

Abstract
Advances in hydrogel design have revolutionized the way biomaterials are applied to address biomedical needs. Hydrogels were introduced in medicine over 50 years ago and have evolved from static, bioinert materials to dynamic, bioactive microenvironments, which can be used to direct specific biological responses such as cellular ingrowth in wound healing or on-demand delivery of therapeutics. Two general classes of mechanisms, those defined by the user and those dictated by the endogenous cells and tissues, can control dynamic hydrogel microenvironments. These highly tunable materials have provided bioengineers and biological scientists with new ways to not only treat patients in the clinic but to study the fundamental cellular responses to engineered microenvironments as well. Here, we provide a brief history of hydrogels in medicine and follow with a discussion of the synthesis and implementation of dynamic hydrogel microenvironments for healthcare-related applications.
Keywords: Hydrogels, biomaterials, tissue engineering, cellular microenvironment, dynamic
Introduction
Hydrophilic polymer networks or hydrogels are emerging as diverse material platforms designed to stimulate and react to specific cellular responses on the molecular level. These sophisticated materials are being developed not only to interact with the body in a clinical setting and promote natural healing, but also to probe fundamental interactions between cells and material interfaces. Hydrogels are a broad class of cross-linked polymeric networks, which often swell extensively in water or biological fluids but maintain their three-dimensional structure. The high water content of hydrogels is appealing for numerous biomedical applications because it is reminiscent of the highly hydrated physiological extracellular matrix environment in soft tissues. Hydrogels as biomaterials have evolved from static implants and devices to dynamic, bioresponsive scaffolds, drug delivery vehicles, and cell culture platforms.
The development of contact lenses provides an illustrative example of the evolution of one hydrogel biomaterial. In 1508 Leonardo da Vinci sketched out several ideas that described the basic principles of a contact lens. These concepts were finally applied in the field of optometry in the 1880s with the invention of the glass contact lens. However, from the 1880s to 1935, no significant progress on lens design was made. In 1936, optometrist William Feinbloom introduced the first plastic lenses[1]. This marks the beginning of a time period that is often referred to as the first generation of biomaterials or the “surgeon hero” era. Following World War II, new metal, ceramic and polymeric materials were available, and surgeons began implanting early biomaterials, such as Teflon, nylon, methacrylate polymers, titanium and stainless steel in high-risk procedures[2]. These materials were selected “off the shelf” with the goal of achieving suitable mechanical properties and minimal toxicity because materials had not yet been designed specifically for biomedical applications. The first plastic contact lenses were made of poly(methyl methacrylate) (PMMA), a material used to make airplane cockpit canopies during World War II. The lenses were rigid, not gas permeable and bioinert[2]. When the first hydrogel synthesized specifically for biomedical applications, poly(2-hydroxyethylmethacrylate) (pHEMA), was described in 1960, it was quickly incorporated into and patented for soft contact lenses[3].
Soft contact lens technologies advanced quickly to increase oxygen permeability by copolymerizing existing hydrogel components with hydrophobic components, such as PDMS and perfluoropolyethers (PFPE). Creative designs that addressed the morphology of the copolymer phases led to the creation of mobile lenses for extended-wear and treatment of complicated optical problems[4]. Although static, inert contact lens systems have successfully corrected vision in increasingly more ophthalmically compatible ways; dynamic, bioresponsive designs are currently under development for new biomedical applications. For example contact lenses are now being studied for ophthalmic drug delivery[5] and measuring and responding to ophthalmic pressure[6] or glucose levels[7].
From first principles, hydrogel precursors can be of natural or synthetic origin. Natural hydrogels are typically composed of extracellular matrix (ECM) components, such as polysaccharides or proteins. Natural components are inherently biocompatible and can be easily recognized and remodeled by cells. Hyaluronic acid (HA) is an example of a polysaccharide that has been widely used to form drug releasing particles and macroscopic networks for tissue regeneration[8]. Collagen is the most prevalent protein in the ECM and has also been widely used to create hydrogel scaffolds for tissue engineering[9]. The methods of preparation and application of hydrogels based on natural polymers in tissue engineering has been reviewed extensively[10]. Despite the advantages of biocompatibility and cellular recognition, natural hydrogels contain endogenous signals that are difficult to isolate and control[11]. The presentation of defined mechanical and biochemical cues is not facile when starting with natural components. Moreover, when these materials are purified from animal tissues, there can be significant batch-to-batch variability and a risk of immunogenicity and disease transmission. To overcome these challenges, engineers and scientists are developing strategies to give biofunctionality to synthetic hydrogels[12], and these strategies are the focus of this review.
In particular, this review begins with a brief history of the application of hydrogels in medicine and then focuses on the development of dynamic hydrogel microenvironments. We specifically examine the synthesis and implementation of synthetic hydrogels engineered with defined, dynamic properties for biomedical applications. Hydrogels have evolved from static, bioinert materials to dynamic, bioactive microenvironments that can be used as scaffolds for tissue regeneration, as well as drug and cell delivery devices that present specific cues to elicit cellular responses in real-time. Further, advances in the design of hydrogels for three-dimensional culture have led to new ways for biological scientists and bioengineers to probe and answer fundamental questions related to how cells receive information from their extracellular, material environment.
Brief History of Hydrogels in Medicine
The first hydrogel material to be widely used as a biomedical implant was a poly(vinyl alcohol) (PVA) [13] sponge formed with foaming agents, cross-linked with formaldehyde. This material was invented in 1949 and marketed with the trade name Ivalon®. Starting in the 1950s, it was studied for nearly every biomedical application imaginable, including: skin replacement[14], articular cartilage replacement[15, 16], scaffolds for bone regeneration[17], vascular prostheses[18], treatment for tuberculosis[19] and as an embolic material[20]. Even though Ivalon® sponges were easily modeled to construct a variety of shapes, these implants essentially served as static scaffold materials with fixed mechanical properties. PVA and heparinized PVA have also been studied extensively for contact with blood. A suitable blood-contacting material inhibited clot formation by reducing protein and platelet adsorption. Both PVA hydrogels containing entrapped heparin[21] and PVA hydrogels copolymerized with heparin[22] significantly prolonged clotting and were employed as coatings on blood-contacting implants and devices such as arteriovenous (AV) shunts in canines[23]. These materials were also suggested for use as membranes in artificial kidneys[22].
Polyacrylamide hydrogels for electrophoresis provide another example of early hydrogels used in medicine. Polyacrylamide, along with many other hydrogel coatings including pHEMA, ethyl methacrylate (EMA) and poly(HEMA-co-EMA), was grafted onto cross-linked poly(dimethylsiloxane) (PDMS) to decrease thrombogenicity for blood contacting applications[24, 25]. Blood contacting surfaces such as poly(ethylene terephthalate) (PET), poly(urethane) (PU) and PVA were also modified with high molecular weight poly(ethylene glycol) (PEG) to reduce the risk of thrombogenicity[26]. Block copolymers of PEG and PET were synthesized for early hemodialysis membranes[27]; however, cellulose, a natural hydrogel, has remained the material of choice for this application[26].
The first hydrogel material formulated specifically for use in healthcare, pHEMA, was described in a publication appearing in 1960[3]. pHEMA was synthesized by copolymerization of 2-hydroxyethyl methacrylate (HEMA) with the cross-linker ethylene dimethacrylate (EDMA) in an aqueous solution. The highly hydrated material resulted in mechanical properties reminiscent of native tissue and was immediately patented for use in soft contact lenses in the United States in 1962 and 1965[2]. Copolymerization of HEMA with more hydrophilic monomers, such as N-vinyl pyrrolidone (NVP) and methacrylic acid (MAA), increased swelling and oxygen permeability of the resulting pHEMA gels [28]. Advances in the chemistry of contact lens design have created more ophthalmically compatible lenses. These lenses have high oxygen transmissibility, move on the eye and do not accumulate significant lipid or protein deposits. Current extended-wear contact lenses are copolymers of siloxanes with hydrophilic monomers to create specifically designed microphases within the lens material to achieve these unique material properties [4].
Coatings for biomaterial implants that minimized cell and protein adhesion were an early application of the synthetic hydrogel poly(ethylene oxide) (PEO), a high molecular weight poly(ethylene glycol) (PEG). PEO and PEG are chemically equivalent but originate from different precursors. It was shown that PEO incorporated into block copolymers [27] or covalently attached to polymer surfaces reduced protein adsorption[29]. A PEO product called Vigilon was cross-linked with radiation and used as a wound covering[30]. Further research, including clinical studies, has shown that PEG and PEG hydrogels are nontoxic and non-immunogenic[31]. Early applications of PEG took advantage of its bioinert properties to process bulk materials or to modify other materials with a PEG surface coating, and the current literature is filled with similar examples exploiting PEG for its unique properties.
The use of hydrogels as a delivery vehicle for drugs was another early application of hydrogels in medicine. Water-soluble cellulose derivatives were the first materials used for controlled release of drugs[32]. Powders of PVA were also used to create swellable tablets for controlled release[33]. The first bio-erodible hydrogels were natural or derivatives of natural polymers including modified chitin, dextran, starch, gelatin and polypeptides[2]. These controlled release devices are amongst the first examples to exploit the responsiveness of hydrogels to engineered dynamic hydrogel environments (i.e., swelling-controlled release of entrapped molecules).
This brief history of hydrogels in medical applications is not comprehensive, but instead, highlights the evolution of hydrogel biomaterials from static to dynamic microenvironments. Figure 1 depicts the applications of some of the prominent hydrogels used in healthcare. The broad and diverse application of the Ivalon® PVA sponge to regenerative medicine in nearly every organ of the human body represents the application of a static, “off-the-shelf” product that was readily implanted without being specifically designed to address a biomedical need. The evolution of hydrogels from bioinert structures to minimize interactions with cells and tissues, for blood-contacting materials, contact lenses, antifouling coatings and drug delivery vehicles, to current trends in materials designed to elicit a controlled response from the environment marks the beginning of the application of bioresponsive design principles and leads to the focus of this review. This contribution will highlight aspects of bioactive and bioresponsive biomaterials design by examining the mechanisms employed to change hydrogel structures when engineering dynamically responsive microenvironments.
Figure 1.

Applications of hydrogels in healthcare. These examples cover a range of hydrogel microenvironments from static to dynamic that have been employed to address clinical needs. This list is not comprehensive but depicts some of the prominent hydrogels used in clinical applications.
Hydrogel Structure and Properties
To engineer dynamically responsive hydrogel microenvironments, it is typically necessary to alter the structure or chemistry of the hydrogel network in real-time. To this end, it is important to first understand how hydrogel structure influences material properties. Hydrogels are cross-linked polymer networks or gels that swell extensively in water. Network gelation is a transition of connectivity among polymer chains. Prior to gelation, the material is a fluid mixture of polydisperse branched finite-sized polymer chains and unreacted monomer or cross-linking molecules. At the gel point, at least one molecule of the polymer must be on the order of the dimensions of the macroscopic sample[34]. Upon gelation a critical number of cross-links form between polymer chains to render an insoluble three-dimensional matrix. Cross-links may be chemical or physical in nature. Chemical cross-links are strong covalent bonds while physical cross-links may be strong crystalline regions or weaker, reversible associations among polymer chains (e.g., electrostatic, hydrophobic or dipole-dipole). In many dynamic hydrogel systems, reverse gelation (i.e., solubilization) is triggered to impart degradation of cell scaffolds, delivery and release of biologics, and/or alteration of material properties [35].
The cross-linking density (ρx), the number of chemical or physical cross-links in a given volume, controls many fundamental hydrogel properties that are important in healthcare applications (Figure 2)[36–38]. These properties include the volumetric swelling ratio (Q), the shear modulus (G) and the diffusion coefficient (D) of entrapped molecules [39]. The volumetric swelling ratio (the ratio of the volume of the water-swollen gel to the volume of dry polymer) is representative of the water content of the swollen hydrogel, and is thermodynamically predicted based on the structure of the gel and the polymer-solvent interaction parameter [40]. The modulus of the gel is directly related to the network crosslinking density by rubber elasticity theory [41, 42], and defines many aspects of the mechanical properties of the gel. The diffusion coefficient is directly related to the network mesh size (ζ), which is a measure of the space that is available between macromolecular chains for the diffusion of cell-secreted molecules for tissue regeneration or drugs intended for delivery[43, 44]. For highly swollen, non-ionic hydrogels, these properties scale with ρx (Table 1). The interdependent nature of these relationships requires compromise to optimize hydrogel properties for specific applications.
Figure 2.

The relationship between cross-linking density and hydrogel properties. Two network structures representative of low and high cross-linking densities are depicted to demonstrate the relationship between cross-linking density and basic hydrogel properties for highly swollen, nonionic gels: shear modulus (G), equilibrium swelling ratio (Q) and diffusivity (D). As cross-linking density increases mesh size (ζ), which is a measure of the space that is available between macromolecular chains for the diffusion decreases.
Table 1.
Many fundamental hydrogel properties are directly related to cross-linking density (ρx). The following relationships are valid for the volumetric swelling ratio (Q), the shear modulus (G) and the diffusion coefficient (D) of highly swollen, nonionic networks.
Beyond the physics of network properties, the chemical composition of the hydrogel precursors regulates the final biochemical properties of the hydrogel including charge, hydrophilicity and bioactivity. These chemical properties influence noncovalent intramolecular within polymer chains and intermolecular interactions among polymer chains in the network and between the network and the solvent. Pendant groups incorporated into the hydrogel backbone can render the material neutral, cationic, anionic or ampholytic[45]. The ionic character and hydrophilicity of the polymer chains affect the degree of swelling of the hydrogel, and allow facile tailoring of pH responsive properties (e.g., depending on the pKa or pKb of pendant functional groups). Since the 1990s, the bioactivity of hydrogels has been tailored by the addition of small peptide ligands to render synthetic gels with biological epitopes to direct specific cell interactions. For example, in 1991, Hubbell et al. demonstrated that immobilization of the Gly-Arg-Gly-Asp (GRGD) ligand on a pHEMA surface promoted mammalian cell adhesion[46]. Now, the manipulation of pendant groups to add or subtract adhesive ligands is also frequently employed to transform dynamic hydrogel microenvironments and direct cell-material interactions.
Control over the physical and chemical properties of hydrogels by a specific stimulus is highly desirable for medical applications. Dynamically responsive moieties can be triggered by user- or cell-controlled mechanisms and can be incorporated into hydrogels in the cross-links or as pendant functional groups. The following sections will provide specific examples of several of these mechanisms currently used to elicit dynamic responses in hydrogel materials and their targeted applications (Figure 3).
Figure 3.

Dynamic hydrogel microenvironments. Hydrogel networks can be tailored to be responsive to a continuum of stimuli ranging from user-defined to cell and tissue-dictated. This schematic highlights a few specific examples of the mechanisms currently used to elicit dynamic responses in hydrogel materials that will be reviewed. The orange macromolecular chain segments represent photodegradable linkages that can be manipulated to degrade a hydrogel (far left) or release bioactive moieties (left middle). The green and purple chains depict a thermoresponsive hydrogel that assembles or disassembles in response to temperature (right middle). The pink macromolecular chain segments illustrate cell-responsive linkages that degrade the hydrogel upon enzyme secretion (right).
User-Controlled Mechanisms to Elicit Dynamic Responses
Several mechanisms can be employed to remotely control dynamic responses in hydrogel materials. The ability to trigger these responses affords temporal control over experimental variables that are stable at initial values and then specifically altered with the introduction of external stimuli. User-controlled mechanisms, such as photoexcitation, are currently a tool for improving the field’s understanding of how cells interact with and receive information from their microenvironments. Other physical external stimuli include temperature or electromagnetic fields and have been translated to clinical applications.
Photopolymerization and Photodegradation
Photopolymerization reactions are versatile light-induced processes that proceed rapidly at ambient conditions. Light is a stimulus that may be remotely controlled by the user both spatially and temporally[47]. Photoresponsive moieties can be incorporated into a hydrogel as a cross-linking molecule or as part of a pendant group. Pendant reactive moieties such as thiol or acrylate groups may participate in photo-initiated reactions to facilitate the addition of bioactive molecules or additional cross-links to a network structure, allowing dynamic and intimate control of the network’s physical and chemical properties. Photodegradable moieties such as o-nitrobenzyl ethers or coumarins contain bonds that cleave upon exposure to specific wavelengths of light under cytocompatible conditions. These molecules can be incorporated into the network structure to control physical properties or used to cage reactive groups that when exposed to light enable biochemical patterning. In this way, photopolymerization and photodegradation reactions can be used to recapitulate aspects of the dynamic biochemical and biophysical cues found in the cellular microenvironment. This can be achieved through light-activated ligand presentation, delivery of soluble factors and physical or mechanical changes in the polymer network.
In an early example of spatially and temporally controlled biochemical patterning within a hydrogel, Shoichet and colleagues incorporated nitrobenzyl-photocaged thiols into an agarose hydrogel that were subsequently released with focused laser light and spontaneously reacted with maleimide-peptides that were present in the solvent[48]. This technique was more recently extended to immobilize two stem-cell differentiation proteins (sonic hedgehog (SHH) and ciliary neurotrophic factor (CNTF)) in a spatial orientation reminiscent of their presentation in vivo (Figure 4a)[49]. Again, an agarose gel was modified with photocaged-thiols, and upon two-photon irradiation, the reactive groups were liberated. In this example thiols were photocaged using coumarin moieties. One part of each of two orthogonal binding pairs, Barnas-barstar and streptavidin-biotin, was maleimide-functionalized and sequentially immobilized with spatiotemporal control within the gel. The candidate proteins containing the appropriate binding partners were then swollen into the gel to bind to the pre-defined pattern. Adult neural precursor cells were seeded onto and migrated along immobilized-SHH-gradients patterned using this technique[49].
Figure 4.

User-defined, photoresponsive dynamic hydrogels. a) Photocleavage of courmarin-caged-thiols enables spatial and temporal patterning of multiple growth factors in 3D. Reprinted by permission from Macmillan Publishers Ltd: Nature Materials [47], copyright 2011. b) Photodegradable hydrogel networks containing-nitrobenzyl ether moieties in the cross-linking molecules can be used to guide cellular outgrowth. Scale bar, 100 μm. Reprinted by permission from Macmillan Publishers Ltd: Nature Chemistry [50], copyright 2011. c) Photoisomerization of azobenzene cross-linkers facilitates delivery of small therapeutic molecules modeled by fluorescein release upon UV irradiation. Reproduced in part from [53] with permission of The Royal Society of Chemistry.
The West lab has also exploited microfabrication techniques that allow post-gelation presentation of bioepitopes in 3D. Here, “blank” hydrogels were formed via radical chain photopolymerization of PEG diacrylate (PEGDA). Post-gelation, a small number of free acrylate functionalities remained for subsequent conjugation to acrylated-bioepitopes. Two distinct peptides derived from fibronectin (Fn) were modified with reactive functional groups (acryl-PEG-RGDS and acryl-PEG-CS-1) and were swollen into the network with a photoinitiator in series. Two-photon absorption laser scanning lithography (TPA-LSL) was used to pattern small volumes selectively within the hydrogel with each epitope. This technique affords user-defined fabrication of pattern sizes ranging from 1 μm up to 1 mm with precise spatial control. Ligand density can also be controlled with laser intensity and scan speed[50].
The Anseth research group increased the level of user-defined control of biochemical patterning by designing a fully cytocompatible hydrogel system that combines two bioorthogonal chemical reactions, which facilitate reversible presentation of bioepitopes. Cytocompatibliity enables the researcher to encapsulate and culture cells within these gels and was achieved through a copper-free click reaction. Hydrogels were formed by a strain promoted, azide-alkyne cycloaddition (SPAAC) reaction between a PEG macromer and a peptide, which contained pendant, photoreactive alkenes. The bioactive molecules of interest were synthesized to contain both a thiol group for the initial photocoupling reaction and a photolabile o-nitrobenzyl moiety to enable photocleavage. Peptides, proteins and other small bioactive molecules were patterned via a thiol-ene reaction photoinitiated with visible light and subsequently removed with UV irradiation [51]. Similar networks, which included photolabile moieties in the cross-linking macromer, also enabled physical degradation for cellular guidance (Figure 4b)[52].
Photoresponsive hydrogels also facilitate spatiotemporally controlled-release of therapeutics. Kros and colleagues copolymerized dextran functionalized with acrylate-modified o-nitrobenzyl moieties and dithiolated PEG via a Michael addition reaction. Triggered release of protein was demonstrated using green fluorescent protein (GFP) as a model molecule[53]. A photoresponsive acrylated ortho-nitrobenzylether (o-NBE) was also conjugated to fluorescein and incorporated into a PEG hydrogel to yield a photoreleasable drug mimic. The release of fluorescein was related to light intensity, duration of exposure and wavelength at biocompatible levels[54]. Photodegradable, acrylated o-NBE has also been incorporated into the cross-linking molecule of microparticle systems capable of entrapping and delivering bioactive molecules with spatial and temporal control for cell culture applications[55]. These models suggest that photoreleaseable hydrogels could allow on demand, real-time delivery of small molecules.
Photoisomerization is a reversible, repeatable process. Azobenzene, one of the most commonly used moieties for photoisomerization, can be isomerized from the trans to cis configuration by irradiation with UV light and returned to the trans form by exposure to visible light or heating. Kros et al. took advantage of this system to create a supramolecularly cross-linked hydrogel using a complex of trans azobenzene modified dextran synthesized through a thiol-maleimide reaction and β-cyclodextrin to create a photoresponsive hydrogel. The gel was loaded with GFP and irradiated with UV light. As a result, the cis azobenzene dissociated from the β-cyclodextrin and released the model protein (Figure 4c)[56].
Biophysical cues, such as matrix stiffness, in cellular microenvironment are influential in tissue development and disease progression, and these ideas have been translated to cell culture. For example, matrix stiffness has been demonstrated to direct lineage commitment of human mesenchymal stem cells (hMSCs). Polyacrylamide cell culture substrates were formed that spanned the range of elasticities of several tissues (e.g., Ebrain~0.1–1 kPa, Emuscle~8–17 kPa, Ecollagenous bone~25–40 kPa) and directed hMSC commitment to the corresponding cell phenotype[57]. Stiffening of the ECM has recently been implicated as a contributing factor to pathogenesis in fibrotic disease progression, not simply an outcome of fibrosis[58]. A photocleavable PEG-based hydrogel was developed[59, 60] and used to study how in situ changes in substrate modulus influence the fibroblast-myofibroblast transition of vavular interstitial cells (VICs)[61]. In this hydrogel, crosslinking can be decreased both temporally and spatially with UV light exposure. Decreasing the substrate modulus from a high value which activated the VICs to a lower value reversed myofibroblast activation. This material platform is an example of a dynamic substrate that can be used to improve our fundamental knowledge of cellular responses in heart disease progression.
The West lab has applied their photopolymerization techniques to create PEGDA hydrogels with dynamically patterned rigidity. Here, the material modulus was increased spatially and temporally. Hydrogels were again formed by a photoinitiated radical chain polymerization of PEGDA with incomplete conversion., but instead of bioepitopes, a low-molecular-weight PEGDA was swollen into the network with a photoinitiator. Controlled UV irradiation through a photomask increased cross-linking density in exposed areas to increase the modulus from E ~ 1 kPa to ~ 3 kPa[62].
Burdick and colleagues build on this concept and combine natural components of the ECM with photoresponsive moieties to create photocrosslinkable, bioactive hydrogels. A step-wise approach to photocrosslink HA hydrogels was developed to fabricate hydrogels that stiffen (e.g., ~3–30 kPa) in the presence of cells. The HA macromers were functionalized with methacrylates, which react with both thiols and radicals. Initial hydrogels were formed via a Michael-type addition reaction with dithiothreitol (DTT). Stiffening was achieved by radical polymerization of remaining methacrylate groups with user-defined spatial and temporal control[63]. The stiffening of tissue can be a consequence of wound healing or disease progression, and this cell culture platform is advantageous for investigating cellular responses to this process.
Topographic cues from the surrounding microenvironment are also dynamic cues that cells receive from the ECM. Researchers are currently developing microfabrication techniques to produce dynamic substrate textures in two and three dimensions. Kasko et al. synthesized an o-NBE cross-linking macromer with a coumarin conjugate. The macromer was incorporated into a PEG hydrogel and resulted in increased sensitivity to two-photon degradation and fluorescent imaging[64]. These hydrogels were degraded to create a variety of surface textures[64]. Recent efforts in the Anseth group have focused on exploiting a newly designed photolabile-hydrogel cell culture platform that allows users to control cell morphology in situ through the presentation of a series of precisely engineered microtopographies to improve our understand of how stem cells sense and respond to microtopographic cues by performing dynamic experiments. Khademhosseini et al. have developed a two-step photo-cross-linking approach to fabricating 3D hydrogel structures. As proof of concept, arrays of hydrogels with prepatterned internal microchannels were fabricated by photolithography and assembled into a tubular construct with a sequential cross-linking step to build a bioinspired microvasculature. Self-assembling peptide amphiphiles that contain RGD were conjugated to a photocleavable nitrobenzyl moiety. These molecules in the absence of the photolabile group can be induced to self-assemble into high-aspect-ratio fibers by charge screening by the addition of salts. With the addition of nitrobenzyl moieties, the peptide amphiphiles assembled into nanospheres and upon irradiation underwent a sol-to-gel transition resulting in a structural conversion from nanospheres to nanofibers in 3D[65].
A limitation to the application of current photopolymerization and photodegradation techniques is in vivo photoexcitation. The penetration depth of light into tissues is maximized between 800–1300 nm; however, even in this near-infrared (IR) range, an Nd:YAG laser emitting at 1064 nm only penetrates 2–6 mm in typical tissue[66]. Excitation in the UV range that is widely used as a light source can be beneficial for user-defined control of hydrogels treating external organs such as the skin and eye. As a way to overcome this limitation, Burdick et al. have developed a near-IR light responsive polymer-nanorod composite that when activated triggers the release of small molecule drugs[67]. As photosensitive chemistries advance, these techniques will translate from tools for fundamental studies on how cells interact with their microenvironment to clinical applications.
Temperature, Electromagnetic Fields and Ultrasound
Triggers, such as temperature changes induced by injection into the physiological environment or the application of electromagnetic fields and stimulation by ultrasound, are currently the most clinically feasible user-defined mechanisms to dynamically alter hydrogels. Temperature-responsive hydrogels can be designed to gel, swell or deswell upon heating or cooling. Tuning the composition of the polymer network controls these processes[68]. Lower critical solution temperature (LCST) hydrogels exhibit a delicate hydrophobic-hydrophilic balance. Below the LCST, enthalpy, which is attributed to hydrogen bonding between polar groups and water molecules, dominates the thermodynamic interactions and results in a more swollen hydrogel [69]. Raising the temperature above the LCST results in an entropic gain caused by the network excluding water, aggregating hydrophobic groups and deswelling (Figure 5a)[70]. Poly(N-isopropylacrylamide) (PNIPAAm) hydrogels demonstrate LCST behavior near physiological temperature, as the NIPAAm repeating unit contains a hydrophilic amide group and a hydrophobic isopropyl pendant group. These hydrogels have been widely studied for healthcare applications.
Figure 5.

User-defined dynamic mechanisms in hydrogels that respond to temperature and electromagnetic fields. a) Poly(N-isopropylacrylamide) (PNIPAAm) swells or deswells in response to temperature changes near its lower critical solution temperature (LCST), which is near physiological temperature. b) Pluronic F127 is a triblock co-polymer that exhibits thermoreversible, physical gelation in response. c) A dually responsive system of drug-loaded polypyrrole nanoparticles in a thermoresponsive hydrogel matrix enable gelation upon injection and drug release triggered by an electric field.
Okano et al. have exploited the LCST swelling behavior of PNIPAAm hydrogels to create cell culture substrates that enable the removal of continuous cell sheets for tissue engineering. Recently, they developed a grafted, comb-type PNIPAAm surface that accelerated hydration of the cell culture substrates upon lowering the temperature and accelerated cell sheet detachment [71]. This technique has been used to fabricate 3D stacks of functional cardiac tissues[72], which have been implanted into rat myocardial infarction models and showed improvement in systolic function and neovascularization[73]. Mikos and colleagues have synthesized macromers for in situ hydrogel formation from a combination of pentaerythritol diacrylate, NIPAAm, acrylamide and hydroxyethyl acetate that produced a range of LCSTs near that of traditional NIPAAm hydrogels (~32°C). Here, the LCST transition was used as a secondary gelation mechanism. The higher transition temperatures, which were closer to physiological temperature (~37°C), supported increased cell viability[74]. MSCs encapsulated in these macromers in vitro remained viable for up to three weeks[75]. These in situ gel-forming systems are promising for cell delivery applications. Thermo-responsive PNIPAAm-g-methylcellulose hydrogels have also been used for encapsulation of a chondrogenic cell line and supported increased synthesis of glycosoaminoglycans[76].
Pluronic F127, is a tri-block copolymer of PEG, poly(propylene glycol) (PPG) and PEG that exhibits thermo-reversible gelation (Figure 5b). Seliktar and colleagues synthesized nanostructured polymer conjugates of these poloxamers and a fibrinogen protein. Altering the temperature at which these materials were chemically cross-linked between free reactive acrylate moieties resulted in changes in cross-linking density, and therefore mechanical properties and diffusivity, throughout the hydrogel. This dual cross-linking, temperature-sensitive hydrogel network enabled the encapsulation of cells in networks of the same chemistry with different mechanical properties, allowing for fundamental studies of cellular responses to these cues, and has the potential to be used as a cell delivery vehicle[77]. A chemically cross-linked Pluronic F127 dimethacrylate modified with RGD was also developed as 3D culture platform for chondrocytes. Again the cross-linking density and material modulus were thermo-responsive and the construct could be used to deliver cells in vivo via injection. The hydrogel modulus was modulated from 1.3 kPa at 15°C to 8.9 kPa at 37°C[78].
External methods to regulate temperature beyond simple changes from ambient conditions to 37°C are also the subject of much research. The development of on-demand drug delivery technologies with remote triggers might enable patients to determine the frequency and dosage of drug delivery for chronic conditions. Iron oxide nanoparticles have been incorporated into PEG methacrylate and PNIPAAm hydrogel matrices, which are temperature-responsive. Stimulation of the iron oxide nanoparticles with an alternating magnetic field results in locally increased temperatures that induce hydrogel deswelling. This type of delivery system is one such example of an approach that could be used as a remote-controlled drug delivery system[79, 80]. Likewise, the heating of cancerous tissue increases the efficacy of chemotherapy. The chemotherapeutic, Paclitaxel, and iron oxide nanoparticles have been entrapped in a poly(β-amino ester) (PBAE) biodegradable hydrogel matrix. Remote heating of the hydrogel composite was achieved through the application of an alternating magnetic field to oscillate the iron nanoparticles. Heating was applied as the bulk hydrogel degraded through hydrolysis of ester bonds, releasing the chemotherapy drug while increasing the local temperature[81]. Drug compounds have also been encapsulated in polypyrrole nanoparticles that were suspended in a temperature-responsive hydrogel, which undergoes a sol-gel transition at body temperature (Figure 5c). This combination was subcutaneously injected into mice and drug release was triggered by exposure to a weak, external direct current electric field[82]. Heating through the LCST to elicit dynamic behavior has also been achieved through the creation of polymer-metal nanoparticle composites[67, 83]. Gold nanoparticles are particularly interesting for this application, because irradiation with near-IR light, which penetrates tissue more readily than UV light, results in local temperature increases[83].
The targeted delivery of thrombolytic drugs such as plasminogen activators could reduce adverse side effects, such as complications arising from bleeding. Ultrasound-responsive nano-gelatin complexes were synthesized that immobilized a thrombolytic drug stabilized by zinc ions, which complex and suppress the drug’s activity. Dissociation and activation of the thrombolytic therapeutic were triggered by exposure to ultrasound. This delivery system could be used concurrently with diagnostic imaging in a variety of biomedical applications[84]. Liposomes containing a model drug were encapsulated in a dextran-based hydrogel along with micro-bubbles, and drug release was triggered by application of ultrasound to increase cavitation. This particular drug delivery system was systemically delivered, and application of ultrasound triggered release at specific locations in a subcutaneous rat model. Adjusting the liposomal drug loading concentration, liposomal concentration in the hydrogel, micro-bubble concentration and/or ultrasound intensity controlled the delivery dose and rate[85].
Electromagnetic stimulation has also been investigated as an external trigger to control hydrogel properties. Copolymerized nanomagnets were encapsulated in vinyl-functionalized carbon shells and embedded in pHEMA to create a magnetically-responsive hydrogel. Cross-linking nanomagnets into the hydrogel backbone reduced magnet migration and produced a material that could be used as a soft, muscle-like actuator in a medical device[86]. External stimuli such as electromagnetic fields and ultrasound can be applied in a clinical setting to trigger dynamic changes in hydrogels implanted in the body and afford user-defined, remote-controlled responses. As current research examples have shown, these dynamic changes have been used to induce delivery of small molecules and proteins or form injectable scaffolds for cell delivery and tissue regeneration. These triggers are currently being tested in vivo in animal models and have the potential to translate to clinical applications.
Cell or Tissue-Dictated Stimuli for Dynamic Hydrogel Responses
Although it is beneficial for a dynamic response to be triggered remotely, it is not always known when to induce a specific response, and other times, it is highly desirable for the material to respond to local cellular events. For this reason, materials are being designed that respond to endogenous signals from cells and tissues. These materials take advantage of natural signals produced during an event such as wound healing to trigger specific reactions. This approach is highly conducive towards engineering materials to promote healing, while simultaneously affording the basic research community with more physiologically relevant culture systems.
Proteolysis
Proteolysis is the breakdown of proteins into smaller polypeptides or amino acids. Cells naturally produce and secrete enzymes called proteases, which degrade proteins that form biophysical barriers in the surrounding ECM during tissue remodeling and repair. Beyond the obvious exploitation of naturally derived hydrogels (e.g., collagen, matrigel, hyaluronan), synthetic hydrogel scaffolds that contain mimics of cleavable proteins in the ECM afford dynamic responsiveness to cellular remodeling, ingrowth and migration. One of the first examples of this type of approach was reported by West and Hubbell [83], who synthesized hydrogels from telechelic block copolymers of PEG and oligopeptides, specifically selected for their susceptibility to degradation by target enzymes (e.g., collagenase and plasmin) involved in cell migration. This work illustrated the feasibility of targeted, proteolysis of hydrogels via a cell-directed degradation mechanism. The matrix metalloproteinases (MMPs) are a class of proteases that have been implicated in the cleavage of nearly all ECM components at specific sites. Lutolf et al. created hydrogels that recapitulated aspects of natural MMP-mediated invasion (Figure 6). These networks were synthesized through a Michael addition reaction of 4-arm-PEG-tetravinyl sulfone with a bi-cysteine MMP-sensitive sequence and cysteine-containing integrin-binding peptides[87]. The MMP-responsive hydrogels were loaded with bone morphogenic protein (BMP-2) to target acceleration of local bone formation in rat cranial defects. All MMP-responsive scaffolds promoted cell invasion and bone formation after 4 weeks, but the healing response was dependent on the degree of MMP sensitivity of the cross-linker[87]. For this reason, a library of 17 different MMP-cleavable cross-linkers was evaluated for degradation kinetics and faster, more sensitive peptide substrates were identified[88].
Figure 6.
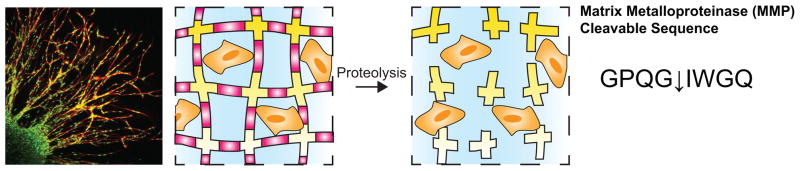
Cell-dictated degradation of a hydrogel. Cells naturally produce and secrete enzymes called matrix metalloproteinases (MMP). Incorporation of MMP-cleavable sequences into a hydrogel allows cell migration throughout the construct such as fibroblast migration from a cell-loaded fibrin clot depicted here. Scale bar, 150 μm. Reprinted from [87], copyright (2003) with permission from National Academy of Sciences, U.S.A.
Building on these findings, Healy et al. created injectable, environmentally responsive hydrogels composed of NIPAAm and acrylic acid (AAc) that were cross-linked with enzymatically degradable peptide cross-linkers[89]. The peptide sequence contained the MMP-13-labile sequence Gln-Pro-Gln-Gly-Leu-Ala-Lys (QPQGLAK). Both peptide-cross-linked networks and semi-IPNs (sIPNs) were synthesized using these components. The sIPNs were synthesized by simultaneously cross-linking the NIPAAm and AAc components with the MMP-13-degradable peptide in the presence of RGD-modified p(AAc) linear chains[90]. Both of these artificial ECM scaffolds were temperature and pH responsive and enzymatically degradable. With increasing sophistication, Heilshorn and colleagues expressed proteolytically labile proteins in Escherichia coli (E. coli) that were crosslinked into hydrogels using a bifunctional disuccinimidyl suberate, which is amine-reactive. By controlling the spatial distributions of biodegradable polymer segments with varied degradation rates, cell-secreted proteases revealed 3D patterns within the hydrogel[91]. These gels could present biophysical guidance cues reminiscent of those available in vivo to cells that are remodeling and migrating through their local matrix. The composition of matrices containing these degradable proteins and the cell-adhesive ligand RGD were tailored to optimize cell adhesion, neural differentiation and neurite outgrowth[92].
Proteolytically degradable hydrogels have also been studied for cell-mediated delivery of growth factors and therapeutic agents. Vascular endothelial growth factor (VEGF) has been implicated in angiogenesis, the physiological formation of new blood vessels. However, therapeutic dosing has proved to be a challenge due to complications associated with high concentrations of VEGF. The Hubbell research group conjugated VEGF to a cell-cleavable matrix to create cell-demanded growth factor delivery. Matrices were completely remodeled into mature vascularized tissue when implanted subcutaneously in rats[93]. Other instances where MMP-triggered degradation has been explored is when delivering chemotherapeutics, when it is often desired for release to occur in response to metastasis, angiogenesis, and invasion-promoting proteases (e.g., MMP-2 and MMP-9). Toward this goal, PEGDA-based hydrogel networks have been formed with pendant complexes of model chemotherapeutic agents and MMP-degradable peptide linkers. Exposure to MMPs triggered drug release and inhibited cell proliferation and/or induced cell death[94, 95].
pH
Physiological stimuli, such as the naturally low pH of the stomach, have been used to trigger dynamic responses in materials, such as swelling-controlled release of drugs from hydrogel carriers. Sensitivity to pH can be tuned by controlling the hydrophilicity and ionic character of pendant functional groups within a hydrogel network. Hydrogels containing acidic moieties have been shown to swell upon pH increases as the acid groups become deprotonated[96]. Conversely, cationic groups impart higher levels of swelling under lower pH conditions. In principle, pH levels at or near the pKa or pKb of the pendant moieties in the network lead to increases in fixed charges in the hydrogel that in turn result in more electrostatic repulsion and an internal increase in osmotic pressure[97].
Carboxylic acid-derived monomers such as acrylic (pKa~4.25) and methacrylic acid (pKa~5) exhibit pH sensitivity at pH values lower than the physiologically relevant pH of 7.4. For example, Peppas et al. demonstrated that poly(methacrylic acid g-ethylene glycol) (PMAA/PEG) collapse at low pH where hydrogen bonding can occur between PMAA and its PEG grafts, but exhibit large changes in swelling in response to increased pH when the PMAA becomes charged [98]. They further demonstrated the reversibility of these processes as one cycles through multiple shifts in pH. Hydrogels containing these carboxylic-acid derived monomers have been designed for targeted delivery of therapeutics to the stomach[99]. The oral delivery of several therapeutic agents, including proteins like insulin, has been impeded by the low pH and proteolytic activity in the stomach. To overcome this major obstacle, Peppas and colleagues developed the first pH-responsive hydrogel complexes of poly(itaconic acid) (PIA) and PEG. Hydrogen bonding between the carboxylic acid groups in PIA and the ether oxygens in PEG leads to a compact network structure at acidic pH levels protecting insulin from exposure to the stomach environment[100]. Instead, the hydrogel carriers swell and release their contents at a more neutral pH in the intestine, which results in increased retention of bioactivity and adsorption.
Kopececk et al. have studied IPNs of a pH-sensitive network containing acrylic acid pendant groups and a hydrolyzable network as a delivery vehicle for peptides and proteins to the colon. These networks were synthesized to create a more linear swelling profile in response to the abrupt pH change that occurs between the stomach and the colon and in turn control the burst effect[101]. Similarly, the pH-sensitivity of poly((2-dimethylamino) ethyl methacrylate-glycidyl methacrylate) (P(DMAEMA-GMA) was combined with the biocompatibility of a natural polymer, alginate, to form a semi-IPN hydrogel with improved properties for drug delivery[102].
Beyond targeting release of molecules in response to normal pH variations that occur in certain regions of the body, regions of local acidosis can also develop in wounds, tumor sites and sites of ischemia. Hoffman and colleagues have developed copolymers of NIPAAm and propylacrylic acid (PAA) with higher pKa values compared to those typically used for delivery to areas of low pH such as the stomach[98], and used this chemistry to form hydrogels in the presence of diseased tissue, deliver their therapeutic payload, and dissolve once a normal physiological pH is restored. Such physically cross-linked, pH-responsive hydrogel complexes materials are widely studied as potential drug delivery systems. Physically cross-linked hydrogels facilitate both drug release and elimination from the body[69]. The copoly(NIPAAm PAA) system exhibited gelation and pH-dependent, sustained release of VEGF at physiological temperatures and pHs[103].
Likewise, ischemic heart disease, which can lead to myocardial infarction, also generates a locally acidic microenvironment (~pH 6–7). Copolymers of NIPAAm, PAA and butyl acrylate (BA) were synthesized as precursors to hydrogels that are liquid at pH 7.4 and 37°C but form a physical gel at pH 6.8 and 37°C. Toward the goal of improving angiogenesis in infarcted myocardium, these hydrogel solutions were loaded with basic fibroblast growth factor (bFGF) and injected into a rat myocardial infarction model. Results demonstrated increased capillary and arteriolar densities and relative blood flow to infarct region[104].
In the management of disease, it is often critical to quickly detect, diagnose and treat a condition. To this end, dynamic, pH-responsive hydrogels are being engineered to serve as biological sensors to detect symptoms of disease under physiological conditions. For example, high partial pressure of carbon dioxide (CO2) in the stomach can be indicative of gastrointestinal ischemia. van den Berg and colleagues have developed a sensor that can measure physiologically relevant partial pressures[105, 106]. In this device, CO2 diffuses through a gas permeable membrane where a reaction with an electrolyte produces a concentration-dependent pH decrease. A thin, pH-responsive hydrogel was embedded within the sensor, such that it triggered a pressure sensor when swollen by the pH decrease[105]. This sensor has the potential to be translated to a clinical diagnostic tool by incorporation into a catheter. Similarly, VanBlarcom and Peppas have designed biodegradable, pH-responsive hydrogels of poly(methacrylic acid) crosslinked with polycaprolactone diacrylate that were incorporated as sensing components in silicon-based cantilever microsensors. The hydrogel composition was tuned to produce volume changes in response to pathological pH values, which in turn caused beam deflection to transduce the pH stimulus to an optical signal. The biodegradability of these sensors makes them potential candidates for short-term in vivo assays, such as identifying a condition like ischemia in a transplanted tissue that requires rapid intervention or monitoring the progression of wound healing[107].
Protein Interactions
Proteins, peptides and other poly(amino acid)-based polymers have been incorporated into hydrogels to yield dynamic transformations via changes in secondary (e.g., helix assembly and β-sheet assembly) and tertiary protein conformations[108]. Protein conformational changes in the context of hydrogels can translate to changes in cross-linking density and subsequent cross-linking dependent properties. Over 200 proteins undergo well-characterized conformational changes upon ligand binding[108]. This library of molecules is available to create dynamic hydrogel systems, which respond to endogenous stimuli, including pH or specific ligand binding.
Using this approach, Tirrell and colleagues have created dynamic, artificial protein hydrogels via leucine zipper aggregation and disulfide bond formation. The artificial protein components contained two terminal leucine zipper domains and a random coil central domain that were modified to include cysteine residues. Leucine zipper aggregation initially formed a physical gel that was then crosslinked through disulfide bond formation. The helical structure of the leucine zipper domains preserves reversibility of gelation, i.e., the network dissolves at pH 12.2 whereas the helical structure is disrupted and reforms under acidic conditions[109]. The composition of the central domain in these artificial proteins can be tailored to control network properties[110]. Leucine zipper domain-based hydrogels are both pH and salt-responsive. The stress relaxation time of the network increases when the pH is decreased or when ionic strength is increased, stabilizing the leucine zipper domain aggregates[111]. Similarly designed telechelic proteins form physical hydrogels that exhibit shear-thinning behavior along with rapid recovery of full elasticity [112]. Such networks provide benefits as cell delivery vehicles that gel in situ.
Murphy et al. pioneered the concept of drug delivery from dynamic hydrogels based on a protein conformational change induced by a specific biochemical ligand[113]. Here, the dynamic protein calmodulin (CaM) was conjugated to terminal PEG chains (PEG-CaM-PEG), which were photo-cross-linked to form a hydrogel. Upon binding the ligand trifluoperazine (TFP), CaM exhibits a hinge motion that dynamically changes the cross-linking density and in turn the mesh size of the network[114, 115]. These networks also demonstrated tunable changes in optical transparency that could be exploited to create label-free biosensors[114]. Tailoring the ratio of PEG to CaM and the initial loading concentration modulated the release of VEGF from the hydrogel construct[115]. These materials have also been processed into microspheres loaded with VEGF and BMP-2 using an aqueous two-phase suspension polymerization[116]. Therapeutic proteins were released in a temporally controlled manner via ligand-induced conformational changes in the network.
Kiick and co-workers have also designed growth factor delivery systems that respond to specific ligand binding by taking advantage of the natural interactions that occur between heparin and heparin-binding peptides to form hydrogels[117, 118]. Here, a non-covalent assembly between VEGF and heparinized PEG was used to create a reservoir of VEGF that erodes in response to the vascular endothelial growth factor receptor 2 (VEGFR-2). In this system, VEGF serves as a targeting molecule, a cross-linker and a potential therapeutic that is delivered in a receptor-responsive manner. The specific release of VEGF was confirmed by the fact that porcine aortic endothelial cells overexpressing VEGFR-2 showed increased proliferation in response to these gels, while cells without the receptor did not[119].
This concept has been exploited in numerous examples of biosensors that are traditionally based on molecular recognition events and use natural recognition elements, such as antibodies and enzymes. However, these naturally derived sensing elements can be high cost, are potentially immunogenic and do not exhibit long-term stability, which has stimulated interest in alternative strategies. Molecularly imprinted polymers are fully synthetic systems formed by the crosslinking of a polymer network in the presence of a template molecule that have been developed for sensing applications[120]. Kryscio and Peppas recently reviewed the field of molecularly imprinted polymers in detail[120]. Many molecularly imprinted polymer networks are dynamic hydrogel microenvironments. Polymer networks of poly(ethylene glycol)dimethacrlyate (PEGDMA) and acrylamide were designed to detect D-glucose through non-covalent complexation[121]. Molecularly imprinted polymers have also been micropatterned into various configurations, along with control substrates, to prove the feasibility of using these networks in sensing devices. Similarly, molecularly imprinted networks have been shown to be effective drug delivery vehicles. The dynamic loading and release characteristics of molecularly imprinted polymers have the potential to serve as biosensing systems that also deliver therapeutics[122].
Challenges for the Future of Hydrogels in Healthcare
Since the description of the first hydrogel over the 50 years ago, significant progress has been made in the systematic design of hydrogel microenvironments for healthcare applications. Design paradigms have shifted from static, bioinert networks to dynamic, bioactive microenvironments. Both user-defined and cell- or tissue-dictated mechanisms enable dynamically tunable hydrogel properties, as highlighted here. Despite this unprecedented level of control, the challenge to maintain the delicate balance of material properties required to recapitulate physiological conditions remains. To tailor hydrogel microenvironments to present the properties and signals that are necessary to promote healing, induce tissue regeneration, facilitate delivery of therapeutics and monitor health, requires achieving balance between opposite ends of several continuums. These include biochemical versus biophysical cues, synthetic versus natural compositions, nano- versus macro-size scales, and pre-engineered versus cell-dictated dynamic mechanisms.
Progress toward achieving ideal material properties has been made with hybrid hydrogel networks that combine inputs from both the user and the surrounding environment. West and colleagues have combined their TPA-LSL biochemical patterning technique, a user-defined mechanism, with enzymatically degradable hydrogel scaffolds to create a microenvironment where cues introduced by the experimenter induce a cell-defined response, i.e., degradation and migration throughout the matrix. Here, confocal images from tissues were used to guide the TPA-LSL patterning of bioepitopes to recapitulate the endogenous capillary network structure. When encapsulated human umbilical vein endothelial cells (HUVECs) and mesenchymal progenitor 10T1/2 cells organized into complex tubule networks that mimicked the patterns within the hydrogels[123]. Schmidt et al. have also developed hybrid hydrogel scaffolds that consist of an IPN of collagen and a photopolymerizable, glycidyl-methacrylate-modified HA (GMHA) formed through sequential polymerization. In this system, the collagen component first forms a fibrillar network in the presence of a GMHA solution. Next GMHA can be locally cross-linked by exposure to UV light in the presence of an initiator resulting in spatial control of cross-linking density, which in turn dictates local properties such as modulus, diffusivity and swelling[124]. Dual cross-linked HA scaffolds have also enabled anisotropic swelling that facilitates user-defined shape control within cell-degradable scaffolds[125]. These gels provide both user-defined control of cross-linking density and cell-dictated degradation through proteolysis to facilitate the presentation of biophysical cues to guide cellular ingrowth into scaffold[125]. Further developing these advanced networks will continue to improve our ability to characterize and understand the fundamental interactions between cells and dynamic hydrogel microenvironments, which may provide new criteria for the rational design of new biomaterial scaffolds for healthcare applications. High-throughput screening techniques are currently being developed to more quickly assess cell-material interactions. For example, Lutolf and colleagues have designed artificial niche microarrays in which biophysical (e.g,. substrate modulus) and biochemical (e.g., adhesive ligand) cues can be examined as regulators of stem cell fate in a combinatorial fashion[126].
One particularly notable application for dynamic hydrogels in healthcare could be the monitoring of, reporting on and intervention in biological crisis. To achieve this ambitious goal, it is necessary to integrate the monitoring of multiple inputs, both biochemical and biophysical, into a system that can transduce these signals into a recordable output and also release therapeutics at an appropriate concentration in a timely fashion. There is a clinical need for this type of biosensing system. For example, during the progression of heart disease, valvular interstitial cells (VICs), the most prevalent cell type in the native heart valve leaflet, respond to both chemical and physical signals. In response to injury, these cells begin to differentiate from a quiescent fibroblast phenotype to an activated myofibroblast [127]. Calcification related to persistent VIC activation is a common problem related to progression of aortic heart valve disease. Research shows that cytokines like TGFβ[128], the mechanical properties of the microenvironment[61] and the surrounding ECM[129] all influence calcification. Cell culture substrates that allow us to further characterize function and pathobiology related to substrate stiffness and chemical cues will lead to a better understanding of disease progession in a complex and dynamic microenvironment, as well as provide new insights and strategies for tissue engineered therapeutics.
Acknowledgments
The authors acknowledge the National Institute Of Arthritis And Musculoskeletal And Skin Diseases, National Institutes of Health (F32AR061923) for fellowship funding to C.M.K. This work was made possible by financial support from the National Institutes of Health (DE16523) and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schaeffer J, Beiting J. Review of Optometry. Jobson Medical Information LLC; 2009. The Early History of Contact Lenses. [Google Scholar]
- 2.Ratner BD, editor. A history of biomaterials. San Diego, CA: Elsevier Academic Press; 2004. [Google Scholar]
- 3.Wichterle O, Lim D. Nature. 1960;185:117. [Google Scholar]
- 4.Nicolson PC, Vogt J. Biomaterials. 2001;22:3273. doi: 10.1016/s0142-9612(01)00165-x. [DOI] [PubMed] [Google Scholar]
- 5.Xinming L, Yingde C, Lloyd AW, Mikhalovsky SV, Sandeman SR, Howel CA, Liewen L. Contact Lens and Anterior Eye. 2008;31:57. doi: 10.1016/j.clae.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Leonardi M, Pitchon EM, Bertsch A, Renaud P, Mermoud A. Acta Ophthalmologica. 2009;87:433. doi: 10.1111/j.1755-3768.2008.01404.x. [DOI] [PubMed] [Google Scholar]
- 7.Badugu R, Lakowicz JR, Geddes CD. Current Opinion in Biotechnology. 2005;16:100. doi: 10.1016/j.copbio.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu X, Jha AK, Harrington DA, Farach-Carson MC, Jia XQ. Soft Matter. 2012;8:3280. doi: 10.1039/C2SM06463D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cen L, Liu W, Cui L, Zhang W, Cao Y. Pediatr Res. 2008;63:492. doi: 10.1203/PDR.0b013e31816c5bc3. [DOI] [PubMed] [Google Scholar]
- 10.Van Vlierberghe S, Dubruel P, Schacht E. Biomacromolecules. 2011;12:1387. doi: 10.1021/bm200083n. [DOI] [PubMed] [Google Scholar]
- 11.Cushing MC, Anseth KS. Science. 2007;316:1133. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 12.Patterson J, Martino MM, Hubbell JA. Mater Today. 2010;13:14. [Google Scholar]
- 13.Peppas NA, Merrill EW. J Appl Polym Sci. 1977;21:1763. [Google Scholar]
- 14.Hogemann KE, Gustafson G, Bjorlin G. Acta Chir Scand. 1961;121:83. [PubMed] [Google Scholar]
- 15.Bray JC, Merrill EW. Journal of Biomedical Materials Research. 1973;7:431. doi: 10.1002/jbm.820070506. [DOI] [PubMed] [Google Scholar]
- 16.Peppas NA, Merrill EW. Journal of Biomedical Materials Research. 1977;11:423. doi: 10.1002/jbm.820110309. [DOI] [PubMed] [Google Scholar]
- 17.Lewin-Epstein J. British Journal of Oral Surgery. 1964;2:115. doi: 10.1016/s0007-117x(64)80025-1. [DOI] [PubMed] [Google Scholar]
- 18.Harrison JH. The American Journal of Surgery. 1958;95:16. doi: 10.1016/0002-9610(58)90736-0. [DOI] [PubMed] [Google Scholar]
- 19.HURST A, GROW JB, LEVINE S, PERLMUTTER HM. Diseases of the Chest. 1951;20:134. doi: 10.1378/chest.20.2.134. [DOI] [PubMed] [Google Scholar]
- 20.Tadavarthy SM, Moller JH, Amplatz K. Am J Roentgenol Radium Ther Nucl Med. 1975;125:609. doi: 10.2214/ajr.125.3.609. [DOI] [PubMed] [Google Scholar]
- 21.Goosen MFA, Sefton MV. Journal of Biomedical Materials Research. 1983;17:359. doi: 10.1002/jbm.820170212. [DOI] [PubMed] [Google Scholar]
- 22.Merrill EW, Salzman EW, Wong PSL, Ashford TP, Brown AH, Austen WG. Journal of Applied Physiology. 1970;29:723. doi: 10.1152/jappl.1970.29.5.723. [DOI] [PubMed] [Google Scholar]
- 23.Cholakis CH, Zingg W, Sefton MV. Journal of Biomedical Materials Research. 1989;23:417. doi: 10.1002/jbm.820230404. [DOI] [PubMed] [Google Scholar]
- 24.Kearney JJ, Amara I, McDevitt NB, editors. Mechanically Stable Non-thrombogenic Hydrogels. New York and London: Plenum Press; 1975. [Google Scholar]
- 25.Ratner BD, Hoffman AS, Hanson SR, Harker LA, Whiffen JD. Journal of Polymer Science: Polymer Symposia. 1979;66:363. [Google Scholar]
- 26.Tsuruta T, Hayashi T, Kataoka K, Ishihara K. Biomedical Applications of Polymeric Materials. Boca Raton, Florida: CRC Press; 1993. [Google Scholar]
- 27.Lyman DJ, Loo BH, Crawford RW. Biochemistry. 1964;3:985. doi: 10.1021/bi00895a025. [DOI] [PubMed] [Google Scholar]
- 28.Tighe BJ. British Polymer Journal. 1976;8:71. [Google Scholar]
- 29.Gombotz WR, Guanghui W, Horbett TA, Hoffman AS. Journal of Biomedical Materials Research. 1991;25:1547. doi: 10.1002/jbm.820251211. [DOI] [PubMed] [Google Scholar]
- 30.Peppas NA, editor. Hydrogels in Medicine and Pharmacy. Boca Raton, Florida: CRC Press; 1986. [Google Scholar]
- 31.Marchant RE, Anderson JM, Dillingham EO. Journal of Biomedical Materials Research. 1986;20:37. doi: 10.1002/jbm.820200105. [DOI] [PubMed] [Google Scholar]
- 32.Lapidus H, Lordi NG. Journal of Pharmaceutical Sciences. 1968;57:1292. doi: 10.1002/jps.2600570803. [DOI] [PubMed] [Google Scholar]
- 33.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. International Journal of Pharmaceutics. 1983;15:25. [Google Scholar]
- 34.Rubinstein M, Colby RH. Polymer Physics. New York: Oxford University Press; 2003. [Google Scholar]
- 35.Sperling LH. Introduction to Physical Polymer Science. Hoboken, NJ: John Wiley & Sons, Inc; 2006. [Google Scholar]
- 36.Metters AT, Bowman CN, Anseth KS. AIChE Journal. 2001;47:1432. [Google Scholar]
- 37.Mason MN, Metters AT, Bowman CN, Anseth KS. Macromolecules. 2001;34:4630. [Google Scholar]
- 38.Metters AT, Anseth KS, Bowman CN. The Journal of Physical Chemistry B. 2001;105:8069. [Google Scholar]
- 39.Bryant SJ, Anseth KS. Photopolymerization of Hydrogel Scaffolds. In: Ma PX, Elisseeff J, editors. Scaffolding in Tissue Engineering. Boca Raton, FL: CRC Press; 2006. p. 71. [Google Scholar]
- 40.Flory PJ, Rehner J. J Chem Phys. 1943;11:521. [Google Scholar]
- 41.Shen M, Blatz PJ. J Appl Phys. 1968;39:4937. [Google Scholar]
- 42.Flory PJ, Rehner J. J Chem Phys. 1943;11:512. [Google Scholar]
- 43.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Advanced Materials. 2006;18:1345. [Google Scholar]
- 44.Lustig SR, Peppas NA. J Appl Polym Sci. 1988;36:735. [Google Scholar]
- 45.Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zhang J. Annu Rev Biomed Eng. 2000;2:9. doi: 10.1146/annurev.bioeng.2.1.9. [DOI] [PubMed] [Google Scholar]
- 46.Massia SP, Hubbell JA. Ann NY Acad Sci. 1990;589:261. doi: 10.1111/j.1749-6632.1990.tb24251.x. [DOI] [PubMed] [Google Scholar]
- 47.Bowman CN, Kloxin CJ. AIChE Journal. 2008;54:2775. [Google Scholar]
- 48.Luo Y, Shoichet MS. Nat Mater. 2004;3:249. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 49.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat Mater. 2011;10:799. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 50.Hoffmann JC, West JL. Soft Matter. 2010;6:5056. [Google Scholar]
- 51.DeForest CA, Anseth KS. Angewandte Chemie International Edition. 2012;51:1978. doi: 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeForest CA, Anseth KS. Nat Chem. 2011;3:925. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng K, Tomatsu I, van den Broek B, Cui C, Korobko AV, van Noort J, Meijer AH, Spaink HP, Kros A. Soft Matter. 2011;7:4881. [Google Scholar]
- 54.Griffin DR, Patterson JT, Kasko AM. Biotechnology and Bioengineering. 2010;107:1012. doi: 10.1002/bit.22882. [DOI] [PubMed] [Google Scholar]
- 55.Tibbitt MW, Han BW, Kloxin AM, Anseth KS. J Biomed Mater Res Part A. 2012;100A:1647. doi: 10.1002/jbm.a.34107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng K, Tomatsu I, Kros A. Chemical Communications. 2010;46:4094. doi: 10.1039/c002565h. [DOI] [PubMed] [Google Scholar]
- 57.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 58.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. The Journal of Cell Biology. 2010;190:693. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kloxin AM, Tibbitt MW, Anseth KS. Nat Protoc. 2010;5:1867. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kloxin AM, Kasko AM, Salinas CN, Anseth K. Science. 2009;234:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kloxin AM, Benton JA, Anseth KS. Biomaterials. 2010;31:1. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nemir S, Hayenga HN, West JL. Biotechnology and Bioengineering. 2010;105:636. doi: 10.1002/bit.22574. [DOI] [PubMed] [Google Scholar]
- 63.Guvendiren M, Burdick JA. Nat Commun. 2012;3:792. doi: 10.1038/ncomms1792. [DOI] [PubMed] [Google Scholar]
- 64.Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 2010;43:2824. [Google Scholar]
- 65.Muraoka T, Koh CY, Cui HG, Stupp SI. Angew Chem-Int Edit. 2009;48:5946. doi: 10.1002/anie.200901524. [DOI] [PubMed] [Google Scholar]
- 66.Prasad PN. Introduction to Biophotonics. Hoboken, NJ: John Wiley & Sons, Inc; 2003. [Google Scholar]
- 67.Hribar KC, Lee MH, Lee D, Burdick JA. ACS Nano. 2011;5:2948. doi: 10.1021/nn103575a. [DOI] [PubMed] [Google Scholar]
- 68.Brazel CS, Peppas NA. Macromolecules. 1995;28:8016. [Google Scholar]
- 69.Jeong B, Kim SW, Bae YH. Advanced Drug Delivery Reviews. 2002;54:37. doi: 10.1016/s0169-409x(01)00242-3. [DOI] [PubMed] [Google Scholar]
- 70.Tekin H, Sanchez JG, Tsinman T, Langer R, Khademhosseini A. AIChE Journal. 2011;57:3249. doi: 10.1002/aic.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang ZL, Akiyama Y, Yamato M, Okano T. Biomaterials. 2010;31:7435. doi: 10.1016/j.biomaterials.2010.06.040. [DOI] [PubMed] [Google Scholar]
- 72.Haraguchi Y, Shimizu T, Sasagawa T, Sekine H, Sakaguchi K, Kikuchi T, Sekine W, Sekiya S, Yamato M, Umezu M, Okano T. Nat Protoc. 2012;7:850. doi: 10.1038/nprot.2012.027. [DOI] [PubMed] [Google Scholar]
- 73.Masumoto H, Matsuo T, Yamamizu K, Uosaki H, Narazaki G, Katayama S, Marui A, Shimizu T, Ikeda T, Okano T, Sakata R, Yamashita JK. Stem Cells. 2012;30:1196. doi: 10.1002/stem.1089. [DOI] [PubMed] [Google Scholar]
- 74.Klouda L, Hacker MC, Kretlow JD, Mikos AG. Biomaterials. 2009;30:4558. doi: 10.1016/j.biomaterials.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klouda L, Perkins KR, Watson BM, Hacker MC, Bryant SJ, Raphael RM, Kasper FK, Mikos AG. Acta Biomaterialia. 2011;7:1460. doi: 10.1016/j.actbio.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sa-Lima H, Tuzlakoglu K, Mano JF, Reis RL. J Biomed Mater Res Part A. 2011;98A:596. doi: 10.1002/jbm.a.33140. [DOI] [PubMed] [Google Scholar]
- 77.Frisman I, Shachaf Y, Seliktar D, Bianco-Peled H. Langmuir. 2011;27:6977. doi: 10.1021/la104695m. [DOI] [PubMed] [Google Scholar]
- 78.Lee H, Choi BG, Moon HJ, Choi J, Park K, Jeong B, Han DK. Macromol Res. 2012;20:106. [Google Scholar]
- 79.Meenach SA, Anderson KW, Hilt JZ. J Polym Sci Pol Chem. 2010;48:3229. [Google Scholar]
- 80.Satarkar NS, Zach Hilt J. Acta Biomaterialia. 2008;4:11. doi: 10.1016/j.actbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 81.Meenach SA, Otu CG, Anderson KW, Hilt JZ. International Journal of Pharmaceutics. 2012;427:177. doi: 10.1016/j.ijpharm.2012.01.052. [DOI] [PubMed] [Google Scholar]
- 82.Ge J, Neofytou E, Cahill TJ, Beygui RE, Zare RN. ACS Nano. 2012;6:227. doi: 10.1021/nn203430m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strong LE, West JL. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2011;3:307. doi: 10.1002/wnan.138. [DOI] [PubMed] [Google Scholar]
- 84.Uesugi Y, Kawata H, Saito Y, Tabata Y. J Drug Target. 2012;20:224. doi: 10.3109/1061186X.2011.633259. [DOI] [PubMed] [Google Scholar]
- 85.Epstein-Barash H, Orbey G, Polat BE, Ewoldt RH, Feshitan J, Langer R, Borden MA, Kohane DS. Biomaterials. 2010;31:5208. doi: 10.1016/j.biomaterials.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fuhrer R, Athanassiou EK, Luechinger NA, Stark WJ. Small. 2009;5:383. doi: 10.1002/smll.200801091. [DOI] [PubMed] [Google Scholar]
- 87.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proceedings of the National Academy of Sciences. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patterson J, Hubbell JA. Biomaterials. 2010;31:7836. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 89.Kim S, Healy KE. Biomacromolecules. 2003;4:1214. doi: 10.1021/bm0340467. [DOI] [PubMed] [Google Scholar]
- 90.Kim S, Chung EH, Gilbert M, Healy KE. J Biomed Mater Res Part A. 2005;75A:73. doi: 10.1002/jbm.a.30375. [DOI] [PubMed] [Google Scholar]
- 91.Straley KS, Heilshorn SC. Advanced Materials. 2009;21:4148. [Google Scholar]
- 92.Straley KS, Heilshorn SC. Soft Matter. 2009;5:114. [Google Scholar]
- 93.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmokel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. Faseb J. 2003;17:2260. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 94.Tauro JR, Gemeinhart RA. Bioconjugate Chemistry. 2005;16:1133. doi: 10.1021/bc0501303. [DOI] [PubMed] [Google Scholar]
- 95.Tauro JR, Lee BS, Lateef SS, Gemeinhart RA. Peptides. 2008;29:1965. doi: 10.1016/j.peptides.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuhn W, Hargitay B, Katchalsky A, Eisenberg H. Nature. 1950;165:514. [Google Scholar]
- 97.Toepke MW, Murphy WL. 1.132 - Dynamic Hydrogels. In: Paul D, editor. Comprehensive Biomaterials. Oxford: Elsevier; 2011. p. 577. [Google Scholar]
- 98.Klier J, Scranton AB, Peppas NA. Macromolecules. 1990;23:4944. [Google Scholar]
- 99.Kim B, Peppas NA. J Biomater Sci-Polym Ed. 2002;13:1271. doi: 10.1163/156856202320893000. [DOI] [PubMed] [Google Scholar]
- 100.Betancourt T, Pardo J, Soo K, Peppas NA. J Biomed Mater Res Part A. 2010;93A:175. doi: 10.1002/jbm.a.32510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chivukula P, Dusek K, Wang D, Duskova-Smrckova M, Kopeckova P, Kopecek J. Biomaterials. 2006;27:1140. doi: 10.1016/j.biomaterials.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 102.Gao CM, Liu MZ, Chen J, Chen C. J Biomater Sci-Polym Ed. 2012;23:1039. doi: 10.1163/092050611X570653. [DOI] [PubMed] [Google Scholar]
- 103.Garbern JC, Hoffman AS, Stayton PS. Biomacromolecules. 2010;11:1833. doi: 10.1021/bm100318z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garbern JC, Minami E, Stayton PS, Murry CE. Biomaterials. 2011;32:2407. doi: 10.1016/j.biomaterials.2010.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Herber S, Bomer J, Olthuis W, Bergveld P, van den Berg A. Biomedical Microdevices. 2005;7:197. doi: 10.1007/s10544-005-3026-5. [DOI] [PubMed] [Google Scholar]
- 106.Herber S, Olthuis W, Bergveld P, van den Berg A. Sens Actuator B-Chem. 2004;103:284. [Google Scholar]
- 107.VanBlarcom DS, Peppas NA. Biomedical Microdevices. 2011;13:829. doi: 10.1007/s10544-011-9553-3. [DOI] [PubMed] [Google Scholar]
- 108.King WJ, Murphy WL. Polym Chem. 2011;2:476. [Google Scholar]
- 109.Shen W, Lammertink RGH, Sakata JK, Kornfield JA, Tirrell DA. Macromolecules. 2005;38:3909. [Google Scholar]
- 110.Shen W, Kornfield JA, Tirrell DA. Soft Matter. 2007;3:99. doi: 10.1039/b610986a. [DOI] [PubMed] [Google Scholar]
- 111.Shen W, Kornfield JA, Tirrell DA. Macromolecules. 2007;40:689. [Google Scholar]
- 112.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010;43:9094. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sui ZJ, King WJ, Murphy WL. Advanced Materials. 2007;19:3377. [Google Scholar]
- 114.Sui ZJ, King WJ, Murphy WL. Adv Funct Mater. 2008;18:1824. doi: 10.1002/adfm.200800218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.King WJ, Mohammed JS, Murphy WL. Soft Matter. 2009;5:2399. [Google Scholar]
- 116.King WJ, Pytel NJ, Ng K, Murphy WL. Macromolecular Bioscience. 2010;10:580. doi: 10.1002/mabi.200900382. [DOI] [PubMed] [Google Scholar]
- 117.Kim SH, Kiick KL. Peptides. 2007;28:2125. doi: 10.1016/j.peptides.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang L, Furst EM, Kiick KLJ. Control Release. 2006;114:130. doi: 10.1016/j.jconrel.2006.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim SH, Kiick KL. Macromol Rapid Commun. 2010;31:1231. doi: 10.1002/marc.201000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kryscio DR, Peppas NA. Acta Biomaterialia. 2012;8:461. doi: 10.1016/j.actbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 121.Hilt JZ, Byrne ME, Peppas NA. Chem Mat. 2006;18:5869. [Google Scholar]
- 122.Byrne ME, Hilt JZ, Peppas NA. J Biomed Mater Res Part A. 2008;84A:137. doi: 10.1002/jbm.a.31443. [DOI] [PubMed] [Google Scholar]
- 123.Culver JC, Hoffmann JC, Poché RA, Slater JH, West JL, Dickinson ME. Advanced Materials. 2012;24:2344. doi: 10.1002/adma.201200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Suri S, Schmidt CE. Acta Biomaterialia. 2009;5:2385. doi: 10.1016/j.actbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 125.Zawko SA, Suri S, Truong Q, Schmidt CE. Acta Biomaterialia. 2009;5:14. doi: 10.1016/j.actbio.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 126.Gobaa S, Hoehnel S, Roccio M, Negro A, Kobel S, Lutolf MP. Nat Meth. 2011;8:949. doi: 10.1038/nmeth.1732. [DOI] [PubMed] [Google Scholar]
- 127.Masters KS, Shah DN, Leinwand LA, Anseth KS. Biomaterials. 2004;26:2517. doi: 10.1016/j.biomaterials.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 128.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Circulation Research. 2004;95:253. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 129.Benton JA, Kern HB, Anseth KS. J Heart Valve Dis. 2008;17:689. [PMC free article] [PubMed] [Google Scholar]