

Abstract
Several 2-(2-phenylethyl)chromones have been shown to possess neuroprotective activity. However, limited synthetic methods have been disclosed to construct the 2-(2-phenylethyl)chromone skeleton. Herein we report a straightforward 3-step preparation of five naturally occurring 2-(2-phenylethyl)chromones utilizing the Claisen condensation as the key step.
Keywords: 2-(2-phenylethyl)chromones, Claisen condensation, Neuroprotection, Chromenes
Natural products have been of great interest for many years, and have provided modern medicine with numerous useful drug leads.1,2 One family of natural products being widely explored for their biological activity are those containing the chromone skeleton (1, Figure 1).3 Of particular interest are those chromones with central nervous system (CNS) activity as they may provide new therapeutics for CNS related disorders.4 Perhaps the most widely studied chromones are the flavonoids (2, Figure 1), which bear a phenyl group at C-2.5
Figure 1.
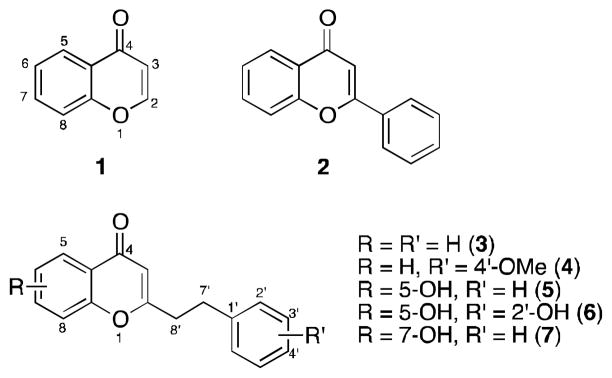
Chromone and flavonoid skeletons, and several biologically active 2-(2-phenylethyl)chromones.
Recently, five chromones were isolated from Imperata Cylindrica and Aquilaria Malaccensis (3–7, Figure 1).6–8 Unlike flavonoids, these chromones have a phenylethyl substituent at C-2 position, which is quite uncommon.9 In addition to their unique structural feature, chromones within this family have demonstrated neuroprotective activity, a property for potentially treating neurodegenerative disorders.6,10 As previously indicated the typical substituent at C-2 for most of chromones is an aromatic ring; thus, most literature is targeted towards the synthesis of chromones like 2.11 These methodologies however are not necessarily transferable to the synthesis of chromones bearing the phenylethyl substituent at C-2. Thus, given their interesting pharmacology and uncommon substitution pattern we sought to develop an efficient and rapid methodology to arrive at the 2-(2-phenylethyl)chromone skeleton. Such a methodology would be beneficial to aid in our future structure activity relationship studies for this class of chromones.
Retrosynthetic analysis of chromones 3–7 indicates the key intermediate 8 or 12 prepared through the corresponding aldol or Claisen condensation should be suitable to afford the desired chromones (Scheme 1). Past literature has shown that the condensation of aromatic aldehydes with 2′-hydroxyacetophenones (9) under aldol conditions leads to spontaneous cyclization to afford intermediates as of 11.12 This domino reaction was considered advantageous as oxidation of 11 would yield the desired 2-(2-phenylethyl)chromone in as few as two steps. Accordingly, 2′-hydroxyacetophenone 9 was condensed with hydrocinnamaldehyde (13) to afford the cyclized product 11 in 50% yield (Scheme 2). Though in our initial trial it was anticipated that oxidation of 11 with iodine in refluxing DMSO13 would afford the desired product, such conditions gave only a complex mixture. Other methodologies to affect this transformation were not pursued, as they would each involve multiple reaction steps.
Scheme 1.
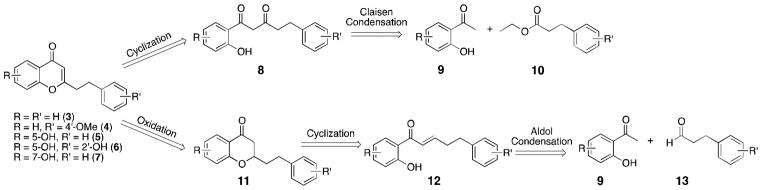
Retrosynthetic analysis for 2-(2-phenylethyl)chromones 3–7.
Scheme 2.
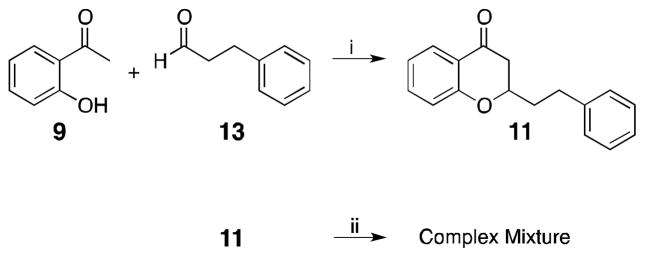
Attempted synthesis of 3 using aldol condensation conditon. Reagents and conditions: (i) Piperdine (cat), EtOH, reflux, 24h, 50% (ii) DMSO, I2, 140 oC 1 h.
Given the difficulties associated with oxidation of 11, the alternative route utilizing the cyclization of 1,3-diones (8) was pursued. The construction of these diones for chromone synthesis is typically accomplished using the Baker-Venkataraman rearrangement.11 While this can be an effective strategy, the synthesis of the precursor benzoate ester is not trivial when the dihydroxyacetophenone needed for the synthesis of 5, 6 and 7 are adopted as the starting reagent. Therefore, we sought to use the direct Claisen condensation of acetophenone 9 with ester 10. This idea had been explored in a previous report on the synthesis of 5 but only very low yields (< 9%) of the condensation product were obtained.14 Thus, this work sought to improve the application of the Claisen condensation for the synthesis of natural products 3–7.
Due to the enolizable nature of both the ketone and ester, it was expected that optimal yields of the Claisen condensation would be achieved if preformation of the enolate from 9 were accomplished. Consequently, 9 was added drop wise to a refluxing slurry of sodium hydride (NaH). After refluxing for 1 h the solution was allowed to cool to room temperature and the ester (10) was added drop wise and the solution stirred overnight (Scheme 3). Based on previous literature we anticipated that the crude Claisen condensation product (8) could be used directly for cyclization without purification.15 Hence, the efficiency of the Claisen condensation would be judged on the yield of the final product. Accordingly, the crude 1,3-dione (8) was cyclized by refluxing in acetic acid with trace amounts of HCl to yield 3 in 73% yield after purification (Table 1, Entry 1).
Scheme 3.

Synthesis of 2-(2-phenylethyl)chromones 3 and 4 utilizing the Claisen condensation. Reagents and conditions: (i) NaH, THF, reflux, 1 h. (ii) 10, THF, rt, 24 h (iii) AcOH or MeOH, HCl (cat.) reflux 45 min.
Table 1.
Reaction conditions explored to optimize yields.
| Entry | Base | Acetopheone | Ester (10) R′ = | Claisen Condensation Conditions | Cyclization Conditions | Product | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | NaH | 9 | H | A | HCl/AcOH/Reflux | 3 | 73 |
| 2 | NaH | 9 | 4-OMe | A | HCl/MeOH/Reflux | 4 | 55 |
| 3 | NaH | 14 | H | A | HCl/AcOH/Reflux | 5 | 43 |
| 4 | NaH | 16 | H | A | HCl/AcOH/Reflux | 5 | 52 |
| 5 | NaH | 14 | H | B | HCl/AcOH/Reflux | 5 | 24 |
| 6 | NaH | 17 | H | A | - | 5 | - |
| 7 | NaOMe | 14 | H | C | - | 5 | - |
| 8 | LDA | 14 | H | C | - | 5 | - |
| 9 | KOtBu | 14 | H | C | - | 5 | - |
| 10 | NaH | 14 | H | A | HCl/MeOH/Reflux | 5 | 40 |
| 11 | NaH | 14 | H | A | HCl(xs)/MeOH/rt | 5 | 15 |
| 12 | NaH | 16 | 2-OMOM | A | - | 6 | - |
| 13 | NaH | 16 | 2-OMOM | D | HCl/AcOH/Reflux | 6 | - |
| 14 | NaH | 14 | 2-OMOM | D | HCl/MeOH/Reflux | 6 | 83 |
| 15 | NaH | 14 | H | D | HCl/MeOH/Reflux | 5 | 80 |
| 16 | NaH | 15 | H | D | HCl/MeOH/Reflux | 7 | 73 |
Continuing with this approch, attention was turned to the synthesis of 4. The highly acidic and elevated temperatures used for the cyclization to provide 3 would very likely lead to the hydrolysis of the methoxy substituent of 4.16 On the other hand, another cylization strategy using refluxing methanol with trace HCl has been reported to give comparable yields to that of refluxing acetic acid.17 When applying this methedology, 4 was prepared in 55% yield over 3 steps after purification (Table 1, Entry 2).
The synthesis of 3 and 4 demonstrate the feasbility of the direct Clasien condensaiton approch for the synthsis of 2-(2- phenylethyl)chromones. The low yields associated with previous attempts on the synthesis of 5 through a direct Clasien condensation approach, were postulated to be the fact of both hydroxyl groups being unprotected. Therefore we hypothesized that incorporation of a protecting group for one or both of the hydroxyl groups should allow a smoother Claisen condensation. Therefore, 2,6-dihydroxyacetophenone 9a was monoprotected as either the methoxymethyl ether 14 or the benzyl ether 16 (Scheme 4).13,18 Benzyl ether 16 was then further protected with MOMCl to provide 17. These protecting groups were selected as they should both be cleaved during the acidic cyclization step simultaneously.16 Following the above described methodology for the Claisen condensation, acetophenone 14 was used to prepare chromone 5 in 43% yield over three steps (Scheme 5, Table 1, Entry 3). Use of the benzyl ether protected acetophenone 15 led to an increase in yield to 52% after purification (Table 1, Entry 4). These results represent a significant improvement over previous attempts utilizing the direct Claisen condensation approach for the synthesis of 2-(2-phenylethyl)chromones.14 It was concluded that the improvement in yield was attributed to the monoprotection of 9. It was hoped then that protection of both hydroxyl groups would further improve the yield. However, when using 17 as the starting acetophenone, the Claisen condensation did not take place (Table 1, Entry 6). This might be due to steric hindrance to the methyl group of the ketone to prevent enolate formation.
Scheme 4.
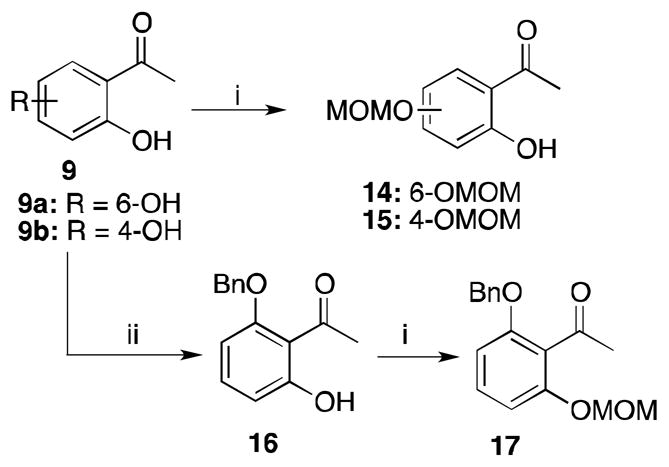
Protection of dihydroxyacetopheones. Reagents and conditions: (i) Diisopropylethylamine, CH2Cl2, MOMCl, rt, 1 h. (ii) BnBr, K2CO3, KI, acetone, reflux, 16 h
Scheme 5.
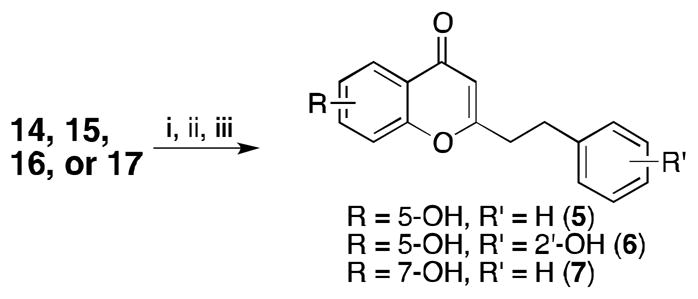
Synthesis of 2-(2-phenylethyl)chromones 5 and 6. Reagents and conditions: (i) NaH, THF, reflux 1 h. (ii) 10, THF, reflux, 4 h (iii) MeOH, HCl (cat.), reflux, 45 min.
Interestingly when the order of addition was changed such that 14 was added to a refluxing solution of 13 a significant decrease in the yield was observed (Table 1, Entry 5).
In addition to changes in the order of addition and introduction of protecting groups, alternative bases were also explored to further improve the yield. Adoption of tBuOK or NaOMe did not provide any of the desired products and only the starting materials were recovered after workup. This may be attributed to the insolubility of these salts in THF. The use of LDA did show consumption of the starting material; however, after reaction workup only a complex mixture of products was obtained. Thus, NaH seemed to be optimal.
In the synthesis of 4 the cyclization solvent was changed from acetic acid to methanol. The impact of this change on yield was also investigated for the synthesis of 5. Refluxing in methanol provided yields comparable to those of acetic acid (Table 1, Entry 10). Cyclization in methanol at room temperature19, however, gave significantly reduced yields (Table 1, Entry 11).
Based on the above results the synthesis of 6 was then undertaken. When the Claisen condensation was carried out at room temperature using acetophenone 16 and the MOM protected ester 10 no reaction was observed. Previous studies have shown that the Claisen condensation can be carried out at elevated temperatures when sterically crowded substrates are used.20 Based on this, after preformation of the enolate, the solution was maintained at reflux and the ester was added drop wise and the reaction mixture allowed to reflux for another 4 h. Thin Layer Chromatography (TLC) of this reaction showed consumption of starting acetophenone 16 and the appearance of a new fluorescent spot, which was presumed to be the condensation product. However, refluxing this crude Claisen condensation product in acetic acid produced an extremely complex mixture by TLC (Table 1, Entry 13). In response, the monoprotected acetophenone 14 was used in the Claisen condensation under refluxing conditions. Again consumption of the starting material was observed along with the appearance of a new fluorescent spot. This crude product was then cyclized in refluxing methanol and upon purification 6 was isolated in 83% yield. The high yield of this reaction prompted a revisit to the synthesis of 5. Utilizing the refluxing Claisen condensation conditions and methanol for the cyclization solvent the yield of 5 was also increased to 80%. Finally, using these optimized conditions 2,4-dihydroxyacetophenone 9b was converted to 2-(2-phenylethyl)chromone 7 in 73% overall yield.
In summary an efficient and rapid route has been developed and applied to the synthesis of five naturally occurring 2-(2-phenylethyl)chromones 3–7. Monoprotection of the starting dihydroxyacetophenones was proven to be critical for improving the yield of the Claisen condensation. Sodium hydride was shown to be the most effective base to execute the Claisen condensation. Carrying out the Condensation at reflux significantly reduced reaction time while simultaneously increasing the yield. Furthermore, this methodology can be used for the synthesis of 2-(2-phenylethyl)chromones bearing substitutions on both phenyl rings. Thus, this approach should allow for the convenient syntheses of a structurally diversified compound library to further study the biological activity of this class of compounds.
General Procedures
Claisen condensation Method A: To a slurry of sodium hydride (4.0 mmol) in refluxing THF was added drop wise the acetophenone 9 (1.0 mmol) dissolved in 10 mL of THF over 10 min. The solution was then allowed to reflux further for 1 h. After cooling to room temperature the ester 10 (1.5 mmol) was added drop wise over 15 min and the resulting solution stirred for 24 h. The reaction mixture was then poured over 50 mL of saturated NH4Cl and extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated to yield the crude product that was used directly without purification for the cyclization step. Method B: A solution of the ester 10 (1.5 mmol) and NaH (4.0 mmol) in THF was brought to reflux while the Acetophenone 9 (1.0 mmol) was added dropwise over 3 min. The resulting solution was refluxed for 4 hours. The reaction mixture was then poured into water and the pH was adjusted to neutral by dropwise addition of 3M HCl. The aqueous layer was extracted three times with dichloromethane (CH2Cl2). The combined CH2Cl2 layers were dried over Na2SO4 and concentrated to yield the crude product that was used directly without purification for the cyclization step. Method C: To a slurry of the corresponding base (3.0 mmol) in THF at 0°C was added drop wise the acetophenone 9 (1.0 mmol). The resulting solution was allowed to stir at room temperature for 1 h. The ester 10 was then added drop wise and the solution allowed to stir at room temperature for 24 h. The reaction mixture was poured over 50 mL of saturated NH4Cl and extracted three times with 25 mL of ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated. Method D: To a slurry of sodium hydride (4.0 mmol) at reflux in THF was added drop wise over 10 min the acetophenone 9 (1.0 mmol) dissolved in 10mL of THF. The solution was then allowed to reflux further for 1 h. While still at reflux the ester 10 (1.5 mmol) was then added drop wise over 15 min and the resulting solution stirred at reflux for 4 hours. The reaction mixture was then poured over 50 mL of saturated NH4Cl and extracted three times with ethyl acetate. The combined organic layers were dried over Na2SO4 and concentrated to yield the crude product that was used directly without purification for the cyclization step.
Cyclization General Procedure
Method A: The crude Claisen Condensation produce was dissolved in 20mL of acetic acid and 15 drops of HCl was then added. The solution was then placed in an oil bath that was preheated to 120°C. The solution began to reflux almost immediately and was refluxed for 45 min. While still hot the solution was poured onto approximately 20 grams of crushed ice. The aqueous layer was extracted with 25 mL ethyl acetate three times. The combined organic layers were then washed with saturated sodium bicarbonate solution followed by brine. After drying over sodium sulfate the organic layer was concentrated to yield the crude product that was then purified in the appropriate manner. Method B: The crude Claisen Condensation produce was dissolved in 20 mL of methanol and 15 drops of HCl was added. The solution was then placed in an oil bath that was preheated to 90°C. The solution began to reflux almost immediately and was refluxed for 45 min. After cooling to room temperature the solution was made neutral by the drop wise addition of saturated Na2CO3. The resulting solution was then diluted with 75 mL of dichloromethane. The organic layer was washed once with 25 mL of sat. NH4Cl, dried over Na2SO4, filtered and concentrated. The cyclized product was then purified in an appropriate manner.
23.2-phenethyl-4H-chromen-4-one (3)
Purified by column chromatography eluting with 70/30 hexanes/ethyl acetate. IR cm−1 3075, 2923, 1642, 1600, 1499, 1463, 1379; 1H-NMR (400MHz, CD3CN): δ, 8.04-8.02 (1H, dd, J = 1.6 and 7.8 Hz), 7.71-7.69 (1H, dt J = 1.6 and 6.96 Hz), 7.47 (1H, d, J = 8.4), 7.41-7.37(1H, dt, J = .92 and 7.0 Hz), 7.29-7.12 (5H, m) 6.04 (1H, s), 3.05 (2H, t, J = 7.9 Hz), 2.92 (2H, t, J = 8.0 Hz); 13C-NMR (100MHz, CD3CN) δ 178.4, 169.9, 157.4, 141.4, 134.7, 129.5, 129.4, 127.3, 126.0, 126.0, 124.6, 119.0, 110.7, 36.5, 33.4.
2-(4-methoxyphenethyl)-4H-chromen-4-one (4)
Purified by column chromatography eluting with 70/30 hexanes/ethyl acetate. IR cm−1 2930, 28.34 1648, 1609, 1511, 1463, 1243, 846, 821; 1H-NMR (400MHz, CD3CN): δ 8.03 (1H, dd, J = 1.52 and 7.96 Hz), 7.69-7.64 (1H, dt J = 1.56 and 8.48), 7.44 (1H, d, J = 8.36 Hz), 7.37 (1H, t, J = 7.76 Hz) 7.13 (2H, d, J = 8.56), 6.82 (2H, d, J = 8.6 Hz), 6.03 (1H, s) 3.71 (3H, s), 2.95 (2H, t, J = 7.0 Hz), 2.85 (2H, t, J = 7.72 Hz); 13C-NMR (100MHz, CD3CN) δ 178.4, 170.0, 159.2, 157.4, 134.7, 133.2, 130.4, 126.0, 124.6, 119.0, 114.8, 110.7, 55.8, 36.7, 32.5.
5-hydroxy-2-phenethyl-4H-chromen-4-one (5)
Purified by column chromatography by gradient elution from 50/50 dichloromethane/hexane to 100% dichloromethane. IR cm−1 2934, 1653, 1616, 1480, 1409, 1258, 845, 802; 1H-NMR (400MHz, CDCl3): δ 12.51 (1H, s, 5-OH), 7.44 (1H, t, J = 8.32 Hz), 7.18-7.32 (5H, m), 6.85 (1H, dd, J = 8.4 and 0.8 Hz), 6.76 (1H, dd, J = 8.4 and 0.8 Hz) 6.06 (1H, s), 3.05 (2H, t, J = 7.0 Hz), 2.92 (2H, t, J = 6.8 Hz); 13C-NMR (100MHz, CDCl3) δ 183.5, 169.8, 160.8, 156.7, 139.4, 135.1, 128.7, 128.2, 126.6, 111.2, 110.6, 108.8, 106.6, 36.0, 32.8.
5-hydroxy-2-(2-hydroxyphenethyl)-4H-chromen-4-one (6)
Purification was done by stirring the crude solid in minimal dichloromethane and collecting the undissolved solid. IR cm−1 3153, 2944, 1650, 1618, 1487, 1583, 1259, 845, 801; 1H-NMR (400MHz, DMSO-d6): δ 12.63 (1H, br. s, 5-OH), 9.48 (1H, br. S, 2′-OH), 7.62 (1H, t, J = 8.32 Hz), 7.08-699 (3H, m), 6.78 (2H, t, J = 8.4 Hz), 6.69 (1H, t, J = 7.4 Hz) 6.23 (1H, s), 2.95 (4H, s,); 13C-NMR (100MHz, DMSO-d6) δ 182.9, 171.4, 159.9, 156.2, 155.2, 135.6, 129.8, 127.4, 125.8, 118.8, 114.9, 110.7, 109.8, 108.1, 107.0, 33.5, 27.0.
7-hydroxy-2-phenethyl-4H-chromen-4-one (7)
Purified by column chromatography by elution with 95/5 dichloromethane/MeOH. IR cm−1 3026, 1621, 1548, 1495, 1421, 1256, 852, 823; 1H-NMR (400MHz, acetone-d6): δ 9.45 (1H, s, 7-OH), 7.96-7.90 (1H, m) 7.28-7.18 (5H, m,), 6.94 (1H, d J = 8.68 Hz), 6.87 (1H, s), 6.00 (1H, s), 3.08 (2H, t, J = 7.76 Hz), 2.95 (2H, t, J = 6.08 Hz); 13C-NMR (100MHz, acetone-d6) δ 177.2, 168.7, 163.1, 159.1, 141.2, 129.29, 129.26, 127.7, 127.1, 117.8, 115.2, 110.3, 103.3, 36.2, 33.4.
Acknowledgments
The authors thank Dr. Abraham Yousef of Sweet Briar College for the use of their NMR facilities and helpful discussions about the chemistry. The authors also thank Thaddeus Williams for help in purifying compound 5. This work was supported by faculty start-up funds (DAW) and a faculty development summer research grant (DAW) from Lynchburg College, Virginia Foundation of Independent Colleges Summer Research Grant (CS), and partially by the NIH/NIDA Postdoctoral Training Grant DA007027 (DAW) and DA024022 (YZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Butler MS. Nat Prod Rep. 2005;22:162. doi: 10.1039/b402985m. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Middleton E, Kandaswami C, Theoharides TC. Pharmacol Rev. 2000;52:673. [PubMed] [Google Scholar]
- 4.Vauzour D, Vafeiadou K, Rodriguez-Mateos A, Rendeiro C, Spencer JPE. Genes Nutr. 2008;3:115. doi: 10.1007/s12263-008-0091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cazarolli LH, Zanatta L, Alberton EH, Figueiredo M, Folador P, Damazio RG, Pizzolatti MG, Silva F. Mini-Rev Med Chem. 2008;8:1429. doi: 10.2174/138955708786369564. [DOI] [PubMed] [Google Scholar]
- 6.Yoon JS, Lee MK, Sung SH, Kim YC. J Nat Prod. 2006;69:290. doi: 10.1021/np0503808. [DOI] [PubMed] [Google Scholar]
- 7.Wu B, Lee JG, Lim CJ, Jia SD, Kwon SW, Hwang GS, Park JH. Helv Chim Acta. 2012;95:636. [Google Scholar]
- 8.Konishi T, Konoshima T, Shimada Y, Kiyosawa S. Chem Pharm Bull. 2002;50:419. doi: 10.1248/cpb.50.419. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Xu ZR, Chai XY, Zeng KW, Jia YX, Bi D, Ma ZZ, Tu PF. Eur J Org Chem. 2012:5389. [Google Scholar]
- 10.Yang L, Qiao LR, Xie D, Yuan YH, Chen NH, Dai JG, Guo SX. Phytochemistry. 2012;76:92. doi: 10.1016/j.phytochem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 11.Li NG, Shi ZH, Tang YP, Ma HY, Yang JP, Li BQ, Wang ZJ, Song SL, Duan JA. J Heterocycl Chem. 2010;47:785. [Google Scholar]
- 12.Alcantara AR, Marinas JM, Sinisterra JV. Tetrahedron Lett. 1987;28:1515. [Google Scholar]
- 13.Smith JA, Maloney DJ, Hecht SM, Lannigan DA. Bioorg Med Chem. 2007;15:5018. doi: 10.1016/j.bmc.2007.03.087. [DOI] [PubMed] [Google Scholar]
- 14.Zhang LP, Wang YL. Chem Res Chin Univ. 2010;26:245. [Google Scholar]
- 15.Heilbron IM, Hey DH, Lowe A. J Chem Soc. 1934:1311. [Google Scholar]
- 16.Green T, Wuts P. Protective Groups in Organic Chemistry. New Your: John Wiley & Sons, Inc; 1999. [Google Scholar]
- 17.Ghosh CK, Bhattacharyya S, Ghosh C, Patra A. Journal of the Chemical Society-Perkin Transactions. 1999;1:3005. [Google Scholar]
- 18.Kagawa H, Shigematsu A, Ohta S, Harigaya Y. Chem Pharm Bull. 2005;53:547. doi: 10.1248/cpb.53.547. [DOI] [PubMed] [Google Scholar]
- 19.Nussbaumer P, Lehr P, Billich A. J Med Chem. 2002;45:4310. doi: 10.1021/jm020878w. [DOI] [PubMed] [Google Scholar]
- 20.Nandurkar NS, Bhanushali MJ, Patil DS, Bhanage BM. Syn Commun. 2007;37:4111. [Google Scholar]