

Abstract
Advanced prostate cancer is responsive to hormone therapy that interferes with androgen receptor (AR) signalling. However, the effect is short-lived, as nearly all tumours progress to a hormone-refractory (HR) state, a lethal stage of the disease. Intuitively, the AR should not be involved because hormone therapy that blocks or reduces AR activity is not effective in treating HR tumours. However, there is still a consensus that AR plays an essential role in HR prostate cancer (HRPC) because AR signalling is still functional in HR tumours. AR signalling can be activated in HR tumours through several mechanisms. First, activation of intracellular signal transduction pathways can sensitize the AR to castrate levels of androgens. Also, mutations in the AR can change AR ligand specificity, thereby allowing it to be activated by non-steroids or anti-androgens. Finally, overexpression of the wild-type AR sensitizes itself to low concentrations of androgens. Therefore, drugs targeting AR signalling could still be effective in treating HRPC.
Keywords: prostate cancer, hormone therapy, androgen receptor, hormone-refractory prostate cancer
Introduction
Advanced prostate cancer is initially hormone sensitive (HS) and can be effectively treated. The most common treatment option is hormone therapy, which is initiated by reducing the concentration of circulating androgens through surgical or medical castration 1 and/or by administering anti-androgens such as flutamide or bicalutamide 2. However, after an average of 12–18 months, almost all patients begin to fail the treatment and show the emergence of hormone-refractory (HR) disease (Figure 1). Clinically, the appearance of HR prostate cancer (HRPC) is detected by an increasing prostate-specific antigen (PSA) level either alone, in asymptomatic patients, or with widespread metastases 3.
Figure 1.
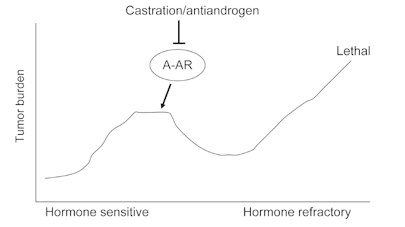
Progression from hormone-sensitive to hormonerefractory prostate cancer (HRPC). Advanced prostate cancer is initially hormone sensitive and can be effectively treated by hormone therapy, with either castration and/or anti-androgens. However, almost all patients progress to HRPC, a lethal state of the disease.
Hormone therapy targets the androgen receptor (AR) because androgen-AR signalling is required for growth and survival of hormone-sensitive prostate cancer (HSPC) cells. The AR is an androgen-activated transcription factor and a member of the steroid receptor subfamily of the nuclear receptors. The human AR is a protein of 919 amino acids in length that consists of four functionally independent domains, including the N-terminal domain, the central DNA-binding domain, the hinge region and the C-terminal domain (Figure 2A). Testosterone (T), which is produced mainly in the testes, and its more potent metabolite dihydrotestosterone (DHT) are androgens that bind and activate the AR 4. The androgen-bound AR (liganded-AR) dissociates from the heat shock protein chaperone complex, exposes the nuclear localization signal and translocates from the cytoplasm to the nucleus through interaction with the importins 5. Through the central DNA-binding domain, the liganded-AR binds to the AR response elements (AREs) that are located in the promoter or enhancer of its target genes, resulting in transcriptional activation (Figure 2B). Two regions of the AR are responsible for transcriptional activation: activation function 1 (AF1) and activation function 2 (AF2) 6, 7. AF1, located at the N-terminus, contributes to most of the AR-mediated transcriptional activation. AF2, located at the C-terminus, has a weak transcriptional activation function.
Figure 2.
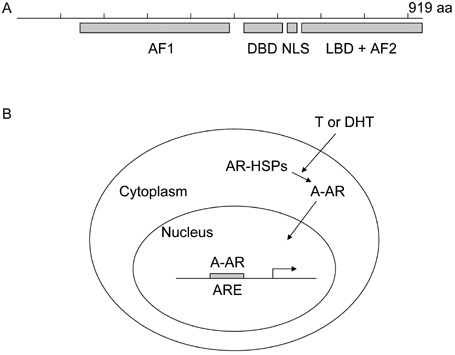
The androgen recepter (AR) is a ligand-activated transcription factor. (A): Schematic map of the major functional units in the AR, a 919-amino-acid protein. The AR contains the activation function 1 (AF1) at the N-terminus, the DNA-binding domain (DBD) in the middle, the ligand-binding domain (LBD) and the activation function 2 (AF2) at the C-terminus, and the nuclear localization signal (NLS) in the hinge region between the DBD and LBD. (B): AR activation. Androgens such as testosterone (T) and dihydrotestosterone (DHT) bind to the AR and result in dissociation from the heat shock protein chaperone complex (AR-HSPs). Androgen-bound AR (A-AR) translocates into the nucleus, binds to AR response elements (AREs) and activates the transcription of its target genes.
Both castration and administration of anti-androgens block androgen-AR signalling and inhibit AR transcriptional activity. Castration inhibits AR transcriptional activity by reducing the concentration of the ligands. Surgical castration removes the testes, which is the major T-producing organ. Therefore, this surgery reduces the supply of androgens. Medical castration uses luteinizing hormone-releasing hormone (LHRH) agonists/antagonists or estrogens to modulate the function of the hypothalamic–pituitary–gonadal axis, which is required for the production of T by the testes. This treatment also reduces the production of androgens. Anti-androgens inhibit AR transcriptional activity through direct competition with androgen binding.
A growing body of literature indicates that the AR plays a causative role in progression of prostate cancer. An important indicator for the emergence of HRPC is an increasing PSA concentration in patients after hormone therapy 3. As PSA expression is tightly regulated by the AR, an increasing PSA level means that the AR is activated in HRPC tumours. Consistent with this idea, expression levels of AR-regulated genes are maintained in HRPC cells 8. Other lines of evidence also support the idea that the AR or AR signalling is important in progression of prostate cancer. For example, about one-third of patients who progress on hormone therapy respond to second- and third-line hormonal treatments 9, indicating that blocking AR function is still beneficial for patients with HRPC. In addition, AR protein is expressed in virtually all clinical tumour samples examined so far. Thus, how is the AR activated in HRPC?
Ligand-independent activation of the AR in HRPC
HRPC was once categorized as androgen-independent prostate cancer (AIPC), and this term is still used in the literature. The name comes from the clinical definition of castration as a process that either removes the major production organ of androgens or blocks the production of androgens in patients. Therefore, prostate cancer that relapses after castration should not have enough androgens to activate the AR, indicating that the disease is androgen independent. However, can the AR be activated in HRPC without the help of androgens? It was proposed that the AR could be activated in HRPC through crosstalk with other kinase signalling pathways. Increased mitogen-activated protein kinase signalling, mediated by peptide growth factors or oncogenes such as Her2/neu, was postulated to cause ligand-independent activation of the AR.
By using prostate cancer cell lines as a model system, it was demonstrated that several peptide growth factors could activate the AR under low levels of androgens. In the experiment, charcoal-stripped serum was used 10. Charcoal stripping reduces the concentration of androgens, leaving approximately 10% of androgens remaining in the serum. Epidermal growth factor (EGF), insulin-like growth factor-1 (IGF-1) and keratinocyte growth factor (KGF) can all activate the AR under conditions of charcoal-stripped serum. An AR-responsive reporter or PSA production was used as readout for these experiments. Based on these data, it was concluded that the AR could be activated in the absence of androgens. These data provide a mechanism for the development and progression of HRPC because increased expression of IGF-1 and IGF-1 receptor is associated with HRPC progression 11. However, charcoal stripping does not deplete all androgens in the serum. Therefore, another explanation for this phenomenon is that peptide growth factors sensitize the AR to low levels of androgens.
Using prostate cancer xenograft models, it was demonstrated that Her2/neu, a tyrosine kinase receptor of the EGF receptor family, is upregulated in HRPC 12. Forced expression of Her2/neu maintained growth of prostate cancer cells in charcoal-stripped serum and promoted growth of xenograft tumours in castrated mice. These data indicated that Her2/neu initiates and promotes progression of HRPC. The Her2/neu-promoting effect was proposed to result from androgen-independent activation of the AR, because Her2/neu activated AR transcriptional activity when charcoal-stripped serum was used. However, as described above, charcoal-stripped serum still has low concentrations of androgens, and AR activation may have resulted from increased sensitivity of the AR when Her2/neu is overexpressed. Consistent with this explanation, low concentrations of androgens synergized with Her2/neu to hyperactivate the AR. The increased sensitivity of the AR caused by Her2/neu is because of stabilization of the AR protein and enhancement of AR binding to ARE 13. These results with Her2/neu may explain how peptide growth factors activate AR transcriptional activity in an environment with castrate levels of androgens.
AR activation by promiscuous ligands in HRPC
Another way to activate the AR in HRPC is by interacting with non-androgen steroids that fail to normally bind and activate the AR. In this situation, the AR can still be activated, even in the absence of androgens. Under normal conditions, the AR can bind to and be activated only by T and DHT. In order to bind to and be activated by other ligands, the ligand-binding pocket of the AR has to be altered such that it has lower binding specificity. This alteration in the AR may be achieved through mutations in the receptor. Therefore, HRPC may harbour AR mutations that alter the ligand specificity.
The first AR mutation was identified in a commonly used prostate cancer cell line, LNCaP 14. The LNCaP cell line was derived from the lymph node of a patient who had undergone long-term estrogen treatment 15. The AR in this cell line has a mutation at codon 877, which changes the amino acid from a threonine to an alanine (T877A) 16. The T877A mutation decreases the ligand specificity of the AR, and, as a result, the mutated AR can bind to and be activated by progestagens, oestradiol and anti-androgens. Therefore, some HRPC tumours may thrive on this mutation, which allows them to benefit from other ligands not affected by castration and/or to escape anti-androgen treatment. Indeed, this mutation was found in 5 of 16 examined HRPC patients who received long-term combined hormone therapies (LHRH for castration and flutamide as an anti-androgen) 17. The transcriptional activity of the mutant AR was strongly stimulated by the anti-androgen flutamide.
Other AR mutations were also found in HRPC tumours. These mutations are mostly located in the ligand-binding domain, suggesting that these mutations may alter the ligand specificity. Thus, HRPC tumours may utilize these AR mutations to maintain growth in conditions of castration and/or anti-androgen treatment. In the above-mentioned study, in which 16 tumours from HRPC patients were examined, one patient had a T877S mutation that changed the amino acid from a threonine to a serine, and another patient had a D890N mutation 17. In another study, point mutations were found in 5 of the 10 examined patients with metastatic prostate cancer in relapse after ablation by castration 18. The mutations spread across the ligand-binding domain from residue 647 to 896, suggesting that these mutations may alter the ligand specificity. Indeed, using an AR-responsive reporter, it was shown that two of the mutations H877S and H874Y can be activated by progesterone and oestradiol 18.
Other studies also identified AR mutations in HRPC tumours or cells, and these mutations can also decrease the binding specificity of AR with its ligand 19. Depending on the treatment options, patient selection, tumour heterogeneity and tissue sources, mutation type and frequency may vary from study to study. However, the mutations identified in HRPC are mostly located in the ligand-binding domain, and these mutations are present in approximately 10%–50% of patients with prostate cancer who experience treatment failure with hormone therapy. Therefore, other mechanisms must exist for activation of the AR in the majority of HRPC cases.
AR activation by overexpression in HRPC
A third way to activate the AR in HRPC tumours is by increasing the concentration of AR in the cells. There will be equilibrium among free androgens, holo-AR and androgen-bound AR. The quantity of androgen-bound AR, the activated form of the AR, depends on the available amount of free androgens and holo-AR. When the concentration of the AR increases, it will drive the balance to form more androgen-bound AR, as long as androgens are not limiting. High levels of AR expression were observed in tumours from HRPC patients when cDNA was used to identify AR mutations 18. In this study, cDNA derived from tissue samples of different HRPC patients was subjected to polymerase chain reaction (PCR) analysis, and the intensity of the PCR product was semi-quantitated by Southern blot analysis. All tissues examined had higher levels of AR than the LNCaP cell line, which expresses a considerable amount of AR. However, this study examined only five HRPC tumours and no hormone-sensitive tumours were used as controls. Therefore, the results do not lead to the conclusion that HRPC has higher levels of the AR than do hormone-sensitive tumours.
A direct comparison between HSPC and HRPC tumours was performed using comparative genomic hybridization (CGH) to screen for alterations in the DNA sequence copy number 20. Of nine HRPC tumours examined, four had an increased copy number of the Xq11–q13 region, a chromosomal region for AR. This DNA alteration was not present in HSPC tissues. Fluorescence in situ hybridization (FISH) was used to further study whether the AR gene was amplified in HRPC tumours by comparing untreated primary tumours and recurrent HRPC tumours from the same patients. Significant AR amplification was observed in 7 of 23 HRPC tumours but in none of the tissues from the same patients prior to hormone therapy. Another study examining HRPC tumours from 54 patients also detected 28% of them having AR amplification, but none of the untreated primary tumours contained this alteration 21. These results suggest that AR amplification is a common phenomenon in HRPC tumours and that AR amplification emerges during hormone therapy.
To determine whether gene amplification results in AR overexpression in HRPC tumours, real-time quantitative reverse transcription-PCR was used to examine a large series of tumours representative of different clinical stages 22. HRPC tumours with AR gene amplification had, on average, a twofold higher level of AR expression than their counterparts without gene amplification. These data indicate that the amplified AR gene is transcriptionally active, and that gene amplification can lead to AR overexpression in HRPC tumours. In addition, all HRPC tumours expressed AR and had an average of sixfold higher expression than hormone-sensitive tumours and benign prostate hyperplasia.
Higher levels of AR expression were also observed in HRPC xenograft tumours 23. Using a DNA microarray to examine gene expression profiles in seven pairs of prostate cancer xenografts that recapitulated the progression of human prostate cancer, it was discovered that the AR was the only gene consistently overexpressed in all HRPC xenografts. Western blot analysis confirmed that AR protein is overexpressed in HRPC xenografts compared with their HSPC counterparts. These data indicate that AR overexpression is a common phenomenon associated with HRPC progression.
Is AR overexpression a cause or a consequence of HRPC progression? If AR overexpression is a cause, forced overexpression of the AR in hormone-sensitive cells should lead to progression of HRPC, and reduced expression of AR in HRPC cells should result in loss of growth under conditions of hormone therapy. Indeed, when hormone-sensitive cells were forced to express high levels of AR, the time required for HRPC xenograft tumours to emerge was significantly shortened. These data indicate that AR overexpression is sufficient for HRPC progression 23. Conversely, reduction of AR expression in HRPC cells through knockdown technology abrogated tumour growth in castrated mice, indicating that AR is necessary for HRPC progression. Therefore, AR overexpression is a cause of HRPC progression.
Is androgen binding required for the AR to promote HRPC progression? To answer this question, two mutations that selectively impair ligand binding, without interfering with ligand-independent functions, were introduced into the AR. Although overexpression of the wild-type AR could lead to HRPC progression, neither AR mutant could do so 23. These data indicate that HRPC tumours are still ligand-dependent.
How does overexpression of the AR cause HRPC progression? The quantity of activated AR is determined by the concentrations of free androgens and holo-AR. When the concentration of the AR increases, high levels of AR will compensate for the low concentrations of androgens, and enough activated AR would be present to maintain HRPC growth. The AR can also form activated AR, even in the presence of anti-androgens, when AR is overexpressed. Therefore, overexpressed AR will be more sensitive to low levels of androgens, as in castration. Indeed, hormone-sensitive cells with forced overexpression of the AR, but not the control cells, grew in 20 pmol of a synthetic androgen 23. Forced overexpression of the AR also led to growth resistance for HRPC in the presence of anti-androgens. Consistent with these results, HRPC cells with high levels of the AR could grow in femtomolar concentrations of DHT, but hormone-sensitive LNCaP cells could grow only in conditions with androgen concentrations four orders of magnitude higher 24. In addition, the concentrations of DHT in HRPC tissues decreased by 91% in HRPC tumours compared with androgen-stimulated prostate tissues 25.
Implications
Although an AR-independent mechanism has been proposed to explain the emergence and growth of some HRPC tumours 26, 27, most of the data in the literature support the concept that liganded-AR signalling is still required for HRPC tumours. Thus, AR remains a drug target for this lethal form of prostate cancer. Altogether, three mechanisms are proposed as ways of activating the AR in HRPC tumours, including crosstalk with kinase pathways, AR mutations and AR overexpresson. No matter which mechanism is used, ligand binding is still essential for HRPC tumours. Therefore, methods that can regulate AR signalling would be good strategies for treating HRPC cancer. These strategies include reduction of AR expression levels, reduction of androgen levels in the prostate cancer tissue and more efficient androgen competitors. As liganded-AR signalling is regulated by epigenetic factors, including coactivators and corepressors, studies of the epigenetic regulation of AR signalling may provide other targets for treating HRPC cancer.
Acknowledgments
Work in this laboratory is supported by the Chinese Academy of Sciences (KSCX2-YW-R-04), the National Basic Research Program of China (973 Program) (2007CB947900), the Shanghai Pujiang Plan (07pj14097) and the National Natural Science Foundation of China (30870538).
References
- Huggins C, Hodges CV.Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941 J Urol 2002167948–51.; discussion 952. [PubMed] [Google Scholar]
- Labrie F, Dupont A, Belanger A, Cusan L, Lacourciere Y, et al. New hormonal therapy in prostatic carcinoma: combined treatment with an LHRH agonist and an antiandrogen. Clin Invest Med. 1982;5:267–75. [PubMed] [Google Scholar]
- Potvin K, Winquist E.Hormone-refractory prostate cancer: a primer for the primary care physician Can J Urol 20081514–20.; discussion 20. [PubMed] [Google Scholar]
- Freeman ER, Bloom DA, McGuire EJ. A brief history of testosterone. J Urol. 2001;165:371–3. doi: 10.1097/00005392-200102000-00004. [DOI] [PubMed] [Google Scholar]
- Cutress ML, Whitaker HC, Mills IG, Stewart M, Neal DE. Structural basis for the nuclear import of the human androgen receptor. J Cell Sci. 2008;121:957–68. doi: 10.1242/jcs.022103. [DOI] [PubMed] [Google Scholar]
- Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–8. [PubMed] [Google Scholar]
- Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–6. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–24. [PubMed] [Google Scholar]
- Kojima S, Suzuki H, Akakura K, Shimbo M, Ichikawa T, et al. Alternative antiandrogens to treat prostate cancer relapse after initial hormone therapy. J Urol. 2004;171:679–83. doi: 10.1097/01.ju.0000106190.32540.6c. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–8. [PubMed] [Google Scholar]
- Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, et al. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61:6276–80. [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–5. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, et al. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–27. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–40. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–18. [PubMed] [Google Scholar]
- Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, et al. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–9. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6:703–6. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–9. [PubMed] [Google Scholar]
- Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, et al. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–5. [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–8. [PubMed] [Google Scholar]
- Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, et al. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52:6940–4. [PubMed] [Google Scholar]
- Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, et al. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995;55:4438–45. [PubMed] [Google Scholar]