

Abstract
A long-standing problem of conventional chemotherapy is the lack of tumor-specific treatments. Traditional chemotherapy relies on the premise that rapidly proliferating cancer cells are more likely to be killed by a cytotoxic agent. In reality, however, cytotoxic agents have very little or no specificity, which leads to systemic toxicity, causing undesirable severe side effects. Consequently, various “molecularly targeted cancer therapies” have been developed for use in specific cancers, including tumor-targeting drug delivery systems. In general, such a drug delivery system consists of a tumor recognition moiety and a cytotoxic “warhead” connected through a “smart” linker to form a conjugate. When a multi-functionalized nanomaterial is used as the vehicle, a “Trojan Horse” approach can be used for mass delivery of cytotoxic “warheads” to maximize the efficacy. Exploitation of the special properties of fluorine has proven successful in the development of new and effective biochemical tools as well as therapeutic agents. Fluorinated congeners can also serve as excellent probes for the investigation of biochemical mechanisms. 19F-NMR can provide unique and powerful tools for mechanistic investigations in chemical biology. This account presents our recent progress, in perspective, on the molecular approaches to the design and development of novel tumor-targeted drug delivery systems for new generation chemotherapy by exploiting the unique nature of fluorine.
Keywords: Tumor-targeted, Drug delivery system, Anticancer agents, Taxoid, Fluorotaxoid, Fluorescent probe, Fluorine probe, Carbon nanotube, Dendrimers, Disulfide linker, PET
1. Introduction
Cancer is one of the most challenging diseases to fight against in spite of tremendous amounts of clinical and basic research over the decades. Conventional chemotherapy relies on the premise that rapidly proliferating tumor cells are more likely to be destroyed by cytotoxic agents than normal cells. In reality, however, these cytotoxic agents have little or no specificity, which leads to systemic toxicity causing undesirable side effects. Accordingly, the development of tumor-specific drug delivery systems for anticancer agents, differentiating the normal and cancer cells or tissues, is an urgent need to dramatically improve the efficacy of cancer chemotherapy. Various drug delivery systems have been studied over the past few decades to address this problem [1]. Rapidly growing cancer cells overexpress tumor-specific receptors to enhance the uptake of nutrients and vitamins. These receptors can be used as targets for cancer cell – specific delivery of cytotoxic agents through receptor-mediated endocytosis. Furthermore, the characteristic physiology of tumor and cancer cells can be exploited to selectively accumulate and release a cytotoxic agent inside these cells. Monoclonal antibodies, polyunsaturated fatty acids, folic acid, biotin, aptamers, transferrin, oligopeptides, and hyaluronic acid, for example, have been employed as tumor-specific “guiding modules” to construct tumor-targeting drug conjugates [1–6].
In general, tumor-targeted drug delivery systems consist of a tumor-targeting module (TTM) connected to a cytotoxic “warhead” directly or through a suitable “smart” linker (Fig. 1). This drug conjugate should be stable in blood circulation to minimize systemic toxicity and should be effectively internalized inside the target tumor cells. Upon internalization, the drug conjugate should efficiently release the cytotoxic agent without loss of potency [1, 6–9].
Fig. 1.

General structures of tumor-targeted drug delivery systems
Fluorine-containing anticancer agents can certainly serve as potent “warheads” for the “guided molecular missiles”. In addition, fluorine can be strategically incorporated to create novel biochemical tools that facilitate the development of such tumor-targeted drug conjugates [7]. For example, since the fluorine nucleus is virtually non-existent in biological systems, 19F NMR can provide techniques to directly observe time-dependent processes in complex biological systems [10]. As 18F is a common radioisotope used for positron emission tomography (PET) imaging, selective fluorination for biodistribution studies has found extensive use in diagnostic applications [11–13].
We describe here a progress report of our research on the strategic incorporation of fluorine(s) for the development of taxoid-based tumor-targeted drug delivery systems in perspective. Through rational design, the unique properties of fluorine has been exploited to increase the potency and metabolic stability of taxoid anticancer agents as potent “warheads”, to validate linker release mechanisms, and to construct PET tracers to assess the biodistribution and efficacy of tumor-targeted drug conjugates.
2. New Generation Taxoids, including Fluorotaxoids, as “Warhead”
Paclitaxel (Taxol®) and docetaxel (a “taxoid”) (Fig. 2) have become two of the most important chemotherapeutic agents, which were approved for use in non-small cell lung cancer (NSCLC), breast cancer, advanced ovarian cancer, hormone-refractory prostate cancer, advanced gastric cancer, head and neck cancer, and Kaposi’s sarcoma [14]. In many of these indications, a taxane is combined with one or more additional anticancer agents, including prednisone, doxorubicin, cyclophosphamide, cis-platin, and 5-fluorouracil. Recently, another taxoid, cabazitaxel (Fig. 2), has been approved by the FDA in combination with prednisone as a second-line treatment for hormone-refractory prostate cancer [15]. In addition to these FDA-approved drugs, a number of novel taxoids are in various stages of clinical and preclinical development.
Fig. 2.
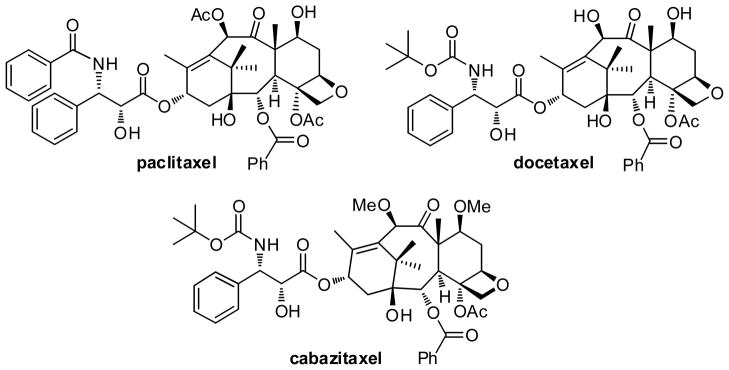
Paclitaxel, docetaxel and cabazitaxel
In spite of the significant impact that these drugs have made on cancer chemotherapy, the utility of these drugs is limited due to their susceptibility to multidrug resistance (MDR) and lack of tumor specificity. Albeit effective against many solid tumors, these drugs exhibit little to no efficacy against melanoma, colon, pancreatic and renal cancers [16]. Human colon carcinoma is intrinsically multidrug-resistant, as it overexpresses ATP-binding cassette transporter proteins, such as the P-glyocoprotein (Pgp) [17]. The Pgp binds hydrophobic molecules, including paclitaxel and docetaxel, and acts as an efflux pump that effectively removes these drugs from the cell, reducing their intracellular concentration and conferring MDR [18]. Through extensive structural-activity relationship (SAR) studies, we have developed a series of highly potent new generation taxoids, including fluorine-containing taxoids, “fluorotaxoids” [19–24]. Many of these taxoids exhibit improved activity in drug-sensitive cell lines as well as increases in potency of two to three orders of magnitude against drug-resistant cell lines in vitro [19–21].
2.1. 3′-Difluoromethyltaxoids and 3′-trifluoromethyltaxoids
In the course of our extensive SAR studies of the taxoid anticancer agents, we have synthesized a good number of fluorotaxoids by means of the β-lactam synthon method to investigate the effects of fluorine on cytotoxicity and metabolic stability [25–27]. Along this line, new generation taxoids possessing difluoromethyl and trifluoromethyl groups at the 3′-position were synthesized and their biological activity evaluated [22, 28]. The cytotoxicity of selected fluorotaxoids in vitro against various cancer cell lines is shown in Table 1. These fluorotaxoids possess substantially higher potencies than those of paclitaxel and docetaxel against drug-sensitive cancer cell lines and their potency against multidrug-resistant cell lines is more impressive (two orders of magnitude more potent than paclitaxel in average). The potency of 3′-CF2H-taxoids against MCF7-S and LCC6-WT appears to be higher and more uniform with different substitution patterns as compared to that of 3′-CF3-taxoids. On the contrary, 3′-CF3-taxoids exhibit more uniform potency against multidrug-resistant MCF7-R and LCC6-MDR cell lines than 3′-CF2H-taxoids. At any rate, it is clear that these new generation fluorotaxoids can serve as potent “warheads” for tumor-targeted drug conjugates.
Table 1.
In vitro cytotoxicity (IC50 nM)a of selected 3′-CF2H-taxoids and 3′-CF3-taxoids
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| taxoid | Rf | R | X | MCF7-Sb (breast) | NCI/ADRc (ovarian) | LCC6-WTd (breast) | LCC6-MDRe (breast) | H460f (lung) | HT-29g (colon) |
| paclitaxel | Ph | Ac | H | 1.7 | 300 | 3.1 | 346 | 4.9 | 3.6 |
| docetaxel | Ph | H | H | 1.0 | 215 | … | … | … | 1.0 |
| SB-T-12841-1 | CF2H | Ac | N3 | 0.32 | 1.68 | 0.22 | 1.57 | 0.48 | 0.57 |
| SB-T-12842-2 | CF2H | Et-CO | F | 0.53 | 7.24 | 0.88 | 4.63 | 0.41 | 0.86 |
| SB-T-12843-1 | CF2H | Me2N-CO | MeO | 0.45 | 4.51 | 0.69 | 7.06 | 0.40 | 0.43 |
| SB-T-12844-3 | CF2H | MeO-CO | Cl | 0.26 | 2.08 | 0.13 | 1.82 | 0.25 | 0.29 |
| SB-T-12821-2 | CF3 | Ac | F | 0.45 | 5.58 | 0.38 | 5.93 | 0.49 | 1.11 |
| SB-T-12822-4 | CF3 | Et-CO | N3 | 0.38 | 1.61 | 1.09 | 2.56 | 0.20 | 0.40 |
| SB-T-12823-3 | CF3 | Me2NCO | Cl | 0.12 | 1.02 | 0.27 | 2.55 | 0.42 | 0.45 |
| SB-T-12824-1 | CF3 | MeOCO | MeO | 0.17 | 2.88 | 0.27 | 3.99 | 0.38 | 0.53 |
Concentration of compound that inhibits 50% (IC50, nM) of the growth of human tumor cell line after a 72 h drug exposure.
MCF7: human breast cancer cell line.
NCI/ADR: Adriamycin-resistant human ovarian cancer cell line (Pgp+) (originally designated as “MCF7-R).
LCC6-WT: human breast cancer cell line (Pgp−).
LCC6-MDR: mdr1 transduced LCC6 cell line (Pgp+).
Human non-small cell lung cancer cell line.
Human colon cancer cell line.
2.2. 3′-Difluorovinyltaxoids
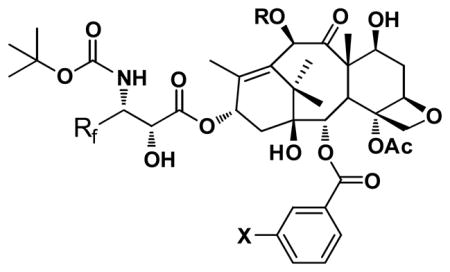
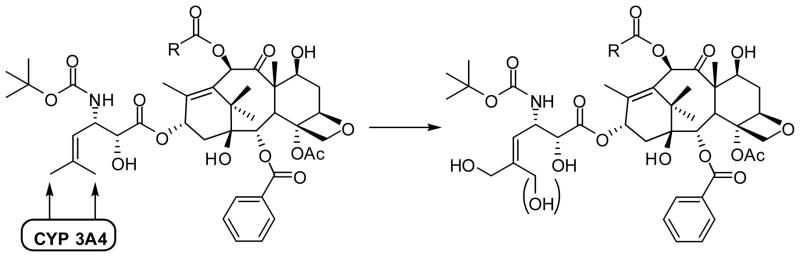
The metabolism study of 2nd-generation taxoids that bear a 3′-isobutenyl group has revealed that these taxoids are primarily hydroxylated by CYP3A4 at the allylic methyl groups (Fig. 3) [29]. Thus, in order to prevent this allylic hydroxylation a difluorovinyl group has been strategically introduced in lieu of the 3′-isobutenyl group [23]. A series of novel 3′-difluorovinyltaxoids were synthesized through the Ojima-Holton coupling of 4-difluorovinyl-β-lactam with various 2,10-modified baccatins (Scheme 1) [23].
Fig. 3.
Primary sites of hydroxylation on the second-generation taxoids by the P450 family of enzyme (CYP 3A4)
Scheme 1.
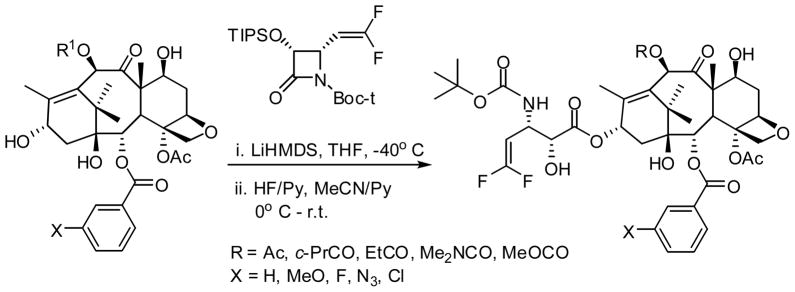
Synthesis of 3′-difluorovinyl-taxoids through the Ojima-Holton coupling
In our recent study on the metabolic stability of difluorovinyltaxoids against P-450 family enzymes, almost no appreciable metabolites were detected [30]. The results suggests that not only the metabolism at C3′ is effectively blocked, but also oxidative metabolism on other parts of the taxoid molecule, including the C3′N-t-Boc and C6 methylene moieties, which are major metabolism sites for docetaxel and paclitaxel [31–33], is suppressed [30]. Thus, our strategic incorporation of a difluorovinyl group in place of an isobutenyl group at C3′ has been proven to be successful in blocking the observed metabolism of the isobutenyl moiety in 2nd-generation taxoids (see Fig. 3).
3′-Difluorovinyltaxoids exhibit impressive activities in vitro. These taxoids initiate apoptosis primarily via the activation of caspases 2, 8 and 9 [34]. As Table 2 shows, 3′-difluorovinyltaxoids exhibit one order of magnitude and up to three orders of magnitude higher potency as compared to that of paclitaxel against drug-sensitive and drug-resistant cancer cell lines, respectively. Thus, with their very high potency and metabolic stability, novel 3′-difluorovinyltaxoids are excellent choices for “warheads” in tumor-targeted drug delivery systems.
Table 2.
In vitro cytotoxicity (IC50 nM)a of 3′-difluorovinyl-taxoids
| ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | taxoid | R | X | MCF7-Sb (breast) | MCF7-Rc (breast) | R/Sd | HT-29e (colon) | PANC-1f (pancreatic) |
| 1 | paclitaxel | 1.2 | 300 | 250 | 3.6 | 25.7 | ||
| 2 | SB-T-12851 | Ac | H | 0.099 | 0.95 | 9.6 | 0.41 | 1.19 |
| 3 | SB-T-12852 | c-Pr-CO | H | 0.12 | 6.0 | 50 | 0.85 | 5.85 |
| 4 | SB-T-12853 | Et-CO | H | 0.12 | 1.2 | 10 | 0.34 | 0.65 |
| 5 | SB-T-12854 | Me2N-CO | H | 0.13 | 4.3 | 33 | 0.46 | 1.58 |
| 6 | SB-T-12852-1 | c-Pr-CO | MeO | 0.092 | 0.48 | 5.2 | … | … |
| 7 | SB-T-12855-1 | MeO-CO | MeO | 0.078 | 0.50 | 6.4 | … | … |
| 8 | SB-T-12851-3 | Ac | N3 | 0.092 | 0.34 | 3.7 | … | … |
| 9 | SB-T-12852-3 | c-Pr-CO | N3 | 0.092 | 0.45 | 4.9 | … | … |
| 10 | SB-T-12855-3 | MeO-CO | N3 | 0.078 | 0.40 | 5.3 | … | … |
See footnotes of Table 1.
Resistance factor = (IC50 for drug resistant cell line, R)/ (IC50 for drug-sensitive cell line, S).
Human colon cancer cell line.
human pancreatic cancer cell line.
2.3. Remarkable activities of 2nd-generation taxoids and 3′-difluorovinyltaxoids against cancer stem cells (CSCs)
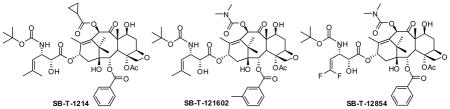
Second-generation taxoid, SB-T-1214, has demonstrated remarkable efficacy in drug-resistant cancers both in vitro and in vivo. SB-T-1214 was found to exhibit excellent activity against spheroids derived from highly drug-resistant cancer stem cells (CSCs) [35]. As CSCs are believed to be responsible for tumor reoccurrence and metastasis [36], this finding is quite significant. A fluorotaxoid, SB-T-12854, was also found to be highly potent against CSC-enriched HCT-116 human colon cancer cells. Comparison of potencies between conventional anticancer drugs and new generation taxoids is exemplified in Table 3 [37]. The ability of new generation taxoids such as SB-T-1214 and SB-T-12854 to critically damage CSC populations clearly indicates the merit in the use of these taxoids as “warheads” for tumor-targeted drug delivery systems.
Table 3.
Preliminary cytotoxicity (IC50 nM) data for standard anticancer drugs and new generation taxoids against CSC-enriched (CD133++) HCT-116 human colon cancer cell line
| |
|---|---|
| Anticancer agent | IC50 (nM) |
| cisplatin | 4,540±276 |
| doxorubicin | 78.0±28.2 |
| methotrexate | 32.7±11.2 |
| paclitaxel | 33.8±3.33 |
| topotecan | 451±12 |
| SB-T-1214 | 0.28±0.10 |
| SB-T-121602 | 0.24±0.13 |
| SB-T-12854 | 0.14±0.05 |
3. Polyunsaturated fatty acid (PUFA)-taxoid conjugates
Polyunsaturated fatty acids, most notably omega-3 PUFAs such as docosahexaenoic acid (DHA), α-linolenic acid (LNA) and eicosapentaenoic acid (EPA), are essential nutrients found in vegetable oils, cold-water fish and meat [38]. They are readily incorporated into cellular membranes, catabolized as an energy resource, and produce metabolites that act as signaling molecules, modulating various intracellular processes ranging from inflammatory response to cellular proliferation [39]. Compared to normal tissues, the intake of PUFAs by tumor tissue is significantly enhanced, as demonstrated by a single-arterial perfusion study [40]. PUFAs are readily bound by human serum albumin (HSA) and drug conjugation to PUFAs greatly alters the PK profile of the parent compound, leading to tumor-selective accumulation of the drug conjugate [41]. Currently, a number of PUFA-drug conjugates are undergoing preclinical and clinical evaluations [42].
HSA is the most abundant blood plasma protein and the primary carrier for PUFAs in vivo [43]. It has been demonstrated that conjugation of drugs to PUFAs enhances plasma protein binding relative to the parent drug [44]. DHA-paclitaxel (Taxoprexin®), a PUFA-drug conjugate currently in a Phase III clinical trial for metastatic melanoma, has been shown to exhibit greater HSA binding, leading to improved PK parameters such as longer half-life and smaller volume of distribution [41, 44]. Tumor-selective accumulation of the drug conjugate has been observed, resulting in a wider therapeutic window [41]. This is likely attributed to the enhanced permeability and retention (EPR) effect based on the nano size of the HSA-DHA-drug complex as well as the enhancement of tumor-selective transcytosis [45] based on efficient binding of HSA to gp60 overexpressed at the interface of blood vessel and tumor [46]. The latter effect is similar to that demonstrated by Abraxane® [47], FDA-approved paclitaxel-HSA nanoparticle formulation [48].
Although DHA-paclitaxel has demonstrated good clinical promise, it uses paclitaxel as the “warhead”, and therefore is subject to all of the limitations mentioned above, regarding paclitaxel treatment and drug resistance. As new generation taxoids and fluorotaxoids possess up to three orders of magnitude greater potency than paclitaxel against drug-resistant cancer cell lines, PUFA conjugates of these taxoids are not likely to suffer from the same drawbacks. Thus, DHA and LNA conjugates of these taxoids have been synthesized and assayed in vivo, of which several have demonstrated outstanding preclinical results [49]. For example, SCID mice bearing DLD-1 colon cancer xenografts were treated with DHA-SB-T-1214, resulting in complete tumor remission for over 190 days in all five mice tested [49]. DHA-SB-T-1214 is currently undergoing extensive late-stage preclinical evaluation and an Investigational New Drug application will be filed to the US FDA in the near future. Since 3′-difluorovinyltaxoids exhibit even greater potency than SB-T-1214 and much higher metabolic stability, DHA and LNA conjugates of SB-T-12854 (Fig. 4) have been synthesized to exploit these favorable properties, and their efficacy in vivo is currently under active investigation. A preliminary in vivo efficacy study on LNA-SB-T-12854 against highly metastatic MX-1 human breast tumor xenograft in nude mice has shown promising results, wherein complete eradication of tumor was achieved using q7d x n regimen at 7.5 mg/kg/dose. The results will be published elsewhere.
Fig. 4.

Omega-3 polyunsaturated fatty acid – fluorotaxoid conjugates
4. Self-immolative disulfide linkers for tumor-targeted drug delivery
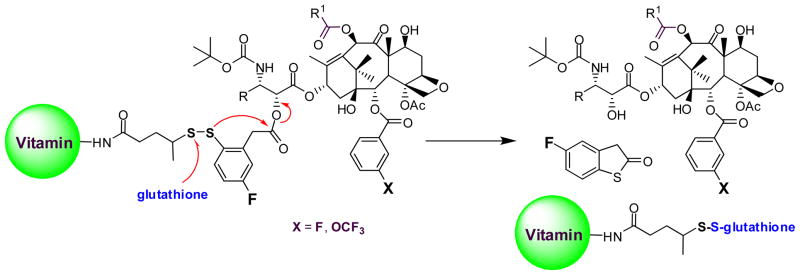
Cleavable disulfide linkers have been widely employed in the development of targeted drug conjugates, including those bearing vitamins and mAbs as TTMs [50, 51]. As the concentration of glutathione (GSH) is three orders of magnitude lower in the blood stream than cancer tissues, disulfide linkers are relatively stable in circulation, yet readily cleave upon internalization of the conjugate [52]. Previously, we synthesized anti-EGFR mAb-taxoid conjugates, bearing an aliphatic disulfide linker, formed via thiol-disulfide exchange reaction between mAb-SH and the methyldisulfanylpropanoyl group at C10 of a 2nd-generation taxoid [53]. These conjugates showed excellent selectivity in vitro and remarkable antitumor activity in vivo against A431 human squamous tumor xenografts in SCID mice, resulting in eradication of the tumor without appreciable systemic toxicity [53]. However, the modification at C10 of the taxoid resulted in 8-fold loss of potency relative to the parent taxoid [53]. Accordingly, the 2nd-generation self-immolative linker system was designed and developed to allow the release of the unmodified taxoid with uncompromised potency.
The 2nd-generation disulfide linker system contains a strategically incorporated phenylacetate moiety adjacent to the disulfide bond that undergoes a thiolactonization to release a drug via cleavage of the disulfide linkage (Fig. 5). First, the attack of an intracellular free thiol such as GSH on the disulfide bond results in the formation of an intermediate thiophenolate or sulfhydrylphenyl group. This species rapidly undergoes an intramolecular nucleophillic attack on the ester group, resulting in ring closure and drug release. The validity of this self-immolative drug-release mechanism has been proven in a model system using fluorine-labeling and monitoring by 19F NMR spectroscopy (Fig. 6) [7], as well as in a real system with cancer cells using fluorescence-labeling and confocal fluorescence microscopy (CFM) [9]. These self-immolative disulfide linkers have been successfully incorporated to various tumor-targeting drug conjugates and their efficacy evaluated in cancer cells [8, 9, 54].
Fig. 5.

Second-generation self-immolative disulfide linker
Fig. 6.
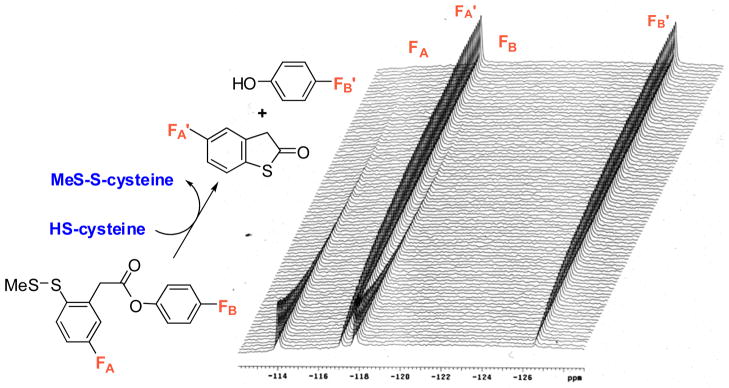
Time-dependent monitoring of disulfide cleavage and thiolactonization by 19F NMR in a model system [Adapted from Ref. 7]
5. Vitamins as Tumor-Targeting Modules
Vitamins are essential for the growth and development of all living cells, but cancer cells depend on certain vitamins, such as those essential for cell division, to sustain their rapid growth. Receptors for these vitamins, including those for folic acid (vitamin M, vitamin B9), biotin (vitamin H, vitamin B7, coenzyme R) and vitamin B12, are overexpressed on the cancer cell surface, providing useful targets for tumor-targeted drug delivery [55]. Biotin is a water-soluble vitamin, which serves as a coenzyme for five biotin-dependent carboxylases and plays a significant role in epigenetic regulation, fatty acid synthesis, energy production and the metabolism of fats and amino acids [56, 57]. Biotin receptors had not been studied until 2004 when vitamin receptor-targeted rhodamine polymers indicated that receptors for biotin were even more overexpressed on the surface of cancer cells than those for folic acid and vitamin B12 [55]. Thus, the biotin receptor has emerged as a novel target for tumor-targeted drug delivery in addition to the well-established folate receptors.
Fig. 7 illustrates a general mechanism for the internalization of a tumor-targeting drug conjugate through receptor-mediated endocytosis (RME), drug release, and the binding of the released drug to the target protein (taxoid binds microtubules) [8]. The course of events for taxoid conjugates bearing a vitamin as the tumor-targeting module is as follows: The vitamin moiety binds to its cell-surface receptor, initiating a signaling cascade and an invagination of the membrane. The drug conjugate is internalized to form intracellular vesicles, which fuse with the early endosome. The highly reducing environment and abundance of GSH would cleave the disulfide linker, initiating thiolactonization and drug release. The released taxoid binds the microtubule target to exert its cytotoxic effect.
Fig. 7.

Schematic representation of the RME of a drug conjugate, drug release and drug-binding to the target protein [adapted from Ref. 9].
The cellular internalization via RME, drug release via disulfide bond cleavage and binding of the released taxoid “warhead” to microtubules were validated by CFM analysis of three fluorescent probes, i.e., (a) biotin-FITC conjugate, (b) biotin-linker-coumarin conjugate and (c) biotin-linker-taxoid(fluorescein) [6, 9, 54]. Also, the internalization of a fluorescently labeled taxoid was quantified by fluorescence activated cell sorting (FACS) flow cytometry [6, 9, 54]. In addition, biotin-linker-taxoid conjugate was synthesized and assayed in vitro against L1210 (mouse lymphocytic leukemia), L1210FR (folate and biotin receptors overexpressed L1210 leukemia) and WI38 (normal human lung fibroblastoma) cells to examine the efficacy of the biomarker-specific targeting of the conjugate [6, 9, 54]. The IC50 values of the conjugate (taxoid = SB-T-1214) against L1210FR, L1210 and WI38 cell lines were 8.80 nM, 522 nM and 570 nM, respectively. The results clearly indicate the high biomarker-specificity of the drug conjugate, which is consistent with the RME-based internalization and drug release observed by CFM and flow cytometry using fluorescent probes. Accordingly, this tumor-targeted drug delivery system bearing a vitamin as the tumor-targeting module is ready to utilize highly potent fluorotaxoids as “warheads”.
5.1. Vitamin-linker-taxoid drug conjugates with fluorine probes
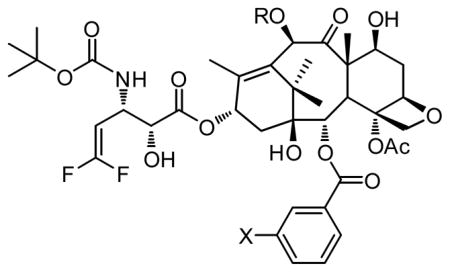
Besides introducing highly potent fluorotaxoids as “warhead” into the vitamin-linker-taxoid conjugates, we have launched the exploration of “fluorine probes” for the assessment of plasma stability of those conjugates as well as monitoring of drug release using 19F NMR analysis. Since the background level of 19F is virtually negligible in multi-component biological media, including blood plasma, cells and tissues, this technique may provide a powerful tool to investigate the behavior of drug conjugates in biological systems. Thus, we set out to synthesize biotin-linker-taxoid conjugates bearing two strategically placed fluorine atoms or groups (Fig. 8). First, we synthesized a conjugate with two fluorine atoms, one in the linker and the other at the 3-position of the C2-benzoate (Fig. 8: X = F, R = isobutenyl, R1 = cyclopropyl). The stability of the conjugate in various biological media is currently being investigated using time-dependent 19F NMR. Usually, such stability assessment is performed by using HPLC or LC-MS analyses. However, often there are many overlapping substances in the chromatograms of biological systems, which hamper accurate analysis. We will also explore 19F NMR analysis in living cells. Of course, the limitation of 19F NMR, in turn, is the sensitivity, and thus the use of a high field instrument (>600 MHz) is necessary.
Fig. 8.
Fluorine-labeled vitamin-linker-drug conjugate that can be used to monitor the drug release by 19F NMR
5.3. Vitamin-linker-taxoid conjugates bearing an imaging arm for PET analysis
As a part of our approach to developing taxoid-based novel “theranostics”, i.e. combination of diagnostics and therapy, we designed vitamin-linker-taxoid conjugates bearing an imaging arm for PET analysis (Fig. 9). In this novel drug conjugate, a vitamin TTM and taxoid “warhead” are linked via a 1,3,5-triazine splitter with a water-solubilizing triethylene glycol (PEG3) spacer and a self-immolative disulfide linker. The imaging arm is attached to the triazine splitter using click chemistry. The 18F-(PEG3)- moiety can be introduced either through the click reaction of 18F-(PEG3)-N3 with the propargylamino-triazine conjugate or 18F fluorination of the MsO-(PEG3)-triazole, linked to the triazine splitter in the drug conjugate, with BuN18F. For a feasibility study, we have successfully synthesized a prototype conjugate with a 19F-(PEG3)-arm, using biotin as TTM and SB-T-1214 as “warhead”. The corresponding “hot” chemistry experiment will be performed in collaboration with Dr. Joanna Fowler’s laboratory at the Brookhaven National Laboratory, shortly. Results of this radio-synthesis and biodistribution analysis of the tumor-targeting drug conjugate in real-time will be reported elsewhere. Again, fluorotaxoids would serve as powerful “warheads” for these “theranostics”, as well.
Fig. 9.
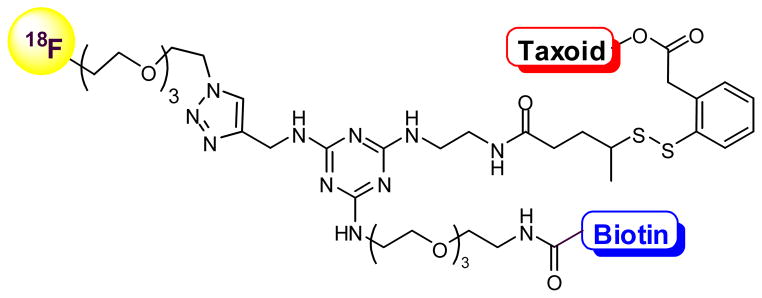
Vitamin-linker-taxoid conjugates bearing an imaging arm for PET analysis
6. Nano-Scale Drug Delivery Scaffolds
6.1. Single-walled carbon nanotubes as vehicles for “Trojan Horse” tumor-targeted drug delivery
Carbon nanotubes have emerged as an efficient tool for transporting and delivering cytotoxic agents [58, 59]. Since functionalized carbon nanotubes (f-CNTs) have displayed low toxicity and are non-immunogenic, these drug delivery systems have been employed as an alternative delivery protocol in targeted chemotherapy. Single-walled carbon nanotubes (SWNTs) functionalized with vitamins as TTM provide a nano-scale and biocompatible platform for tumor-targeted drug delivery. Thus, we designed and synthesized a novel biotin-SWNT-linker-taxoid(fluorescein) conjugate 1 (Fig. 10) to investigate the mass-delivery of payloads to cancer cells, wherein the enhancement of internalization via RME was also expected through multivalent binding of TTM to the vitamin receptors [54].
Fig. 10.
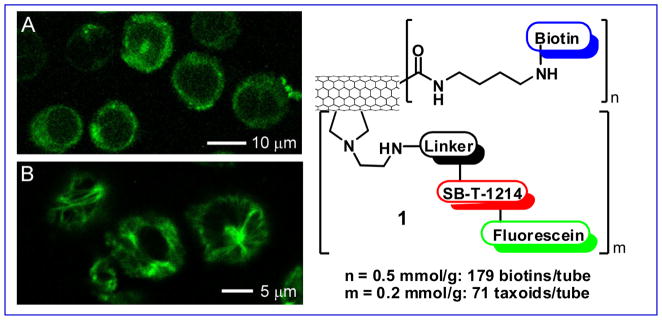
Novel SWNT-based “Trojan Horse” guided molecular missile 1 and the CFM images of L1210FR cells treated with 1 incubated (A) before and (B) after the addition of GSH-ethyl ester. Image B clearly highlights the presence of fluorescent microtubule networks in the living cells generated by the binding of SB-T-1214-fluorescein upon cleavage of the disulfide bond in the linker by either GSH or GSH-ethyl ester. [CFM images were adapted from Ref.{Chen, 2008 #5}]
The internalization via RME, drug release and binding to the target protein (i.e., microtubules) of fluorescein-labeled biotin-SWNT-taxoid conjugate 1 were verified using CFM (Fig. 10) and FACS analysis of L1210FR cells. The results were fully consistent with those for the biotin-linker-taxoid(fluorescein) conjugate mentioned above. The cytotoxicity assay of the conjugate 1 against L1210FR, L1210 and WI38 cell lines (IC50 0.36, >50, and >50 μg/mL respectively) has revealed excellent biomarker-specificity and substantially enhanced potency attributed to mass delivery of the taxoid “warheads” inside the cancer cells [54], assuring the merit of the “Trojan Horse” strategy in tumor-targeting drug delivery. This nano-scale drug delivery system is attractive for the tumor-specific delivery of highly potent fluorotaxoids. In addition to the use of a fluorotaxoid as “warhead”, we plan to investigate possible monitoring of the drug release by 19F NMR, using a fluorine-containing self-immolative disulfide linker, mentioned above, wherein an unmodified “warhead” is used instead of fluorescein-tethered taxoid or fluorotaxoid (Fig. 11).
Fig. 11.

Novel SWNT-based “Trojan Horse” guided molecular missile 2, bearing a fluorine probe
6.2. Asymmetric bowtie dendrimers as vehicles for tumor-targeted drug delivery
Dendrimers are monodisperse, branched polymers with a defined architecture originating from a central core. The high density of surface functional groups allows for polyvalent conjugation of TTMs, drugs, and tracers, making dendrimers attractive candidates for tumor-targeted drug delivery [60]. Recently, an orthogonal diblock strategy for the generation of asymmetric bowtie dendrimers with a polyamido(amine) (PAMAM) backbone and a cystamine core has been developed in our laboratory as well as by others [61]. First, two dendrimers are fully functionalized with TTMs as well as drugs and/or imaging agents. Second, each dendrimer is reductively cleaved to produce two half dendrons that can be asymmetrically joined via a bis-maleimido spacer. This synthetic approach offers the unique advantage of selective modification and quantification of TTMs, drugs, and tracers on each half-dendron prior to coupling. Therefore, this strategy can be adapted to produce tumor-targeting nano-scale drug conjugates with well-defined drug loading and enhanced tumor specificity.
We have designed a novel asymmetric bowtie dendrimer scaffold 3, bearing a 1,3,5-triazine splitter in the bis-malenimide linker module to introduce an imaging module (Fig. 12). We set out to construct a prototype dendrimer conjugate wherein the vitamin TTM is biotin, the “warhead” is fluorotaxoid SB-T-12854, and the imaging module is fluorine (19F for cold material and 18F for PET). In this prototype, an asymmetric bow-tie dendrimer, consisting of the G3 and G1 PAMAM dendrons, is used. The G3 dendron allows the conjugation of 16 biotin molecules, while the G1 dendron holds 4 taxoids. We have successfully synthesized key components and the final assembly to the designed conjugate is in progress. Synthesis and evaluation of this dendrimer-based tumor-targeted drug delivery system will be reported in due course.
Fig. 12.
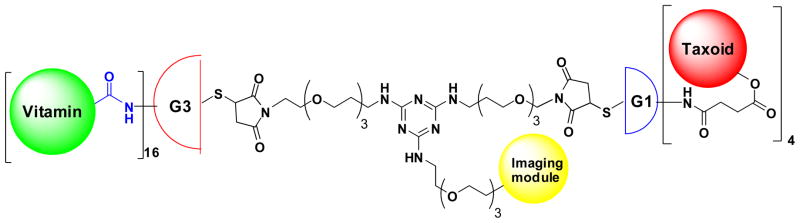
Tumor-targeted drug delivery system based on an asymmetric bowtie PAMAM dendrimer designed for the targeted delivery of new generation taxoids, bearing an imaging arm
7. Concluding Remarks
Tumor-targeted drug delivery systems have been successfully designed and constructed. These novel drug conjugates consist of tumor-targeting modules, mechanism-based self-immolative disulfide linkers and “warheads”. For the “warheads”, highly potent fluorotaxoids with excellent metabolic stability have been designed and developed in addition to new generation taxoids. The omega-3 PUFA–taxoid conjugates and mAb-taxoid conjugates have exhibited remarkable efficacy against human tumor xenografts in animal models with minimal systemic toxicity. Fluorotaxoids are obviously highly potent “warheads” for these tumor-targeting drug conjugates. CFM studies using fluorescent and fluorogenic probes have unambiguously confirmed the designed internalization of drug-conjugates via RME and drug release via glutathione-triggered self-immolation of the disulfide linker. In addition to these fluorescence-based probes, we have been exploring the potential of “fluorine probe” approach based on 19F NMR as well as 18F-tracer for PET analysis. The use of multi-functionalized SWNTs as a drug delivery system bearing multiple-“warheads” and multiple-targeting modules has shown highly promising results on the benefit of mass-delivery (“Trojan Horse” strategy) of anticancer drug molecules to cancer cells with high specificity. For this nano-scale drug delivery system, we have been introducing fluorotaxoids as well as exploring the “fluorine probe” for monitoring drug release by 19F NMR. We have also been constructing another nano-sacle drug delivery system based on asymmetric bowtie PAMAM dendrimers, bearing an imaging module, including 18F tracer for PET. Our future efforts will be concentrated on the design and synthesis of “tailor made nano-medicines”, consisting of a well-defined vehicle, multiple targeting modules (including dual targeting molecules), self-immolative smart linkers, highly potent cytotoxic “warheads” (e.g., fluorotaxoids), and imaging modules (for MRI, PET, fluorescence imaging and 19F NMR), which would enable us to perform diagnostics and therapy in the real time.
Highlights for review.
A series of 3′-difluoromethyltaxoids and 3′-trifluoromethyltaxoids C10 modifications as well as those with C2 and C10 modifications were synthesized, which exhibit substantially higher potencies than those of paclitaxel and docetaxel, especially against multidrug-resistant cell lines (two orders of magnitude more potent than paclitaxel in average).
3′-Difluorovinyl taxoids exhibit several to 16 times better activity against MCF7, HT-29 and PANC-1 cell lines and up to three orders of magnitude higher potency against multi-drug resistant NCI/ADR cell line as compared to paclitaxel.
Those highly potent novel 3′-Rf-taxoids (Rf = CF2H, CF3, CH=CF2) serve as excellent “warheads” in the tumor-targeted drug delivery systems.
Vitamin-linker-drug conjugates bearing a self-immolative disulfide linker have been designed and developed. Using fluorescent probes, the receptor-mediated endocytosis, drug release and drug binding to the target protein (microtubules) have been verified by confocal fluorescence microscopy (CFM) and flow cytometry. This is an excellent system to incorporate a fluorotaxoid as “warhead”.
Nano-scale tumor-targeted drug delivery systems based on single-walled carbon nanotubes (SWNTs) as well as asymmetric bowtie PAMAM dendrimers have been designed and developed. Highly potent fluorotaxoids serve as excellent “warheads” for these “Trojan Horse” molecular missiles. Those drug delivery systems, bearing 18F PET tracer module as well as 19F fluorine probe for 19F NMR analysis for drug release, have been designed and prototype conjugates synthesized.
Acknowledgments
This work has been supported by grants from the National Institutes of Health (GM42798 and CA103314 to I.O.). Generous support from Indena SpA, is gratefully acknowledged
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaracz S, Chen J, Kuznetsova L, Ojima I. Bioorganic Medicine Chemistry Letters. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 2.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proceedings of the National Academy of Sciences, USA. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu TC, Marks JW, III, Lavery LA, Faulkner S, Rosenblum MG, Ellington AD, Matthew Levy M. Cancer Research. 2006;66:5989–5992. doi: 10.1158/0008-5472.CAN-05-4583. [DOI] [PubMed] [Google Scholar]
- 4.Ducry L, Stump B. Bioconjugate Chemistry. 2010;21:5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 5.Xia W, Low P. Journal of Medicinal Chemistry. 2010;53:6811–6824. doi: 10.1021/jm100509v. [DOI] [PubMed] [Google Scholar]
- 6.Ojima I, Zuniga E, Berger W, Seitz J. Future Medicinal Chemistry. 2012;4:33–50. doi: 10.4155/fmc.11.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ojima I. ChemBioChem. 2004;5:628–635. doi: 10.1002/cbic.200300844. [DOI] [PubMed] [Google Scholar]
- 8.Ojima I. Accounts of Chemical Reseach. 2008;41:108–119. doi: 10.1021/ar700093f. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Zhao X, Chen J, Chen J, Kuznetsova L, Wong S, Ojima I. Bioconjugate Chemistry. 2010;21:979–987. doi: 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobb S, Murphy C. Journal of Fluorine Chemistry. 2009;130:132–143. [Google Scholar]
- 11.Reivich M, Kuhl D, Wolf A, Greenberg J, Phelps M, Ido T, Casella V, Fowler J, Hoffman E, Alavi A, Som P, Sokoloff L. Circulation Research. 1979;44:127–137. doi: 10.1161/01.res.44.1.127. [DOI] [PubMed] [Google Scholar]
- 12.Ametamey S, Honer M, Schubiger P. Chemical Reviews. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 13.Kimura Y, Simeon FJ, Atazawa H, Mozley P, Pike V, Innis R, Fugita M. European Journal of Nuclear Medicine and Molecular Imaging. 2010;37:1943–1949. doi: 10.1007/s00259-010-1447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowinsky E. Annual Reviews in Medicine. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 15.Bono JD, Oudard S, Ozguroglu M, Hansen S, Machiels J, Shen L, Matthews P, Sartor A. Journal of Clinical Oncology. 2010;28:s4508. [Google Scholar]
- 16.Bissery M, Nohynek G, Sanderink G, Lavelle F. Anticancer Drugs. 1995;6 doi: 10.1097/00001813-199506000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Fojo A, Ueda K, Slamon D, Poplack D, Gottesman M, Pastan I. Proceedings of the National Academy of Sciences, USA. 1987;84:265–269. doi: 10.1073/pnas.84.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottesman M, Pastan I. Annual Reviews in Biochemistry. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 19.Ojima I, Das M. Journal of Natural Products. 2009;72:554–565. doi: 10.1021/np8006556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pepe A, Kuznetsova L, Sun L, Ojima I. Fluoro-taxoid anticancer agents. In: Ojima I, editor. Fluorine in Medicinal Chemistry and Chemical Biology. Wiley-Blackwell; Chichester: 2009. pp. 117–139. [Google Scholar]
- 21.Ojima I, Chen J, Sun L, Borella C, Wang T, Miller M, Lin S, Geng X, Kuznetsova L, Qu C, Gallagher G, Zhao X, Zanardi I, Xia S, Horwitz S, Clair JS, Guerriero J, Bar-Sagi D, Veith J, Pera P, Bernacki R. Journal of Medicinal Chemistry. 2008;51:3203–3221. doi: 10.1021/jm800086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuznetsova L, Pepe A, Ungureanu I, Pera P, Bernacki R, Ojima I. Journal of Fluorine Chemistry. 2008;129:817–828. doi: 10.1016/j.jfluchem.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuznetsova L, Sun L, Chen J, Zhao X, Seitz J, Das M, Li Y, Veith J, Pera P, Bernacki R, Xia S, Horwitz S, Ojima I. Journal of Fluorine Chemistry. 2012;143:177–188. doi: 10.1016/j.jfluchem.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojima I, Kuduk SD, Slater JC, Gimi RH, Sun CM, Chakravarty S, Ourevitch M, Abouabdellah A, Bonnet-Delpon D, Begue J-P, Veith JM, Pera P, Bernacki Ralph J. Syntheses, biological activity, and conformational analysis of fluorine-containing taxoids. In: Ojima I, McCarthy JR, Welch JT, editors. Biomedical Frontiers of Fluorine Chemistry, ACS Symposium Series 639. American Chemical Society; Washington, D. C: 1996. pp. 228–243. [Google Scholar]
- 25.Ojima I, Inoue T, Chakravarty S. Journal of Fluorine Chemistry. 1999;97:3–10. [Google Scholar]
- 26.Ojima I, Slater J, Michaud E, Kuduk S, Bounaud PY, Vrignaud P, Bissery M, Veith J, Pera P, Bernacki R. Journal of Medicinal Chemistry. 1996;39:3889–3896. doi: 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
- 27.Ojima I, Kuduk S, Slater J, Gimi R, Sun CM. Tetrahedron. 1996;52:209–224. [Google Scholar]
- 28.Ojima I, Slater J. Chirality. 1997;9:487–494. doi: 10.1002/(SICI)1520-636X(1997)9:5/6<487::AID-CHIR15>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 29.Gut I, Ojima I, Vaclavikova R, Simek P, Horsky S, Linhart I, Soucek P, Knodrova E, Kuzetsova L, Chen J. Xenobiotica. 2006;36:772–792. doi: 10.1080/00498250600829220. [DOI] [PubMed] [Google Scholar]
- 30.Ehrlichová M, Ojima I, Václavíková R, Němcová-Fürstová V, Vobořilová J, Šimek P, Horský S, Souček P, Kovár J, Gut I. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2012;385:1035–1048. doi: 10.1007/s00210-012-0785-4. [DOI] [PubMed] [Google Scholar]
- 31.Marre F, Sanderink F-J, Sousa Gd, Gaillard C, Martinet M, Rahmani R. Cancer Research. 1996;56:1296–1302. [PubMed] [Google Scholar]
- 32.Monsarrat B, Mariel E, Cros S, Gares M, Guenard D, Gueritte-Voegelein F, Wright M. Drug Metabolism and Disposition. 1990;18:895–901. [PubMed] [Google Scholar]
- 33.Cresteil T, Monsarrat B, Alvinerie P, Treluyer J, Vleira I, Wright M. Cancer Research. 1994;54:386–392. [PubMed] [Google Scholar]
- 34.Voborilova J, Nemcoa-Furstova V, Neubauerova J, Ojima I, Zanardi I, Gut I, Kovar J. Investigational New Drugs. 2011;29:411–423. doi: 10.1007/s10637-009-9368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Botchkina G, Zuniga E, Das M, Wang Y, Wang H, Zhu S, Savitt A, Rowehl R, Leyfman Y, Ju J, Shroyer K, Ojima I. Molecular Cancer. 2010;9:192–204. doi: 10.1186/1476-4598-9-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li F, Tiede B, Massague J, Kang Y. Cell Research. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 37.Zuniga ES. PhD Dissertation. Stony Brook University; 2012. [Google Scholar]
- 38.Meyer B, Mann N, Lewis J, Milligan G, Sinclair A, Howe P. Lipids. 2003;38:391–398. doi: 10.1007/s11745-003-1074-0. [DOI] [PubMed] [Google Scholar]
- 39.Grammatikos S, Subbaiah P, Victor T, Miller W. British Journal of Cancer. 1994;70:219–227. doi: 10.1038/bjc.1994.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sauer L, Dauchy R, Blask D. Cancer Research. 2000;60:5289–5295. [PubMed] [Google Scholar]
- 41.Bradley M, Webb N, Anthony F, Devanesan P, Wittman P, Hemamalini S, Chander M, Baker S, He L, Horowitz S, Swindell C. Clinical Cancer Research. 2001;7:3229–3238. [PubMed] [Google Scholar]
- 42.Seitz J, Ojima I. Chapter V.9. Drug Conjugates with Polyunsaturated Fatty Acids. In: Kratz F, Senter P, Steinhagen H, editors. Drug Delivery in Oncology - From Research Concepts to Cancer Therapy. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 43.Peters T., Jr Advances in Protein Chemistry. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- 44.Sparreboom A, Wolff A, Verweij J, Zabelina Y, Zomeren Dv, McIntire G, Swindell C, Donehower R, Baker S. Clinical Cancer Research. 2003;9:151–159. [PubMed] [Google Scholar]
- 45.Desai N, Trieu V, Damascelli B, Soon-Shiong P. Translational Oncology. 2009;2:59–64. doi: 10.1593/tlo.09109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schubert W, Frank P, Ranzani B, Park D, Chow C, Lisanti M. Journal of Biological Chemistry. 2001;276:48619–48622. doi: 10.1074/jbc.C100613200. [DOI] [PubMed] [Google Scholar]
- 47.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P. Clinical Cancer Research. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 48.Gradishar W, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. Journal of Clinical Oncology. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 49.Kuznetsova L, Chen J, Sun X, Wu A, Pepe J, Veith P, Pera R, Bernacki R, Ojima I. Bioorganic Medicine Chemistry Letters. 2006;16:974–977. doi: 10.1016/j.bmcl.2005.10.089. [DOI] [PubMed] [Google Scholar]
- 50.Vlahov I, Santhapuram H, Kleindl P, Howard S, Stanford K, Leamon C. Bioorganic Medicine Chemistry Letters. 2006;16:5093–5096. doi: 10.1016/j.bmcl.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 51.Wu A, Senter P. Nature Biotechnology. 2005;23:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 52.Kigawa J, Minagawa Y, Kanamori Y, Itamochi H, Cheng X, Okada M, Oisho T, Terakawa N. Cancer. 1998;82:697–702. doi: 10.1002/(sici)1097-0142(19980215)82:4<697::aid-cncr12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 53.Ojima I, Geng X, Wu X, Qu C, Borella C, Xie H, Wilhelm S, Leece B, Bartle L, Goldmacher V, Chari R. Journal of Medicinal Chemistry. 2002;45:5620–5623. doi: 10.1021/jm025540g. [DOI] [PubMed] [Google Scholar]
- 54.Chen J, Chen S, Zhao X, Kuznetsova L, Wong S, Ojima I. Journal of the American Chemical Society. 2008;130:16778–16785. doi: 10.1021/ja805570f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russell-Jones G, McTavish K, McEwan J, Rice J, Nowotnik D. Journal of Inorganic Biochemistry. 2004;98:1625–1633. doi: 10.1016/j.jinorgbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 56.Zempleni J. Annual Reviews in Nutrition. 2005;25:175–196. doi: 10.1146/annurev.nutr.25.121304.131724. [DOI] [PubMed] [Google Scholar]
- 57.Zempleni J, Wijeratne S, Hassan Y. Biofactors. 2009;35:36–46. doi: 10.1002/biof.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bianco A, Kostarelos K, Prato M. Current Opinions in Chemical Biology. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Prato M, Kostarelos K, Bianco A. Accounts of Chemical Research. 2008;41:60–68. doi: 10.1021/ar700089b. [DOI] [PubMed] [Google Scholar]
- 60.Gillies E, Frechet J. Drug Discovery Today. 2005;10:35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 61.Gaertner H, Cerini F, Kamath A, Rochat AF, Siegrist CA, Menin L, Hartley O. Bioconjugate Chemistry. 2011;22:1103–1114. doi: 10.1021/bc1005653. [DOI] [PubMed] [Google Scholar]