

Abstract
The final step in the direct restoration of the nociceptor threshold by peripheral administration of morphine and dipyrone was recently suggested to result from the opening of ATP-sensitive K+ channels (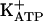 ). This channel is known to be open either directly by cGMP or indirectly via protein kinase G (PKG) stimulation. In the present study, it was shown that the blockade was caused by a specific PKG inhibitor (KT5823) of the antinociceptive effect of morphine and dipyrone on acute hypernociception and of dipyrone on persistent hypernociception. It was also shown that, in both models, KT5823 prevented the peripheral antinociceptive effect of an analogue of cGMP, the nitric oxide (NO) donor (S-nitroso-n-acetyl-d,l-penicilamine). However, in acute hypernociception, KT5823 did not prevent the peripheral antinociceptive effect of diazoxide (a direct
). This channel is known to be open either directly by cGMP or indirectly via protein kinase G (PKG) stimulation. In the present study, it was shown that the blockade was caused by a specific PKG inhibitor (KT5823) of the antinociceptive effect of morphine and dipyrone on acute hypernociception and of dipyrone on persistent hypernociception. It was also shown that, in both models, KT5823 prevented the peripheral antinociceptive effect of an analogue of cGMP, the nitric oxide (NO) donor (S-nitroso-n-acetyl-d,l-penicilamine). However, in acute hypernociception, KT5823 did not prevent the peripheral antinociceptive effect of diazoxide (a direct 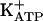 opener). In persistent hypernociception, the sensitization plateau was induced by daily injections of prostaglandin E2 (PGE2, 100 ng) into the rat paw for 14 days. After cessation of PGE2 injections, the pharmacological blockade of persistent hypernociception led to a quiescent phase in which a rather small stimulus restored the hypernociceptive plateau. In this phase, glibenclamide (which specifically closes
opener). In persistent hypernociception, the sensitization plateau was induced by daily injections of prostaglandin E2 (PGE2, 100 ng) into the rat paw for 14 days. After cessation of PGE2 injections, the pharmacological blockade of persistent hypernociception led to a quiescent phase in which a rather small stimulus restored the hypernociceptive plateau. In this phase, glibenclamide (which specifically closes 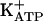 ) fully restored persistent hypernociception, as did injection of PGE2. Thus, the activation of the arginine/NO/cGMP pathway causes direct blockade of acute and persistent hypernociception by opening
) fully restored persistent hypernociception, as did injection of PGE2. Thus, the activation of the arginine/NO/cGMP pathway causes direct blockade of acute and persistent hypernociception by opening 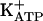 via the stimulation of PKG. Analgesic stimulators of the neuronal arginine/NO/cGMP/PKG/
via the stimulation of PKG. Analgesic stimulators of the neuronal arginine/NO/cGMP/PKG/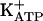 pathway constitute a previously undescribed well defined class of peripheral analgesics with a mechanism of action different from either glucocorticoids or inhibitors of cyclooxygenases.
pathway constitute a previously undescribed well defined class of peripheral analgesics with a mechanism of action different from either glucocorticoids or inhibitors of cyclooxygenases.
The suggestion that the main mechanism of action of aspirin and aspirin-like drugs is due to the prevention of nociceptor sensitization (development of hyperalgesia) is now broadly accepted (1). In contrast with specific cyclooxygenase (COX)-1 and -2 inhibitors, some analgesics, such as morphine, dipyrone, diclofenac, and keterolac are able to directly block ongoing nociceptor sensitization, as shown by its blockade of acute prostaglandin E2- (PGE2) induced hypernociception in rat hind paws (2-5). In 1979, the peripheral antinociceptive effect of opioids was discovered; this discovery led to the understanding that hypernociception (induced by inflammation or directly by inflammatory mediators) was essential for the manifestation of peripheral hypernociception (2, 6). In these studies, it was shown that the analgesic action of encephalin derivatives that did not cross the blood-brain barrier (e.g., BW180c and Tyr-d-Ala-Gly-Phe) was achieved when the drugs were given systemically. At that time and based on these observations, the possibility of developing strong peripheral analgesics was suggested, a suggestion recently rediscovered by Stein et al. (7). These pioneering observations were confirmed over the next decade by Ferreira and coworkers (8-10) and by other scientists, notably Smith and coworkers (11-16). More recently, other groups have become interested in this area (17-19).
Our serendipitous discovery of the opiates' peripheral antinociceptive activity was a corollary of the observation that morphine inhibited the activation of adenylyl cyclase (20), because we had proposed that inflammatory hyperalgesia was due to stimulation of the neuronal adenosine cAMP/Ca2+ pathway (21). In this work, following the idea that in several biological systems guanosine cGMP has the opposite effect of cAMP (22), we showed that dibutyryl cGMP blocked ongoing hypernociception. The discovery of the arginine/nitric oxide (NO)/cGMP pathway (23) and the availability of pharmacological tools to investigate this pathway in vivo prompted us to review our hypothesis. It was concluded that some analgesics that blocked ongoing hypernociception (morphine, dipyrone, diclofenac, and ketorolac) did so by stimulation of this pathway (3, 5, 24-31). By using agonists and antagonists, it was found that other antinociceptive agents, such as the κ-opioid agonist (±)-bremazocine and crotalus durissus terrificus snake venom, caused antinociception via this pathway (32, 33). This mechanism was supported by the observations that the peripheral antinociception achieved with these analgesics was inhibited by NO synthase inhibitors [NG-monomethyl-l-arginine acetate (l-NMMA) and l-N 5-(1-iminoethyl)-ornithine-dihydrochloride) as well as by guanylyl cyclase inhibitors [methylene blue and 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ)]. Moreover, the antinociception brought about by these analgesics was mimicked by NO donors such as S-nitroso-N-acetyl-dl-penicilamine (SNAP) or sodium nitroprusside and potentiated by phosphodiesterase inhibitor My5445 (24-28, 31). The hypothesis also received support from other investigators (34-36).
Recently, Duarte and coworkers (37-40) observed that blockade of PGE2-evoked hypernociception by morphine, dipyrone, sodium nitroprusside, and dibutyryl cGMP was antagonized by specific inhibitors of KATP, glibenclamide, and tolbutamide (37-40). Thus, directly acting peripheral analgesics seem to act by restoring the normal high threshold of nociceptors via the increase of K+ permeability. The missing link in this chain of events is the understanding of how the increased concentration of cGMP promotes the opening of ATP-sensitive K+ channels (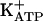 ). It is known that, depending on the biological system, cGMP directly modulates ion channels (41) or acts indirectly via protein kinase G (PKG) stimulation and the opening of
). It is known that, depending on the biological system, cGMP directly modulates ion channels (41) or acts indirectly via protein kinase G (PKG) stimulation and the opening of 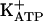 channels (42-44).
channels (42-44).
In the present study, we investigated the relevance of PKG in the mechanism of action of dipyrone and morphine in acute and persistent nociception (which can be considered a model of chronic inflammatory pain). A persistent state of hypernociception is induced by successive daily injections into rat paws of a nociceptor-sensitizing mediator (PGE2 or sympathetic amines) or cytokines that release such mediators as tumor necrosis factor α, IL-1β, and IL-8 (45, 46). This state lasts for >30 days after cessation of treatment. This persistent ongoing hypernociceptive state is blocked by intraplantar injection of analgesics such as morphine or dipyrone, leading to a quiescent phase. This antinociceptive quiescent phase is also long-lasting and, when a low-intensity hypernociceptive stimulus is applied, it fully restores the persistent state (45). We also tested substances that block or restore persistent hypernociception to verify the molecular similarities with acute hypernociception. KT5823 blocked the analgesic effect of morphine, dipyrone, SNAP, or 8-bromoguanosine cGMP (8-Br-cGMP) but not diazoxide (a direct 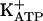 opener). Our results are consistent with the hypothesis that the blockade of ongoing acute or persistent hypernociception is due to stimulation of the arginine/NO/cGMP/PKG/
opener). Our results are consistent with the hypothesis that the blockade of ongoing acute or persistent hypernociception is due to stimulation of the arginine/NO/cGMP/PKG/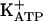 pathway.
pathway.
Materials and Methods
Animals. The experiments were performed on 180- to 200-g male Wistar rats housed in an animal care facility of the University of São Paulo and taken to the testing area at least 1 h before testing. Food and water were available ad libitum. All behavioral testing was performed between 9:00 a.m. and 4:00 p.m. Animal care and handling procedures were in accordance with International Association for Study of Pain guidelines for the use of animals in pain research and with the approval of the Ethics Committee of the School of Medicine of Ribeirão Preto (University of São Paulo). Each experiment used five rats per group. All efforts were made to minimize the number of animals used and any discomfort.
Experimental Protocol for Acute and Persistent Hypernociception.Acute and persistent mechanical hypernociception was tested in rats by using the constant rat-paw pressure test, as described (47). In this method, a constant pressure of 20 mmHg (1 mmHg = 133 Pa) is applied via a syringe piston moved by compressed air to an area of 15 mm2 on the plantar surface of the hind paw and discontinued when the rat presents a “freezing reaction.” The reaction typically comprises a reduction in escape movements (that animals normally make to free themselves), increased vibrissae movements, a variation in the respiratory frequency terminating with a brief apnea concomitant with retraction of the head toward forepaws. The apnea is frequently associated with successive waves of muscular tremor. For each animal, the latency to onset of the freezing reaction is measured before (zero time) and after administration of the hypernociceptive stimuli. In this test, the end point is a behavioral response, the freezing reaction. The constant-pressure rat-paw test over the years has been instrumental in many original observations (2, 4, 6, 48, 49). The drugs were tested by connection to a 100-ml Hamilton microsyringe. The needle was introduced s.c. near the third digit, with its tip reaching the middle of the plantar hind paw. Acute hypernociception was measured 3 h after PGE2 (100 ng) challenge. The dose of PGE2 injected was the smallest dose that evoked maximum acute mechanical hypernociception (50). The drug treatments are described in Figs. 1 and 2 legends. Persistent hypernociception was induced by daily injections of PGE2 (100 ng) over 14 days. After the discontinuation of PGE2 injection, the persistent hypernociception remained for at least 30 days (after treatment; see Figs. 1 and 2). To avoid the local release of prostaglandins triggered by trauma of intraplantar (i.pl.) injections, all animals were treated with indomethacin (2 mg/kg, i.p.) 30 min before i.pl. injections. The intensity of hypernociception was measured before and after the daily i.pl. injections by using the values measured at zero hour on the first experimental day as control reaction times. The intensity of mechanical hypernociception was quantified as the reduction in the reaction time, calculated by subtracting the value of the second measurement from the first (zero time) (47). In a large naïve rat population, the reaction time was 30.2 ± 0.5 s (mean ± SEM; n = 50). This value, after i.pl. injection of saline, changed <2 s during the experimental period.
Fig. 1.

Dose-dependent blockade of the antinociceptive effect of 8-Br-cGMP by KT5823. The measurements, made 3 h after induction of hypernociception by i.pl. injection of PGE2 (100 ng), were significantly inhibited by 8-Br-cGMP (300 μg) injected 1 h beforehand. (*, P < 0.05). Saline (S; 50 μl per paw) and KT5823 pretreatments (0.15, 0.5, or 1.5 μg) were made 10 min before 8-Br-cGMP injection. There was a dose-dependent regression for KT5823 pretreatment (nonlinear regression, R2 = 0.99). The data are the means (±SEM) of five animals per group.
Fig. 2.

Blockade and restoration of PGE2-induced persistent hypernociception. The blockade of PGE2-induced persistent hypernociception was affected by i.pl. administration of dipyrone (Dip, 160 μg, A), SNAP (SNAP; 200 μg, B) 8-Br-cGMP (cGMP, 300 μg, C), and diazoxide (Diaz; 600 μg, D). These treatments were given 5 days after discontinuation of the PGE2 injections and measured 1 h after the i.pl. administration of dipyrone, SNAP, or 8-Br-cGMP and 15 min after diazoxide i.pl. administration. A-C also show the restoration of the persistent hypernociception by i.pl. administration of glibenclamide (Glib; 160 μg); D shows the restoration by i.pl. administration of PGE2 (10 ng) made 5 days after the blockade of persistent hypernociception. Restoration of hypernociception was measured 1 and 3 h after glibenclamide or PGE2 injections, respectively. The data are the means (±SEM) of five animals per group.
Drugs. The agents used in this study were indomethacin (Prodome, Campinas, Brazil) dissolved in Tris buffer (Merck). Dipyrone, glibenclamide, diazoxide, 8-Br-cGMP, PGE2 (Sigma),L-NMMA (Research Biochemicals, Natick, MA), and morphine sulfate (Cristália, São Paulo, Brazil). ODQ and SNAP were purchased from Tocris Cookson (Ballwin, MO) and KT5823 from Calbiochem. Glibenclamide and diazoxide were dissolved in saline and Tween (Sigma, 2%) vehicle. ODQ and KT5823 were dissolved in saline and dimethyl sulfoxide (Sigma, 2%) vehicle. PGE2 was dissolved in saline and ethanol (Merck, 1%) vehicle. Dipyrone, morphine sulfate, SNAP, 8-Br-cGMP, andL-NMMA were dissolved in saline.
Data Analysis. Results are presented as means ± SEM for groups of five animals. One-way ANOVA followed by Bonferroni test was used. The level of significance was set at P < 0.05. The dose response for KT5823 was analyzed by nonlinear regression test.
Results and Discussion
The primary objective of this study was to understand how the increased concentration of cGMP promotes the opening of 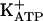 . It is known that, depending on the biological system, cGMP modulates ion channels directly (41) or indirectly (via PKG stimulation and opening of
. It is known that, depending on the biological system, cGMP modulates ion channels directly (41) or indirectly (via PKG stimulation and opening of 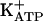 ) (42-44). By using a modification of the classical Randall-Sellito mechanical test, the final pharmacological event in the peripheral antinociceptive effect of morphine, dipyrone, sodium nitroprusside (NO donor), or dibutyryl cGMP resulting from the opening of
) (42-44). By using a modification of the classical Randall-Sellito mechanical test, the final pharmacological event in the peripheral antinociceptive effect of morphine, dipyrone, sodium nitroprusside (NO donor), or dibutyryl cGMP resulting from the opening of 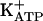 was shown in a series of papers (37-40). Specific blockers of
was shown in a series of papers (37-40). Specific blockers of 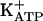 , glibenclamide, and tolbutamide antagonized the antinociceptive effect of these agents, in contrast with the absence of effect of treatments with (i) charybdotoxin (a large conductance Ca2+-activated K+ channel blockers); (ii) apamin (a selective blocker of a small conductance Ca2+-activated K+ channel), and (iii) tetraethylammonium or cesium (nonspecific K+ channel blockers). Thus, peripheral analgesics that block ongoing hypernociception seem to restore the normal high receptor threshold via opening the
, glibenclamide, and tolbutamide antagonized the antinociceptive effect of these agents, in contrast with the absence of effect of treatments with (i) charybdotoxin (a large conductance Ca2+-activated K+ channel blockers); (ii) apamin (a selective blocker of a small conductance Ca2+-activated K+ channel), and (iii) tetraethylammonium or cesium (nonspecific K+ channel blockers). Thus, peripheral analgesics that block ongoing hypernociception seem to restore the normal high receptor threshold via opening the 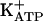 channels with consequent increase in the K+ current. It is our working hypothesis that these analgesics induce antinociception by stimulation of the arginine/NO/cGMP pathway (5, 24-31).
channels with consequent increase in the K+ current. It is our working hypothesis that these analgesics induce antinociception by stimulation of the arginine/NO/cGMP pathway (5, 24-31).
In the first series of experiments with acute hypernociception, it was shown that pretreatment with the specific PKG inhibitor (KT5823) inhibited, in a dose-dependent manner, the antinociceptive effect of 8-Br-cGMP (Fig. 1). An effective dose of KT5823 (1.5 μg) was selected for intraplantar pretreatment in subsequent experiments involving acute and persistent hypernociception. In addition, the i.pl. injection of KT5823 in rats paws pretreated with two injections of saline (50 μl per paw) did not induce acute mechanical hypernociception (Fig. 1). Our results (Table 1) support the previous suggestion that morphine- or dipyrone-induced peripheral antinociception results from the stimulation of the arginine/NO/cGMP pathway (24-31). In fact, pretreatment with the NO synthase inhibitor l-NMMA blocked the antinociceptive effect of morphine and dipyrone but did not affect SNAP, 8-Br cGMP, or diazoxide (a direct opener of 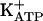 ). Furthermore, the pretreatment of rat paws with an inhibitor of soluble guanylyl cyclase [ODQ (51)] blocked the antinociceptive effects of morphine, dipyrone, and SNAP but not of 8-Br-cGMP or diazoxide (Table 1). Note that the doses of l-NMMA, ODQ, and KT5823 used did not alter the control hypernociceptive effect of PGE2 (Table 1).
). Furthermore, the pretreatment of rat paws with an inhibitor of soluble guanylyl cyclase [ODQ (51)] blocked the antinociceptive effects of morphine, dipyrone, and SNAP but not of 8-Br-cGMP or diazoxide (Table 1). Note that the doses of l-NMMA, ODQ, and KT5823 used did not alter the control hypernociceptive effect of PGE2 (Table 1).
Table 1. Acute hypernociception: Effect of pretreatment with l-NMMA, ODQ, and KT5823 on the antinociceptive effect of morphine, dipyrone, SNAP, 8-Br-cGMP, or diazoxide.
| Pretreatment
|
||||
|---|---|---|---|---|
| Treatment | Saline | l-NMMA | ODQ | KT5823 |
| PGE2+ Control | 16.5 ± 1.0 | 16.7 (ns) ± 0.8 | 16.7 (ns) ± 0.5 | 15.7 (ns) ± 1.0 |
| PGE2+ Morphine | 5.5* ± 0.8 | 13.1† ± 1.1 | 14.5† ± 0.6 | 14.3† ± 0.8 |
| PGE2+ Dipyrone | 4.3* ± 0.8 | 15.0† ± 0.5 | 13.8† ± 1.0 | 13.5† ± 0.9 |
| PGE2+ SNAP | 6.1* ± 1.0 | 7.1 (ns) ± 0.5 | 12.0† ± 0.3 | 13.3† ± 0.2 |
| PGE2+ 8-Br-cGMP | 5.8* ± 0.6 | 7.2 (ns) ± 1.3 | 6.8 (ns) ± 1.3 | 11.7† ± 0.7 |
| PGE2+ Diazoxide | 9.8* ± 0.7 | 11.3 (ns) ± 0.2 | 11.4 (ns) ± 0.7 | 10.0 (ns) ± 0.8 |
Morphine (6 μg), dipyrone (160 μg), SNAP (200 μg), 8-Br-cGMP (300 μg), and diazoxide (600 μg) were injected 2 h after i.pi. injection of PGE2 (100 ng). The i.pl. pretreatments with l-NMMA (50 μg) or ODQ (8 μg) were given 30 min before treatment, with the exception of KT5823 (1.5 μg), which was given 10 min before treatment. Hypernociception was measured 3 h after PGE2 injection. *, P < 0.05 compared with PGE2-control injected rats pretreated with saline. †, P < 0.05 compared with morphine-dipyrone-, SNAP-, 8-Br-cGMP-, or diazoxide-injected paws pretreated with saline, respectively. ns, nonsignificant compared with the respective controls. The intensity of mechanical hypernociception was expressed in seconds. The data are the means (±SEM) of five animals per group.
Table 1 also shows that pretreatment of rat paws with KT5823 antagonized the antinociceptive effect of morphine, dipyrone, SNAP, and 8-Br-cGMP but not of diazoxide. PKG may promote the opening of the 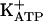 by phosphorylation (42-44). These results, consistent with previous results (discussed above), show that the last stage in the peripheral antinociception of morphine or dipyrone is mediated by the opening of
by phosphorylation (42-44). These results, consistent with previous results (discussed above), show that the last stage in the peripheral antinociception of morphine or dipyrone is mediated by the opening of 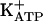 .
.
As in all acute hypernociception experiments, the peripheral administration of morphine or dipyrone elicited identical pharmacological profiles, thus reducing the number of animals; dipyrone was selected for further studies of persistent hypernociception. It is shown in Table 2 that, as in acute hypernociception induced by PGE2, the antinociception after treatment with (i) dipyrone on persistent hypernociception was blocked by pretreatment with l-NMMA, ODQ or KT5823; (ii) SNAP on persistent hypernociception was blocked by pretreatment with ODQ and KT5823; and (iii) 8-Br-cGMP on persistent hypernociception was antagonized by pretreatment with KT5823. The results with SNAP and 8-Br-cGMP show the similarities of the mechanism of blockade of acute and persistent hypernociception and also indicate that the antinociceptive effect of dipyrone, on persistent mechanical hypernociception, is due to stimulation of the arginine/NO/cGMP/PKG.
Table 2. Persistent hypernociception: Effect of l-NMMA, ODQ, or KT5823 on the antinociceptive effect of dipyrone, SNAP, or 8-Br-cGMP.
| Pretreatment
|
|||||
|---|---|---|---|---|---|
| Treatment | Before | After | l-NMMA | ODQ | KT |
| Dip | 14.1 ± 0.6 | 6.5* ± 0.7 | 12.7† ± 0.6 | 13.4† ± 0.5 | 14.1† ± 0.6 |
| SNAP | 15.6 ± 0.7 | 6.4* ± 0.5 | − | 14.0† ± 1.0 | 12.2† ± 1.6 |
| 8-Br-cGMP | 15.2 ± 0.6 | 7.9* ± 0.3 | − | − | 14.4† ± 0.3 |
The blockade of persistent hypernociception was affected by i.pl. treatment with dipyrone (Dip, 160 μg), SNAP (200 μg), or 8-Br-cGMP (300 μg). These treatments were given 5 days after discontinuation of PGE2 injections (Before), and responses were measured 1 h after the i.pl. treatment (After). Pretreatment with l-NMMA (50 μg) or ODQ (8 μg) was given 30 min before treatment, with the exception of KT5823 (KT, 1.5 μg), which was given 10 min before administration of antinociceptive agents. All treatments (Before and After) caused significant inhibition (*, P < 0.05). Pretreatment was significant compared with data after treatments (†, P < 0.05). The intensity of mechanical hypernociception was expressed in seconds. Data are the means (±SEM) of five animals per group.
Reinforcing the importance of the closure of 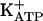 for the maintenance of persistent hypernociception, Fig. 2 shows that, similar to small doses of PGE2, glibenclamide, given in the quiescent phase induced by either dipyrone, SNAP, or 8-Br-cGMP, restores the intensity of the persistent hypernociception (Fig. 2 A-C). In contrast, treatment with diazoxide, like dipyrone, blocks ongoing persistent hypernociception, leading to the antinociceptive quiescent phase of persistent hypernociception. In fact, similar to acute hypernociception (Table 1), persistent hypernociception was blocked by diazoxide (Fig. 2D). Fig. 2D also shows that a small dose of PGE2 restores the intensity of persistent hypernociception. These results are in agreement with the mechanism of the antinociceptive action suggested for dipyrone (and morphine), namely that acute hypernociception is induced by stimulation of the arginine/NO/cGMP/PKG/
for the maintenance of persistent hypernociception, Fig. 2 shows that, similar to small doses of PGE2, glibenclamide, given in the quiescent phase induced by either dipyrone, SNAP, or 8-Br-cGMP, restores the intensity of the persistent hypernociception (Fig. 2 A-C). In contrast, treatment with diazoxide, like dipyrone, blocks ongoing persistent hypernociception, leading to the antinociceptive quiescent phase of persistent hypernociception. In fact, similar to acute hypernociception (Table 1), persistent hypernociception was blocked by diazoxide (Fig. 2D). Fig. 2D also shows that a small dose of PGE2 restores the intensity of persistent hypernociception. These results are in agreement with the mechanism of the antinociceptive action suggested for dipyrone (and morphine), namely that acute hypernociception is induced by stimulation of the arginine/NO/cGMP/PKG/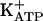 pathway. It must be pointed out, however, that in the past the central analgesic effect of opioids was suggested to be due to inhibition of adenylyl cyclase (52), inhibition today associated with opiate dependency (53, 54) and not with analgesia (55). Furthermore, stressing that the antinociceptive effect of dipyrone is after generation of cAMP, we showed that the dose-dependent increase of mechanical hypernociception induced by DbcAMP was antagonized by dipyrone, an effect potentiated by an inhibitor of phosphodiestarase, MY5445 (56). The central analgesic action of morphine has recently been suggested to involve cGMP release (25, 28, 55).
pathway. It must be pointed out, however, that in the past the central analgesic effect of opioids was suggested to be due to inhibition of adenylyl cyclase (52), inhibition today associated with opiate dependency (53, 54) and not with analgesia (55). Furthermore, stressing that the antinociceptive effect of dipyrone is after generation of cAMP, we showed that the dose-dependent increase of mechanical hypernociception induced by DbcAMP was antagonized by dipyrone, an effect potentiated by an inhibitor of phosphodiestarase, MY5445 (56). The central analgesic action of morphine has recently been suggested to involve cGMP release (25, 28, 55).
Table 3 shows that treatment with l-NMMA, ODQ, or KT5823 during the quiescent phase induced by treatment with dipyrone, SNAP, or 8-Br-cGMP did not alter the intensity of hypernociception. Thus, the quiescent phase of persistent hypernociception (the memory of peripheral hypernociception) does not need continuous activation of the arginine/NO/cGMP/PKG/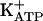 pathway.
pathway.
Table 3. Persistent hypernociception: Effect of l-NMMA, ODQ, and KT5823 on the intensity of hypernociception of the quiescent phase induced by antinociceptive drugs.
| Quiescent phase
|
|||||
|---|---|---|---|---|---|
| Treatment | Before | After | l-NMMA | ODQ | KT |
| Dip | 14.1 ± 0.6 | 6.5* ± 0.7 | 6.5* ± 0.7 | 4.1* ± 1.2 | 6.9* ± 0.3 |
| SNAP | 15.6 ± 0.7 | 6.4* ± 0.5 | − | 6.4* ± 0.5 | 7.2* ± 0.5 |
| 8-Br-cGMP | 15.2 ± 0.6 | 7.9* ± 0.3 | − | − | 8.7* ± 0.7 |
Before and after columns are the controls and values of the treatments shown in Table 2, made in the persistant phase. Ten days after the induction of the quiescent phase by dipyrone (Dip), SNAP, and 8-Br-cGMP, the paws were injected with l-NMMA (50 μg), ODQ (8 μg), and KT5823 (KT, 1.5 μg). Measurements made 90 min after the injections show no significant differences in the quiescent phase between the control values (After) and the inhibitors tested (*, P < 0.05). The data (in seconds) are the means (±SEM) of five animals per group.
Tetrodotoxin-resistant voltage-gated Na+ channels (TTX-R Na+), characteristic of C-fibers involved in inflammatory hypernociception, have been suggested as a main contributor to the development and maintenance of inflammatory as well as of PGE2-evoked hypernociception (57-60). It is generally accepted that hypernociception results from a metabotropic event initiated by activation of adenylyl cyclase/PKA and phopholipase/PKC pathways, which results in the lowering of the nociceptor threshold (21, 61, 62). This nociceptor sensitization may occur because of phosphorylation of Ca2+ and K+ channels. During this initial phase of hypernociception induction, TTX-R Na+ may also be phosphorylated, becoming ready to be activated by receptor-induced membrane voltage variation. In our study, the blockade of acute or persistent hypernociception by morphine or dipyrone stresses the relevance of K+ channels in the modulation of the nociceptor threshold, as illustrated by the blockade and restoration of persistent hypernociception by diazoxide and glibenclamide, respectively. There is evidence that blockade of TTX-R Na+ causes antinociception (60). However, the relevance of this channel on antinociceptive effects of morphine or dipyrone remains to be investigated.
The long quiescent phase of persistent hypernociception induced by i.pl. injections of dipyrone, SNAP, 8-Br-cGMP, and diazoxide (>30 days, Fig. 2) and the restoration of its full intensity by a small hypernociceptive stimulus indicate that the neuron acquires a memory. This peripheral memory of hypernociception may explain the ease of induction of recurrent periods of chronic inflammatory pain. Thus, drugs like peripheral opiates, dipyrone, and diclofenac constitute a class of analgesics different from selective COX inhibitors. It should be understood that our model of acute and persistent hypernociception induced by PGE2 is insensitive to COX inhibitors. Indeed, persistent hypernociception can be induced even in animals treated with indomethacin (see Materials and Methods). Thus, drugs effective in this test, when compared with COX inhibitors, are either more potent or have a quicker onset of effect. Specific drugs that directly stimulate the arginine/NO/cGMP/PKG/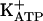 pathway represent an important target for the development of peripheral analgesics. In this context, it is possible that peripheral opiates will be available for clinical use. Thus, the κ-opioid agonist (±)-bremazocine was found to stimulate the arginine/NO/cGMP/
pathway represent an important target for the development of peripheral analgesics. In this context, it is possible that peripheral opiates will be available for clinical use. Thus, the κ-opioid agonist (±)-bremazocine was found to stimulate the arginine/NO/cGMP/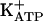 pathway (32). This previously undescribed class of drugs might be an alternative to the nonopiate analgesics, the use of which is limited in patients with gastric, renal, or coagulated disturbances. Even analgesics that directly block ongoing hypernociception have COX-related side effects (e.g., diclofenac and ketorolac) (63, 64).
pathway (32). This previously undescribed class of drugs might be an alternative to the nonopiate analgesics, the use of which is limited in patients with gastric, renal, or coagulated disturbances. Even analgesics that directly block ongoing hypernociception have COX-related side effects (e.g., diclofenac and ketorolac) (63, 64).
It is well established that the arginine/NO/cGMP pathway, depending on the dose and site of administration, may have opposite effects, antinociception or nociception (65-67). In the rat paw, it was shown that, whereas the s.c. (i.pl.) injection of NO donors and dibutyryl cGMP caused mechanical antinociception, as described here, intradermal administration caused the opposite effect, i.e., hypernociception (68). These results suggest there are different subtypes of primary nociceptive neurons in which the arginine/NO/cGMP pathway causes opposite nociceptive effects, antinociception or hypernociception. Thus, it is plausible to assume that the peripheral effect of these drugs will be analgesic in pathologies of deep tissues (trauma, arthritis, etc.) but may even worsen pain if applied superficially, as in some vascular processes or lesions caused by burning. It is interesting to note that, whereas activation of the l-arginine/NO/cGMP pathway promoted opposite modulation in the s.c. and intradermal tissue layers, the administration of PGE2 stimulated the cAMP/PKA/PKC pathway at both sites of administration (21, 61, 62, 69).
Conclusion
We have confirmed that agents stimulating the neuronal arginine/NO/cGMP pathway directly block both acute and persistent hypernociception via opening of 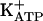 . The activation of PKG is an intermediate in the cGMP-induced opening of the
. The activation of PKG is an intermediate in the cGMP-induced opening of the 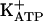 , either in acute or persistent mechanical hypernociception. Also, we showed that the quiescent phase of persistent hypernociception does not depend on the inflammatory stimulus that induced the hypernociception. Stimulation of peripheral neuronal arginine/NO/cGMP/PKG/
, either in acute or persistent mechanical hypernociception. Also, we showed that the quiescent phase of persistent hypernociception does not depend on the inflammatory stimulus that induced the hypernociception. Stimulation of peripheral neuronal arginine/NO/cGMP/PKG/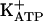 pathway or quiescent-phase inhibitors are interesting targets for new drug discoveries.
pathway or quiescent-phase inhibitors are interesting targets for new drug discoveries.
Acknowledgments
We thank I. R. Santos and S. R. Rosa for excellent technical assistance. This study was supported by grants from the Conselho Nacional de Pesquisa (CNPq) and Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP), Brazil.
Abbreviations: 8-Br-cGMP, 8-bromoguanosine cGMP; COX, cyclooxygenase; i.pl., intraplantar; 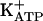 , ATP-sensitive K+ channel; L-NMMA, NG-monomethyl-l-arginine acetate; ODQ, 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; PGE2, prostaglandin E2; SNAP, S-nitroso-N-acetyl-dl-penicilamine; PKG, protein kinase G.
, ATP-sensitive K+ channel; L-NMMA, NG-monomethyl-l-arginine acetate; ODQ, 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; PGE2, prostaglandin E2; SNAP, S-nitroso-N-acetyl-dl-penicilamine; PKG, protein kinase G.
References
- 1.Ferreira, S. H. (1972) Nat. New Biol. 240, 200-203. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira, S. H. & Nakamura, M. (1979) Prostaglandins 18, 191-200. [DOI] [PubMed] [Google Scholar]
- 3.Granados-Soto, V., Flores-Murrieta, F. J., Castaneda-Hernandez, G. & Lopez-Munoz, F. J. (1995) Eur. J. Pharmacol. 277, 281-284. [DOI] [PubMed] [Google Scholar]
- 4.Lorenzetti, B. B. & Ferreira, S. H. (1985) Eur. J. Pharmacol. 114, 375-381. [DOI] [PubMed] [Google Scholar]
- 5.Tonussi, C. R. & Ferreira, S. H. (1994) Eur. J. Pharmacol. 251, 173-179. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira, S. H. & Nakamura, M. (1979) Prostaglandins 18, 201-208. [DOI] [PubMed] [Google Scholar]
- 7.Stein, C., Schafer, M. & Machelska, H. (2003) Nat. Med. 9, 1003-1008. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira, S. H., Molina, N. & Vettore, O. (1982) Prostaglandins 23, 53-60. [DOI] [PubMed] [Google Scholar]
- 9.Molina, N., Vettore, O., Lorenzetti, B. B. & Ferreira, S. H. (1983) Braz. J. Med. Biol. Res. 16, 345-352. [PubMed] [Google Scholar]
- 10.Nakamura, M. & Ferreira, S. H. (1988) Eur. J. Pharmacol. 146, 223-228. [DOI] [PubMed] [Google Scholar]
- 11.Adcock, J. J., Schneider, C. & Smith, T. W. (1988) Br. J. Pharmacol. 93, 93-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark, S. J. & Smith, T. W. (1985) Naunyn-Schmiedeberg's Arch. Pharmacol. 330, 179-183. [DOI] [PubMed] [Google Scholar]
- 13.Follenfant, R. L., Hardy, G. W., Lowe, L. A., Schneider, C. & Smith, T. W. (1988) Br. J. Pharmacol. 93, 85-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith, T. W., Buchan, P., Parsons, D. N. & Wilkinson, S. (1982) Life Sci. 31, 1205-1208. [DOI] [PubMed] [Google Scholar]
- 15.Smith, T. W. (1984) Mol. Aspects Med. 7, 509-545. [DOI] [PubMed] [Google Scholar]
- 16.Smith, T. W. & Buchan, P. (1984) Neuropeptides 5, 217-220. [DOI] [PubMed] [Google Scholar]
- 17.Sbacchi, M., Colombo, M., La Regina, A., Petrillo, P. & Tavani, A. (1988) Life Sci. 42, 2079-2089. [DOI] [PubMed] [Google Scholar]
- 18.Stein, C., Millan, M. J., Shippenberg, T. S. & Herz, A. (1988) Neurosci. Lett. 84, 225-228. [DOI] [PubMed] [Google Scholar]
- 19.Stein, C., Millan, M. J., Yassouridis, A. & Herz, A. (1988) Eur. J. Pharmacol. 155, 255-264. [DOI] [PubMed] [Google Scholar]
- 20.Ferri, S., Santagostino, A., Braga, P. C. & Galatulas, I. (1974) Psychopharmacologia 39, 231-235. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira, S. H. & Nakamura, M. (1979) Prostaglandins 18, 179-190. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg, N. D., Haddox, M. K., Nicol, E. S., Glass, D. B., Sanford, C. H., Kuehl, J. R. & Estensen, R. (1975) in Advances in Cyclic Nucleotides Research, ed. Drummond, G. I. (Raven, New York), pp. 307-330. [PubMed]
- 23.Knowles, R. G., Palacios, M., Palmer, R. M. & Moncada, S. (1989) Proc. Natl. Acad. Sci. USA 86, 5159-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duarte, I. D., dos, S., I, Lorenzetti, B. B. & Ferreira, S. H. (1992) Eur. J. Pharmacol. 217, 225-227. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira, S. H., Duarte, I. D. & Lorenzetti, B. B. (1991) Eur. J. Pharmacol. 201, 121-122. [DOI] [PubMed] [Google Scholar]
- 26.Duarte, I. D. G., Lorenzetti, B. B. & Ferreira, S. H. (1990) in Nitric Oxide from l-arginine: A Bioregulatory System, eds. Moncada, S. & Higgs, E. A. (Elsevier, Amsterdam), pp. 165-171.
- 27.Duarte, I. D., Lorenzetti, B. B. & Ferreira, S. H. (1990) Eur. J. Pharmacol. 186, 289-293. [DOI] [PubMed] [Google Scholar]
- 28.Ferreira, S. H., Duarte, I. D. & Lorenzetti, B. B. (1991) Agents Actions Suppl. 32, 101-106. [DOI] [PubMed] [Google Scholar]
- 29.Duarte, I. D. & Ferreira, S. H. (1992) Eur. J. Pharmacol. 221, 171-174. [DOI] [PubMed] [Google Scholar]
- 30.Ferreira, S. H. & Duarte, I. D. G. (1992) in Biology of Nitric Oxide (Portland Press, London), pp. 314-317.
- 31.Lorenzetti, B. B. & Ferreira, S. H. (1996) Inflamm. Res. 45, 308-311. [DOI] [PubMed] [Google Scholar]
- 32.Amarante, L. H. & Duarte, I. D. (2002) Eur. J. Pharmacol. 454, 19-23. [DOI] [PubMed] [Google Scholar]
- 33.Picolo, G., Giorgi, R. & Cury, Y. (2000) Eur. J. Pharmacol. 391, 55-62. [DOI] [PubMed] [Google Scholar]
- 34.Aguirre-Banuelos, P. & Granados-Soto, V. (1999) J. Pharmacol. Toxicol. Methods 42, 79-85. [DOI] [PubMed] [Google Scholar]
- 35.Brignola, G., Calignano, A. & Di Rosa, M. (1994) Br. J. Pharmacol. 113, 1372-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dambisya, Y. M. & Lee, T. L. (1995) Methods Find. Exp. Clin. Pharmacol. 17, 577-582. [PubMed] [Google Scholar]
- 37.Alves, D. & Duarte, I. (2002) Eur. J. Pharmacol. 444, 47-52. [DOI] [PubMed] [Google Scholar]
- 38.Rodrigues, A. R. & Duarte, I. D. (2000) Br. J. Pharmacol. 129, 110-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soares, A. C., Leite, R., Tatsuo, M. A. & Duarte, I. D. (2000) Eur. J. Pharmacol. 400, 67-71. [DOI] [PubMed] [Google Scholar]
- 40.Soares, A. C. & Duarte, I. D. (2001) Br. J. Pharmacol. 134, 127-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dowling, J. E. (1996) in Comprehensive Human Physiology, eds. Greger, R. & Windhorst, U. (Springer, Heidelberg), pp. 773-788.
- 42.Han, J., Kim, N., Kim, E., Ho, W. K. & Earm, Y. E. (2001) J. Biol. Chem. 276, 22140-22147. [DOI] [PubMed] [Google Scholar]
- 43.Han, J., Kim, N., Joo, H., Kim, E. & Earm, Y. E. (2002) Am. J. Physiol. 283, H1545-H1554. [DOI] [PubMed] [Google Scholar]
- 44.Segawa, K., Minami, K., Shiga, Y., Shiraishi, M., Sata, T., Nakashima, Y. & Shigematsu, A. (2001) Nephron 87, 263-268. [DOI] [PubMed] [Google Scholar]
- 45.Ferreira, S. H., Lorenzetti, B. B. & De Campos, D. I. (1990) Pain 42, 365-371. [DOI] [PubMed] [Google Scholar]
- 46.Sachs, D., Cunha, F. Q., Poole, S. & Ferreira, S. H. (2002) Pain 96, 89-97. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira, S. H., Lorenzetti, B. B. & Correa, F. M. (1978) Eur. J. Pharmacol. 53, 39-48. [DOI] [PubMed] [Google Scholar]
- 48.Cunha, F. Q., Poole, S., Lorenzetti, B. B. & Ferreira, S. H. (1992) Br. J. Pharmacol. 107, 660-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura-Craig, M. & Smith, T. W. (1989) Pain 38, 91-98. [DOI] [PubMed] [Google Scholar]
- 50.Ferreira, S. H., Nakamura, M. & Abreu Castro, M. S. (1978) Prostaglandins 16, 31-37. [DOI] [PubMed] [Google Scholar]
- 51.Moro, M. A., Russel, R. J., Cellek, S., Lizasoain, I., Su, Y., Darley-Usmar, V. M., Radomski, M. W. & Moncada, S. (1996) Proc. Natl. Acad. Sci. USA 93, 1480-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collier, H. O. & Roy, A. C. (1974) Prostaglandins 7, 361-376. [DOI] [PubMed] [Google Scholar]
- 53.Kaplan, G. B., Sethi, R. K., McClelland, E. G. & Leite-Morris, K. A. (1998) Brain Res. 788, 104-110. [DOI] [PubMed] [Google Scholar]
- 54.Deng, H. B., Yu, Y., Wang, H., Guang, W. & Wang, J. B. (2001) Brain Res. 898, 204-214. [DOI] [PubMed] [Google Scholar]
- 55.Levy, R. A., Proudfit, H. K. & Goldstein, B. D. (1983) Pharmacol. Biochem. Behav. 19, 79-84. [DOI] [PubMed] [Google Scholar]
- 56.Duarte, I. D. G., Faccioli, L. H. & Ferreira, S. H. (1992) in Biology of Nitric Oxide, eds. Moncada, S., Marletta, M. A., Hibbs, J. B. & Higgs, E. A. (Portland Press, London), pp. 258-260.
- 57.England, S., Bevan, S. & Docherty, R. J. (1996) J. Physiol. 495, 429-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gold, M. S., Reichling, D. B., Shuster, M. J. & Levine, J. D. (1996) Proc. Natl. Acad. Sci. USA 93, 1108-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gold, M. S., Levine, J. D. & Correa, A. M. (1998) J. Neurosci. 18, 10345-10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khasar, S. G., Gold, M. S. & Levine, J. D. (1998) Neurosci. Lett. 256, 17-20. [DOI] [PubMed] [Google Scholar]
- 61.Aley, K. O. & Levine, J. D. (1999) J. Neurosci. 19, 2181-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aley, K. O., Messing, R. O., Mochly-Rosen, D. & Levine, J. D. (2000) J. Neurosci. 20, 4680-4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ku, E. C., Wasvary, J. M. & Cash, W. D. (1975) Biochem. Pharmacol. 24, 641-643. [DOI] [PubMed] [Google Scholar]
- 64.Jett, M. F., Ramesha, C. S., Brown, C. D., Chiu, S., Emmett, C., Voronin, T., Sun, T., O'Yang, C., Hunter, J. C., Eglen, R. M., et al. (1999) J. Pharmacol. Exp. Ther. 288, 1288-1297. [PubMed] [Google Scholar]
- 65.Prado, W. A., Schiavon, V. F. & Cunha, F. Q. (2002) Eur. J. Pharmacol. 441, 57-65. [DOI] [PubMed] [Google Scholar]
- 66.Sousa, A. M. & Prado, W. A. (2001) Brain Res. 897, 9-19. [DOI] [PubMed] [Google Scholar]
- 67.Tegeder, I., Schmidtko, A., Niederberger, E., Ruth, P. & Geisslinger, G. (2002) Neurosci. Lett. 332, 146-150. [DOI] [PubMed] [Google Scholar]
- 68.Vivancos, G. G., Parada, C. A. & Ferreira, S. H. (2003) Br. J. Pharmacol. 138, 1351-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cunha, F. Q., Teixeira, M. M. & Ferreira, S. H. (1999) Br. J. Pharmacol. 127, 671-678. [DOI] [PMC free article] [PubMed] [Google Scholar]