimulatory cytokines from the bone surface and inducing a cycle of resorption or tumour stimulation, but this hypothesis has not been proven definitively. As the metastasis develops, an imbalance occurs in the regulated, coupled skeletal cycle of bone resorption and bone formation, resulting in accelerated and synchronous bone formation and resorption. This is caused by changes in local cytokine production and interactions (Figure 8).
Figure 8.
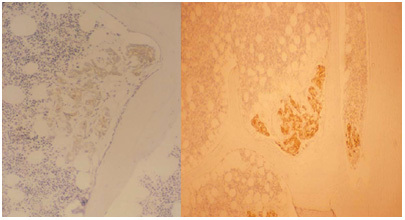
Photomicrographs of two bone biopsies taken from different prostate cancer (PCa) patients. These show the typical appearance of early micro-metastases from PCa. The epithelial cells are stained for prostate-specific angiten (PSA) and appear brown in this image. The cell colonies lie close to the bone surface (uniformly pale), where they stimulate early osteoblastic activity. In the surrounding bone marrow, the fibroblasts are stimulated to induce a desmoplastic reaction. There is no evidence of bone resorption at this stage of the disease process (Reproduced with permission from2)
Many stimulating factors have been identified with respect to osteoblastic metastases in PCa. These mechanisms have been clarified in recent years and point towards the importance of the endothelin axis. There are three types of endothelin (ET-1, -2 and -3), which act through the endothelin receptors ETa and ETb. They are synthesized in vascular endothelial cells and are involved in processes such as vasoconstriction, nociception and the physiological regulation of bone function, amongst others. Effects on bone function are important in relation to PCa. Nelson and Carducci (reviewed by Nelson71) showed that exogenous ET-1 induces PCa proliferation and enhances the mitogenic effects of insulin-like growth factor (IGF) and epidermal growth factor. Regarding PCa metastasis in bone, ET-1 production is a major factor in osteoblast overstimulation72. PECs produce ET-1, and its receptor, ETa, is present throughout the prostate gland73, 74, 75. ET-1 is also produced by PCa cells in a bone environment76. Experiments using an osteoblast mouse model72 showed that tumours producing ET-1 (e.g., PCa) act via ETa receptors on osteoblasts to stimulate accelerated bone formation. This abnormal activity is blocked by the ET-1 inhibitor ABT-627 (Atrasentan)71.
Although ET-1 is important, it is not the only osteoblast stimulator in PCa metastasis. Other factors include up-regulation of the Wnt pathway and production of cytokines, for example, bone morphogenetic protein, TGF-β, IGF, vascular endothelial growth factor, platelet-derived growth factor and MDA-BF77. A further interesting aspect of the cytokine balance in PCa metastasis relates to the IGF axis and parathyroid hormone-related protein (PTHrP), which is produced in PCa bone metastases78. The prostate-specific antigen (PSA), a known protease, cleaves PTHrP and possibly shifts the balance within the immediate milieu of the prostate metastasis from bone resorption to formation79, 80. PSA can also cleave insulin-like growth factor binding protein (IGFBP-3), which in turn increases the levels of IGF-1. This too would have the effect of shifting the axis of stimulation by the metastatic PCa cells towards increased osteoblast activity81. Osteoblast hyperactivity is responsible for the measurable increase in bone volume in PCa bone metastases82, 83 and for the accelerated bone mineralization rate84. Prostate tumour-generated bone in these deposits is formed as abnormal 'woven' bone, characteristic of the bone produced in high-turnover states. This is responsible for the sclerotic appearance measured histomorphometrically83 and seen radiologically in over 90% of patients with advanced metastatic PCa85.
The traditional view of PCa as osteoblastic obscured for many years the fact that the disease is responsible for major bone destruction. Resorptive effects of PCa were initially suggested following histological studies in bone86, and the phenomenon was subsequently confirmed after histomorphometric measurements of metastatic bone biopsies82, 83 and biochemical measurements of bone resorption products in humans87, 88. The paradox of increasing bone volume in the presence of bone resorption is explained by histomorphometric studies showing that the resorption of the existing skeleton is accompanied by synchronous replacement of abnormal woven bone, which itself undergoes further resorption83. This produces a measurable increase in bone volume coincident with wholesale destruction of the normal skeleton.
Molecular mechanisms responsible for this lytic process arise as the consequences of abnormal concentrations of soluble growth factors produced by the invading PCa, which stimulate abnormal osteoclast activity, inducing bone resorption. Osteoclast recruitment, differentiation and activation by tumours are incompletely understood, but are known to be related to the osteoblast stimulation that results from osteoblastic over-expression of NF-κB (RANK ligand) and the production of osteoprotegerin, known to be increased in PCa metastasis89. This effect may also be induced by macrophage colony-stimulating factor, the receptor activator of the RANK ligand and osteoprotegerin90, 91. Osteoblasts secrete the RANK ligand, which then induces osteoclast differentiation by binding to the RANK surface receptor on the osteoclast precursor, which in turn stimulates osteoclastogenesis90. Osteoprotegerin plays a key regulatory role in this process by competing for the RANK-binding site on osteoclast precursors. A co-factor in this process is PTHrP. Cancer cells are unable to express the RANK ligand and therefore cannot stimulate osteoclastogenesis by this route. However, when PTHrP is present (as in murine osteoblasts and haemopoietic progenitors in culture92), osteoclasts differentiate in the absence of other stimulatory agents, suggesting that PTHrP plays a facilitating role. PTHrP is a major factor in bone resorption in breast cancer93 and is expressed in both primary tumours and bone metastases of PCa78.
Conclusion
The molecular mechanisms of metastasis in PCa are complex and involve a number of specific steps and interrelated mechanisms. A more complete understanding of the molecular mechanisms controlling this process will help to develop novel therapies that may enable us to control this progressively fatal condition.
References
- Liotta LA. Tumor invasion and metastases-role of the extracellular matrix: Rhoads Memorial Award lecture. Cancer Res. 1986;46:1–7. [PubMed] [Google Scholar]
- Clarke N, Brown M.Molecular Mechanisms of Metastasis in Prostate CancerIn: Kirby RS, Partin AW, Feneley M, Parsons JK, editors. Prostate Cancer: Principles And Practice London&New York: Taylor and Francis; 2006p383–400. [Google Scholar]
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochem Biophys Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, et al. E-cadherin-mediated cell–cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–85. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemakers MJ, van Moorselaar RJ, Giroldi LA, Ichikawa T, Isaacs JT, et al. Decreased expression of E-cadherin in the progression of rat prostatic cancer. Cancer Res. 1992;52:2916–22. [PubMed] [Google Scholar]
- Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W van Roy F, et al. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–19. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- Näthke IS, Hinck L, Swedlow JR, Papkoff J, Nelson WJ. Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J Cell Biol. 1994;125:1341–52. doi: 10.1083/jcb.125.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–3. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Cheng L, Nagabhushan M, Pretlow TP, Amini SB, Pretlow TG. Expression of E-cadherin in primary and metastatic prostate cancer. Am J Pathol. 1996;148:1375–80. [PMC free article] [PubMed] [Google Scholar]
- Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, et al. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994;54:3929–33. [PubMed] [Google Scholar]
- Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, et al. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992;52:5104–9. [PubMed] [Google Scholar]
- McWilliam LJ, Knox WF, Hill C, et al. E-Cadherin expression fails to predict progression and survival in prostate cancer. J Urol. 1996;155:516A. [Google Scholar]
- Bryden AAG, Freemont AJ, Clarke NW, George NJ. Paradoxical expression of E-cadherin in prostatic bone metastases. BJU Int. 1999;84:1032–4. doi: 10.1046/j.1464-410x.1999.00378.x. [DOI] [PubMed] [Google Scholar]
- Shimoyama Y, Nagafuchi A, Fujita S, Gotoh M, Takeichi M, et al. Cadherin dysfunction in a human cancer cell line: possible involvement of loss of alpha-catenin expression in reduced cell–cell adhesiveness. Cancer Res. 1992;52:5770–4. [PubMed] [Google Scholar]
- Morita N, Uemura H, Tsumatani K, Cho M, Hirao Y. E-cadherin and alpha-, beta- and gamma-catenin expression in prostate cancers: correlation with tumour invasion. Br J Cancer. 1999;79:1879–83. doi: 10.1038/sj.bjc.6690299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murant SJ, Handley J, Stower M, Reid N, Cussenot O, et al. Co-ordinated changes in expression of cell adhesion molecules in prostate cancer. Eur J Cancer. 1997;33:263–71. doi: 10.1016/s0959-8049(96)00418-2. [DOI] [PubMed] [Google Scholar]
- Umbas R, Isaacs WB, Bringuier PP, Xue Y, Debruyne FM, et al. Relation between aberrant alpha-catenin expression and loss of E-cadherin function in prostate cancer. Int J Cancer. 1997;74:374–7. doi: 10.1002/(sici)1097-0215(19970822)74:4<374::aid-ijc2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Voeller HJ, Truica CI, Gelmann EP. Beta-catenin mutations in human prostate cancer. Cancer Res. 1998;58:2520–3. [PubMed] [Google Scholar]
- Cress AE, Rabinovitz I, Zhu W, Nagle RB. The alpha 6 beta 1 and alpha 6 beta 4 integrins in human prostate cancer progression. Cancer Metastasis Rev. 1995;14:219–28. doi: 10.1007/BF00690293. [DOI] [PubMed] [Google Scholar]
- Honn KV, Tang DG. Adhesion molecules and tumor cell interaction with endothelium and subendothelial matrix. Cancer Metastasis Rev. 1992;11:353–75. doi: 10.1007/BF01307187. [DOI] [PubMed] [Google Scholar]
- Nagle RB, Knox JD, Wolf C, Bowden GT, Cress AE. Adhesion molecules, extracellular matrix, and proteases in prostate carcinoma. J Cell Biochem Suppl. 1994;19:232–7. [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–72. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Lokeshwar BL, Selzer MG, Block NL, Gunja-Smith Z. Secretion of matrix metalloproteinases and their inhibitors (tissue inhibitor of metalloproteinases) by human prostate in explant cultures: reduced tissue inhibitor of metalloproteinase secretion by malignant tissues. Cancer Res. 1993;53:4493–8. [PubMed] [Google Scholar]
- Montague R, Hart CA, George NJ, Ramani VA, Brown MD, et al. Differential inhibition of invasion and proliferation by bisphosphonates: anti-metastatic potential of zoledronic acid in prostate cancer. Eur Urol. 2004;46:389–401. doi: 10.1016/j.eururo.2004.04.022. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Hart CA, Scott LJ, Bagley S, Bryden AA, Clarke NW, et al. Role of proteolytic enzymes in human prostate bone metastasis formation: in vivo and in vitro studies. Br J Cancer. 2002;86:1136–42. doi: 10.1038/sj.bjc.6600207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford G, Theodorescu D. Ras superfamily monomeric G proteins in carcinoma cell motility. Cancer Lett. 2003;189:117–28. doi: 10.1016/s0304-3835(02)00510-4. [DOI] [PubMed] [Google Scholar]
- Johnston SR. Farnesyl transferase inhibitors: a novel targeted therapy for cancer. Lancet Oncol. 2001;2:18–26. doi: 10.1016/s1470-2045(00)00191-1. [DOI] [PubMed] [Google Scholar]
- Hu L, Shi Y, Hsu JH, Gera J, Van Ness B, et al. Downstream effectors of oncogenic ras in multiple myeloma cells. Blood. 2003;101:3126–35. doi: 10.1182/blood-2002-08-2640. [DOI] [PubMed] [Google Scholar]
- Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–74. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- Giniger E. How do Rho family GTPases direct axon growth and guidance? A proposal relating signaling pathways to growth cone mechanics. Differentiation. 2002;70:385–96. doi: 10.1046/j.1432-0436.2002.700801.x. [DOI] [PubMed] [Google Scholar]
- Khafagy R, Stephens T, Hart C, et al. In vitro effects of the prenyl transferase inhibitor AZD3409 on prostate cancer epithelial cells. J Clin Oncol. 2004;22:442S. [Google Scholar]
- Heung YM, Walsh K, Sriprasad S, Mulvin D, Sherwood RA. The detection of prostate cells by the reverse transcription-polymerase chain reaction in the circulation of patients undergoing transurethral resection of the prostate. BJU Int. 2000;85:65–9. doi: 10.1046/j.1464-410x.2000.00380.x. [DOI] [PubMed] [Google Scholar]
- Straub B, Müller M, Krause H, Schrader M, Goessl C, et al. Detection of prostate-specific antigen RNA before and after radical retropubic prostatectomy and transurethral resection of the prostate using “Light-Cycler”-based quantitative real-time polymerase chain reaction. Urology. 2001;58:815–20. doi: 10.1016/s0090-4295(01)01351-6. [DOI] [PubMed] [Google Scholar]
- Eschwége P, Dumas F, Blanchet P, Le Maire V, Benoit G, et al. Haematogenous dissemination of prostatic epithelial cells during radical prostatectomy. Lancet. 1995;346:1528–30. doi: 10.1016/s0140-6736(95)92054-4. [DOI] [PubMed] [Google Scholar]
- Price DK, Clontz DR, Woodard WL, 3rd, Kaufman JS, Daniels JM, et al. Detection and clearance of prostate cells subsequent to ultrasound-guided needle biopsy as determined by multiplex nested reverse transcription polymerase chain reaction assay Urology 199852261–6.discussion 266–7. [DOI] [PubMed] [Google Scholar]
- Siddiqua A, Chendil D, Rowland R, Meigooni AS, Kudrimoti M, et al. Increased expression of PSA mRNA during brachytherapy in peripheral blood of patients with prostate cancer. Urology. 2002;60:270–5. doi: 10.1016/s0090-4295(02)01703-x. [DOI] [PubMed] [Google Scholar]
- Hart CA, Brown M, Bagley S, Sharrard M, Clarke NW. Invasive characteristics of human prostatic epithelial cells: understanding the metastatic process. Br J Cancer. 2005;92:503–12. doi: 10.1038/sj.bjc.6602325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre IG, Bhatt RI, Clarke NW. Isolation of epithelial cells from blood and bone marrow of prostate cancer patients. Prostate Cancer Prostatic Dis. 2002;5:S21. [Google Scholar]
- Orr FW, Sanchez-Sweatman OH, Kostenuik P, Singh G. Tumor–bone interactions in skeletal metastasis. Clin Orthop Relat Res. 1995;312:19–33. [PubMed] [Google Scholar]
- Lehr JE, Pienta KJ. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J Natl Cancer Inst. 1998;90:118–23. doi: 10.1093/jnci/90.2.118. [DOI] [PubMed] [Google Scholar]
- Tang DG, Chen YQ, Newman PJ, Shi L, Gao X, et al. Identification of PECAM-1 in solid tumor cells and its potential involvement in tumor cell adhesion to endothelium. J Biol Chem. 1993;268:22883–94. [PubMed] [Google Scholar]
- Zheng Z, Pan J, Chu B, Wong YC, Cheung AL, et al. Downregulation and abnormal expression of E-cadherin and beta-catenin in nasopharyngeal carcinoma: close association with advanced disease stage and lymph node metastasis. Hum Pathol. 1999;30:458–66. doi: 10.1016/s0046-8177(99)90123-5. [DOI] [PubMed] [Google Scholar]
- Voura EB, Sandig M, Siu CH. Cell–cell interactions during transendothelial migration of tumor cells. Microsc Res Tech. 1998;43:265–75. doi: 10.1002/(SICI)1097-0029(19981101)43:3<265::AID-JEMT9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Hartstein ME, Grove AS, Jr, Woog JJ. The role of the integrin family of adhesion molecules in the development of tumors metastatic to the orbit. Ophthal Plast Reconstr Surg. 1997;13:227–38. doi: 10.1097/00002341-199712000-00001. [DOI] [PubMed] [Google Scholar]
- Rabinovitz I, Nagle RB, Cress AE. Integrin alpha 6 expression in human prostate carcinoma cells is associated with a migratory and invasive phenotype in vitro and in vivo. Clin Exp Metastasis. 1995;13:481–91. doi: 10.1007/BF00118187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneath RJ, Mangham DC. The normal structure and function of CD44 and its role in neoplasia. Mol Pathol. 1998;51:191–200. doi: 10.1136/mp.51.4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha M, Raso E, Cai Y, Fazakas Z, Paku S, et al. Role of alphaII(b)beta3 integrin in prostate cancer metastasis. Prostate. 1998;35:185–92. doi: 10.1002/(sici)1097-0045(19980515)35:3<185::aid-pros4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Lang SH, Clarke NW, George NJ, Allen TD, Testa NG. Interaction of prostate epithelial cells from benign and malignant tumor tissue with bone-marrow stroma. Prostate. 1998;34:203–13. doi: 10.1002/(sici)1097-0045(19980215)34:3<203::aid-pros8>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Lang SH, Clarke NW, George NJ, Testa NG. Primary prostatic epithelial cell binding to human bone marrow stroma and the role of alpha 2 beta 1 integrin. Clin Exp Metastasis. 1997;15:218–27. doi: 10.1023/a:1018465213641. [DOI] [PubMed] [Google Scholar]
- Curry FE. Modulation of venular microvessel permeability by calcium influx into endothelial cells. Faseb J. 1992;6:2456–66. doi: 10.1096/fasebj.6.7.1563597. [DOI] [PubMed] [Google Scholar]
- Lewalle JM, Cataldo D, Bajou K, Lambert CA, Foidart JM. Endothelial cell intracellular Ca2+ concentration is increased upon breast tumor cell contact and mediates tumor cell transendothelial migration. Clin Exp Metastasis. 1998;16:21–9. doi: 10.1023/a:1006555800862. [DOI] [PubMed] [Google Scholar]
- Pili R, Corda S, Passaniti A, Ziegelstein RC, Heldman AW, et al. Endothelial cell Ca2+ increases upon tumor cell contact and modulates cell–cell adhesion. J Clin Invest. 1993;92:3017–22. doi: 10.1172/JCI116925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khafagy R, Stephens T, Hart C, et al. The novel oral protein prenyl transferase inhibitor AZD3409 inhibits prostate epithelial cell proliferation and invasion in vitro, without bone marrow toxicity. Eur Urol Suppl. 2005;4:54. [Google Scholar]
- Virtanen SS, Väänänen HK, Härkönen PL, Lakkakorpi PT. Alendronate inhibits invasion of PC-3 prostate cancer cells by affecting the mevalonate pathway. Cancer Res. 2002;62:2708–14. [PubMed] [Google Scholar]
- Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–7. [PubMed] [Google Scholar]
- Müller A, Homey B, Soto H, Ge N, Catron D, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med. 1997;185:111–20. doi: 10.1084/jem.185.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. 2000;106:1331–9. doi: 10.1172/JCI10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T, Möhle R, Hesselgesser J, Hoxie J, Nachman RL, et al. Transendothelial migration of megakaryocytes in response to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J Exp Med. 1998;188:539–48. doi: 10.1084/jem.188.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Tavian M, Cipponi A, Ficara F, Zappone E, et al. Expression of CXCR4, the receptor for stromal cell-derived factor-1 on fetal and adult human lympho-hematopoietic progenitors. Eur J Immunol. 1999;29:1823–31. doi: 10.1002/(SICI)1521-4141(199906)29:06<1823::AID-IMMU1823>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Peled A, Petit I, Kollet O, Magid M, Ponomaryov T, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283:845–8. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, et al. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–11. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Burger M, Glodek A, Hartmann T, Schmitt-Gräff A, Silberstein LE, et al. Functional expression of CXCR4 (CD184) on small-cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene. 2003;22:8093–101. doi: 10.1038/sj.onc.1207097. [DOI] [PubMed] [Google Scholar]
- Koshiba T, Hosotani R, Miyamoto Y, Ida J, Tsuji S, et al. Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: a possible role for tumor progression. Clin Cancer Res. 2000;6:3530–5. [PubMed] [Google Scholar]
- Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462–73. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, et al. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003;550:79–83. doi: 10.1016/s0014-5793(03)00824-x. [DOI] [PubMed] [Google Scholar]
- Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, et al. Prostate cancer cell growth is modulated by adipocyte–cancer cell interaction. BJU Int. 2003;91:716–20. doi: 10.1046/j.1464-410x.2003.04218.x. [DOI] [PubMed] [Google Scholar]
- Brown MD, Hart CA, Gazi E, Bagley S, Clarke NW. Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br J Cancer. 2006;94:842–53. doi: 10.1038/sj.bjc.6603030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JB.Endothelin inhibition: novel therapy for prostate cancer J Urol 2003170S65–7.discussion S67–8. [DOI] [PubMed] [Google Scholar]
- Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–84. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, Opgenorth TJ, et al. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996;56:663–8. [PubMed] [Google Scholar]
- Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995;1:944–9. doi: 10.1038/nm0995-944. [DOI] [PubMed] [Google Scholar]
- Nelson JB, Nguyen SH, Wu-Wong JR, Opgenorth TJ, Dixon DB, et al. New bone formation in an osteoblastic tumor model is increased by endothelin-1 overexpression and decreased by endothelin A receptor blockade. Urology. 1999;53:1063–9. doi: 10.1016/s0090-4295(98)00658-x. [DOI] [PubMed] [Google Scholar]
- Chiao JW, Moonga BS, Yang YM, Kancherla R, Mittelman A, et al. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer. 2000;83:360–5. doi: 10.1054/bjoc.2000.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- Bryden AA, Islam S, Freemont AJ, Shanks JH, George NJ, et al. Parathyroid hormone-related peptide: expression in prostate cancer bone metastases. Prostate Cancer Prostatic Dis. 2002;5:59–62. doi: 10.1038/sj.pcan.4500553. [DOI] [PubMed] [Google Scholar]
- Cramer SD, Chen Z, Peehl DM. Prostate specific antigen cleaves parathyroid hormone-related protein in the PTH-like domain: inactivation of PTHrP-stimulated cAMP accumulation in mouse osteoblasts. J Urol. 1996;156:526–31. doi: 10.1097/00005392-199608000-00076. [DOI] [PubMed] [Google Scholar]
- Iwamura M, Hellman J, Cockett AT, Lilja H, Gershagen S. Alteration of the hormonal bioactivity of parathyroid hormone-related protein (PTHrP) as a result of limited proteolysis by prostate-specific antigen. Urology. 1996;48:317–25. doi: 10.1016/S0090-4295(96)00182-3. [DOI] [PubMed] [Google Scholar]
- Cohen P, Peehl DM, Graves HC, Rosenfeld RG. Biological effects of prostate specific antigen as an insulin-like growth factor binding protein-3 protease. J Endocrinol. 1994;142:407–15. doi: 10.1677/joe.0.1420407. [DOI] [PubMed] [Google Scholar]
- Charhon SA, Chapuy MC, Delvin EE, Valentin-Opran A, Edouard CM, et al. Histomorphometric analysis of sclerotic bone metastases from prostatic carcinoma special reference to osteomalacia. Cancer. 1983;51:918–24. doi: 10.1002/1097-0142(19830301)51:5<918::aid-cncr2820510526>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Clarke NW, McClure J, George NJR. Morphometric evidence for bone-resorption and replacement in prostate-cancer. Br J Urol. 1991;68:74–80. doi: 10.1111/j.1464-410x.1991.tb15260.x. [DOI] [PubMed] [Google Scholar]
- Clarke NW, McClure J, George NJ. Osteoblast function and osteomalacia in metastatic prostate-cancer. Eur Urol. 1993;24:286–90. doi: 10.1159/000474311. [DOI] [PubMed] [Google Scholar]
- Cook GB, Watson FR. Events in the natural history of prostate cancer: using salvage curves, mean age distributions and contingency coefficients. J Urol. 1968;99:87–96. doi: 10.1016/S0022-5347(17)62647-8. [DOI] [PubMed] [Google Scholar]
- Galasko CS. Mechanisms of bone destruction in the development of skeletal metastases. Nature. 1976;263:507–8. doi: 10.1038/263507a0. [DOI] [PubMed] [Google Scholar]
- Clarke NW, McClure J, George NJ. Disodium pamidronate identifies differential osteoclastic bone-resorption in metastatic prostate-cancer. Br J Urol. 1992;69:64–70. doi: 10.1111/j.1464-410x.1992.tb15461.x. [DOI] [PubMed] [Google Scholar]
- Urwin GH, Percival RC, Harris S, Beneton MN, Williams JL, et al. Generalised increase in bone resorption in carcinoma of the prostate. Br J Urol. 1985;57:721–3. doi: 10.1111/j.1464-410x.1985.tb07040.x. [DOI] [PubMed] [Google Scholar]
- Jung K, Lein M, Stephan C, Von Hösslin K, Semjonow A, et al. Comparison of 10 serum bone turnover markers in prostate carcinoma patients with bone metastatic spread: diagnostic and prognostic implications. Int J Cancer. 2004;111:783–91. doi: 10.1002/ijc.20314. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–57. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–8. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Thomas RJ, Guise TA, Yin JJ, Elliott J, Horwood NJ, et al. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140:4451–8. doi: 10.1210/endo.140.10.7037. [DOI] [PubMed] [Google Scholar]
- Guise TA, Mundy GR. Cancer and bone. Endocr Rev. 1998;19:18–54. doi: 10.1210/edrv.19.1.0323. [DOI] [PubMed] [Google Scholar]