

Abstract
The loss of flight ability has occurred thousands of times independently during insect evolution. Flight loss may be linked to higher molecular evolutionary rates because of reductions in effective population sizes (Ne) and relaxed selective constraints. Reduced dispersal ability increases population subdivision, may decrease geographical range size and increases (sub)population extinction risk, thus leading to an expected reduction in Ne. Additionally, flight loss in birds has been linked to higher molecular rates of energy-related genes, probably owing to relaxed selective constraints on energy metabolism. We tested for an association between insect flight loss and molecular rates through comparative analysis in 49 phylogenetically independent transitions spanning multiple taxa, including moths, flies, beetles, mayflies, stick insects, stoneflies, scorpionflies and caddisflies, using available nuclear and mitochondrial protein-coding DNA sequences. We estimated the rate of molecular evolution of flightless (FL) and related flight-capable lineages by ratios of non-synonymous-to-synonymous substitutions (dN/dS) and overall substitution rates (OSRs). Across multiple instances of flight loss, we show a significant pattern of higher dN/dS ratios and OSRs in FL lineages in mitochondrial but not nuclear genes. These patterns may be explained by relaxed selective constraints in FL ectotherms relating to energy metabolism, possibly in combination with reduced Ne.
Keywords: molecular evolutionary rates, effective population size, flight loss, comparative method, mitochondrial genes, selective constraints
1. Introduction
The evolution of flight in insects is a key innovation thought to have in part enabled their diversification [1–3]. Despite the advantages associated with flight, such as the ability to disperse widely and forage, flight has been independently lost within nearly every pterygote order and an estimated thousands of times overall [4,5]. The lower dispersal ability associated with flight loss tends to increase population subdivision [6,7], with population subdivision expected to reduce the effective population size (Ne) of species [8]. Lower dispersal ability may additionally reduce geographical range size (reviewed in [9]) and cause greater fluctuations in population size over time owing to increased susceptibility of (sub)populations to extinction, both of which could limit the Ne of a species [10].
Lineages that differ in Ne over the long term are expected to differ in non-synonymous-to-synonymous substitution (dN/dS) ratios [11,12] owing to the greater strength of genetic drift in smaller populations. Non-synonymous mutations are more likely to have a fitness effect than synonymous mutations, and the majority of mutations that occur are thought to have a neutral or a deleterious effect [11,13]. In smaller populations, slightly deleterious mutations are more easily fixed, because purifying selection is not as effective at eliminating them, and thus can lead to increased fixation of non-synonymous mutations relative to synonymous mutations. Higher non-synonymous (dN) rates may also produce observable increases in overall substitution rates (OSRs) in those lineages. Therefore, if flightless (FL) lineages did have consistently reduced Ne, flightlessness may be a predictor of increased dN/dS ratios and OSRs.
A few previous studies of marine organisms, although including a small number of independent comparisons, have used dispersal ability as the predictor of Ne differences and have indeed observed differences in dN/dS ratios [14,15]. Molecular patterns have been examined for bird flight loss [16], with the expectation of reduced Ne for FL bird lineages, partially as FL birds are more often found on islands. Additionally, Shen et al. [16] expected and observed higher dN/dS ratios in FL bird energy-related genes due to a hypothesized relaxation of selective constraints on those genes following the loss of flight. Although the biology of birds and insects differ greatly (e.g. endothermy versus ectothermy), flying metabolic rate in insects is estimated at 50 times higher than resting rate [17,18], and thus we may also observe similar patterns in insects relating to relaxed constraints on energy metabolism. We would expect energy-related influences to mainly affect mitochondrial and nuclear genes involved in the oxidative phosphorylation (OXPHOS) pathway and effective population size influences to have a genome-wide effect.
In this study, we tested for differences in patterns of molecular evolution between FL and flight-capable (F) insect groups by comparing dN rates, synonymous (dS) rates, dN/dS ratios and OSRs of available protein-coding genes. Although there are many ecological or biological factors associated with flight loss (reviewed in [1]), drawing comparisons from a broad range of taxa may help to reduce biases towards certain life-history or ecological traits that could affect the pattern of molecular substitution rates. We have conducted a test of the effect of flightlessness on the rates and patterns of molecular evolution using 49 phylogenetically independent transitions (table 1; [1,5–7,19–36]) that span a wide range of taxa (moths, flies, beetles, mayflies, stick insects, stoneflies, scorpionflies and caddisflies) and different nuclear and mitochondrial protein-coding genes. We approached the analysis in two complementary ways so as to include as many data as possible: whole-tree analysis (as in [14]), which allows analysis of all transitions within a phylogenetic tree and increases the power for detecting a pattern within a tree, and sister-pair analysis (as in [37]), which allows the investigation of individual transitions as independent data points to assess the consistency of any overall trend. Our results show a significant pattern of higher dN/dS ratios and OSRs across multiple instances of flight loss for mitochondrial genes but not nuclear genes. These results are consistent with our initial hypotheses, although are more favourable towards the idea of relaxed selective constraints on energy metabolism.
Table 1.
Source data for cases of insect flight loss. (The electronic supplementary material gives the species names and GenBank accession numbers, assigned flight status of species and flight state sources. EF-1a, elongation factor 1 α; Wgl, wingless; IDH, isocitrate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; RpS5, ribosomal protein S5; CAD, carbamoyl-P synthetase/aspartate transcarbamylase/dihydroorotase; H3, histone 3; NF1, neurofibromin; COI, cytochrome c oxidase subunit I; COII, cytochrome c oxidase subunit II; cytB, cytochrome b; FFL, female flightless; bsFL, both-sexes-flightless.)
| order, taxonomic level and group, ‘common name’ | type of flight loss, potential biological associations | no. transitions in tree (no. sister comparisons) | sequences |
|---|---|---|---|
| studies/phylogenies containing multiple losses of flight, included in both whole-tree and sister-clade analyses | |||
| Lepidoptera, subfamily Ennominae, ‘winter moths’ [19] | FFL, forest dwelling, winter flight, polyphagy | 7 (7) | EF-1a, Wgl, IDH, GAPDH, RpS5, COI |
| Lepidoptera, tribe Operophterini, ‘winter moths’ [20] | FFL, forest dwelling, winter flight, polyphagy | 2a (2) | EF-1a, Wgl |
| Lepidoptera, genus Thyrocopa [21] | bsFL, windswept areas | 2 (2) | EF-1ab, Wgl, COI |
| Coleoptera, family Lampyridae, ‘fireflies’ [22] | FFL, female neoteny, loss of male spermatophore | 4 (4) | COI |
| Coleoptera, family Geotrupidae, ‘earth-boring dung beetles’ [23] | bsFL, arid or semi-arid conditions | 6 (5) | NF1, COI, COII |
| Diptera, superfamily Hippoboscoidea, ‘tsetse/louse/bat flies’ [24],[25]c | bsFL | 4 (4) | CAD, COI; COIIc, cytBc |
| Phasmatodea, order Phasmatodea, ‘stick insects’ [5] | bsFL and FFL | 13 (5) | H3 |
| Mecoptera, order Mecoptera, ‘scorpionflies/hangingflies’ [26],[27]c | bsFL and FFL | 4 (4) | EF-1a, COII; COIc |
| studies/phylogenies containing single transitions, included in sister-clade analysis | |||
| Trichoptera, clade within genus Nothopsyche [28] | FFL, change from plants to sand in case material | 1 | COI |
| Diptera, genus Chionea, ‘snow flies’ [29] | bsFL, winter adults [1] | 1 | CAD |
| Lepidoptera, genus Orgyia [30,31] | FFL, spring feeding [32] | 1 | DDC, EF-1a, CAD, IDH, MDH, RpS5b, COI |
| Lepidoptera, family Psychidae, sp. [33] | FFL | 1 | CAD, IDH, COI |
| Coleoptera, genus Silpha (within ‘carrion beetles’) [17] | bsFL, shift in feeding habit from necrophagy to predatory [34] | 1 | COI |
| Ephemeroptera, genus Cheirogenesia [35] | bsFL, lack of fish predation [36] | 1 | H3b |
| Plecoptera, Zelandoperla fenestrate [6] | bsFL | 1 | H3b, COIb |
aOne of the three transitions in this study overlaps with [19] and the transition was included in whole-tree analyses for this study [20] for one gene that was not available in [19].
bGene did not have enough amino acid variation, but was included in substitution rates tests.
cStudy or gene has overlapping transition with a whole-tree study, adds gene data to sister-clade analysis.
2. Material and methods
(a). Data
We collected published molecular phylogenetic studies including both FL and F insect species (see table 1 and the electronic supplementary material, text S1 for description of categorization). We avoided including comparisons where another obvious transition potentially affecting population size or molecular rates occurred simultaneously or subsequent to flight loss (e.g. island living and parasitism) to test ‘flightlessness’ itself while minimizing confounding factors. Phylogenetic relationships were taken from the source studies, with preference for those constructed by model-based methods. Transitions to flightlessness were taken from source studies (see the electronic supplementary material, table S2) assuming flight was never regained [38]. Source phylogenies for whole-tree analysis were trimmed in some cases to eliminate clades that were not suitable for inclusion (e.g. owing to severe confounding factors such as parasitism).
Datasets were constructed as per two methods of analysis; whole tree and sister clade. Overall, 49 independent cases of flight loss were included in the analyses, each represented by at least one protein-coding gene. Eight of the source studies contained two or more independent losses of flight, which include both paraphyletic and reciprocally monophyletic relationships between F and FL relatives. These phylogenies represented 42 independent transitions in total. These source trees were analysed with both whole-tree and sister-clade methods. For whole-tree analysis, there were 18 trees represented by one gene each. Whole-tree gene data were not combined per study and gene type (nuclear versus mitochondrial) since different transitions were represented by different genes, except in one case where data for two mitochondrial genes were available for identical taxa. In this latter case, the data were combined using dN and dS substitution counts and numbers of sites as described further below. Not all of the cases of flight loss within a tree could be included in the sister-clade analysis since sister FL and F pairs had to be matched without overlap of the branches that connect the comparisons on the phylogeny. Thirty-three sister-clade comparisons were obtained from the whole-tree sets, with other studies contributing genetic data towards some of these transitions, and an additional seven transitions were obtained from other studies including single transitions.
Protein-coding gene sequences were downloaded from GenBank, obtained from the authors directly or downloaded from the authors’ webpages (GenBank accession nos. are given in the electronic supplementary material, table S1). Nucleotide sequences were aligned in MEGA v. 5.0 [39] using the Clustal function (default settings) and verified by visual inspection and amino acid translation. Whole-tree alignments were included if they possessed at least two amino acid substitutions in the ingroup and if variability occurred between FL : F pairs. Sister-clade dN/dS datasets were included if the ingroup (FL–F lineages) contained amino acid differences, or if the calculated non-synonymous (dN) rate was not 0 for both FL and F lineage types. Sister-clade relative-rate datasets were included if possessing any nucleotide variability between sister lineages. In total, there were 35 dN/dS sister comparisons and 39 relative-rate sister comparisons that met the criteria for analysis; each transition was represented by at least one gene result (one to seven genes).
(b). dN/dS ratio and overall substitution rate analysis
dN/dS ratio analysis and OSR analysis for whole trees, as well as dN/dS ratio analysis for sister clades, were conducted using maximum-likelihood methods in the program PAML [40]. Component codeml was used to examine dN, dS and dN/dS ratios, and component baseml was used to examine OSRs; the same input data were used for the two types of whole-tree analyses. All input and output files for all analyses were deposited in the Dryad Digital Repository (doi:10.5061/dryad.3ps4r). For OSR analysis, the sequence models were estimated in MEGA v. 5.0 using constrained topology (maximum likelihood methods, Bayesian Information Criterion). For whole-tree analysis, only lineage tips were coded to two foreground classes: FL or F, with a background rate for all deeper nodes. We applied this method of coding for whole-tree analysis to avoid biasing the results; dN/dS rates are estimated as higher for tip lineages [41], and deeper nodes tend to be reconstructed as F due to the unidirectional nature of flight loss [38,42].
FL and F lineages in sister-clade dN/dS codings were each assigned a class from their point of divergence onward by coding the entire pathway of branches leading to taxa, not just lineage tips. dS, dN and dN/dS ratios and branch lengths were calculated for each FL and F group of lineages in each dataset. Multiple gene results for the same transition were combined per transition, with nuclear and mitochondrial results separately combined since both the dN and dS rates of mitochondrial genes are expected to be higher than those of nuclear genes. The combination was performed by multiplying the dN rate (site-wise estimate) by the number of non-synonymous (N) sites, and these resultant counts were summed across multiple genes per transition, then divided by the total number of N sites in all the genes. The same procedure was performed for dS and synonymous sites. Average branch lengths for FL and F clades in a comparison were calculated in the same way that is performed in PAML by taking (N × dN + S × dS/(N + S)) × 3.
Sister-clade relative-rate analysis was performed using relative rates tests in the program Phyltest v. 2.0 (http://www.kumarlab.net/publications) with the Kimura-2-Parameter model on FL, F and closest outgroup sequences. A simple model was selected due to the small number of available sequences for estimating the branch lengths for these comparisons. The same number of sequences was chosen for both the FL and F groups, and selected individuals were randomly chosen in a stratified design so as to be phylogenetically dispersed across each group [43]; this was performed as a compromise to avoid the node-density effect and to better characterize the overall rate for the group of sequences [43]. Where multiple gene results existed per transition, the z-values (representing significance of difference between FL and F lineages) were signed based on direction and averaged across genes.
To examine whether FL or F lineages had consistently higher dN, dS, dN/dS or relative rates, we examined the direction of the difference in rates across the multiple transitions with binomial tests. All statistical tests performed are 2-tailed.
(c). Sensitivity and additional testing
Higher variance in rates is expected for sister-clade pairs with low information content [44] and with higher divergences [45]. Since we use a binomial test for analysis of patterns and not a weighted test we consider low information content in the data. The square roots of the sum of the branch lengths for the pairs were plotted against the absolute differences in dN/dS rates. The data were examined for a negative trend, whereby the datasets with the smallest sum of branch lengths would be excluded from binomial pattern testing [44].
To assess whether the node-density effect, i.e. whereby better sampled clades are expected to have longer reconstructed branch lengths, is likely to have produced the observed dN/dS sister-clade results, we examined the prevalence of input comparisons in which the number of tips is greater in the FL or F groups. Additionally, we assessed whether female-flightless (FFL) patterns differed from those comparisons including complete flight loss in both sexes by performing Fisher's exact tests on the nuclear and mitochondrial gene results between these flight loss categories.
3. Results
(a). dN/dS ratios
(i). Whole-tree results
The mitochondrial whole-tree dN/dS results (figure 1a, darker bars) show a significant pattern when the gene results are combined per study; out of the six studies containing mitochondrial genes, all six dN/dS results are higher for the FL than F categories (p = 0.0313). The dN/dS ratios of FL lineages in these six comparisons were approximately double those of the F lineages (median FL/F dN/dS ratio was 2.44). Although nuclear tree results (figure 1a, lighter bars) were not combined due to differing transitions in the trees per study, there is no consistent pattern across the six studies that contain nuclear gene results (two positive (FL > F) results, two negative (F > FL) results and two mixed (both positive and negative gene results for the same study)). Mitochondrial gene dN rates were generally, though not significantly, positive across the studies (five of six studies were positive), and nuclear dN results were not consistent across the studies (two positive, two mixed, one negative and one neutral). dS rates were not consistent across studies for mitochondrial genes, though majority negative (five of six studies negative), nor were they consistent for nuclear genes (three positive, two negative and one mixed). All whole-tree results are given in the electronic supplementary material, table S3.
Figure 1.
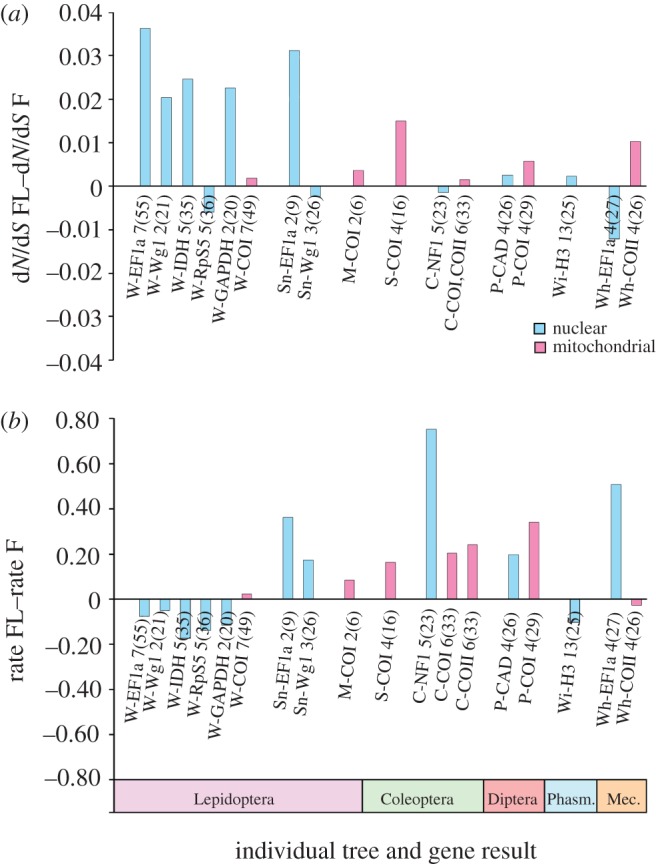
(a) Summary of dN/dS ratio results for whole trees and (b) OSR results for whole trees between FL and F insects in eight studies. Each study (represented by grouped bars) includes phylogenies each containing multiple transitions to flightlessness (42 separate instances of flight loss total). Each result bar represents a gene or genes. The y-axis represents the dN/dS ratio or OSR of the FL lineages minus the dN/dS ratio or OSR of the F lineages; therefore, a positive bar height indicates that the FL lineages have higher dN/dS ratios or longer branch lengths than the F lineages with which it was compared. Bar labels are in the form ‘study’–‘gene’ ‘no. independent cases of flight loss for that gene’ ‘(no. of species in tree used)’. For example, W-EF1a 7(55) is study ‘W’—gene ‘EF1a’, ‘seven’ cases of loss (‘55’ species). Studies: W[19], Sn[20], M[21], S[22], C[23], P[24], Wi[5], Wh[26]. Phasm., Phasmatodea; Mec., Mecoptera. (Online version in colour.)
(ii). Sister-clade results
Thirty-five independent transitions were analysed. Mitochondrial sister-clade dN/dS results (figure 2a) showed a significant pattern; out of 30 independent comparisons, 23 have higher dN/dS ratios in the FL lineages (p = 0.0052). The dN results are also significantly higher in mitochondrial genes with 24 of 30 comparisons being positive (p = 0.0014). dS mitochondrial rates did not show a significant pattern (17 of 30 positive, p = 0.5847). The dN/dS ratios of FL lineages in these 30 comparisons were approximately double those of the F lineages (median FL/F dN/dS ratio was 1.99). The nuclear sister-clade dN/dS rates (figure 2b) did not show a consistent pattern (12 of 26 positive, p = 0.8450) and neither did nuclear dN (12 of 26 positive, p = 0.8450) or dS rates (16 of 26 positive, p = 0.3269). COI was the most common gene available for analysis, and patterns for this gene were consistent with those of the overall mitochondrial results; dN/dS and dN were consistently positive (22 of 27 positive, p = 0.0015 and 21 of 27 positive, p = 0.0059, respectively), whereas dS rates were not (14 of 27 positive, p = 1.000). All dN/dS sister-clade gene results are given in the electronic supplementary material, table S4, and the final combined results are organized in the electronic supplementary material, table S5.
Figure 2.

Comparison of dN/dS ratios in FL and F lineages. (a) Thirty sister-clade comparisons involving mitochondrial genes and (b) 26 sister-clade comparisons involving nuclear genes. Each bar represents an individual instance of flight loss. A positive bar height indicates a sister comparison in which the dN/dS ratio is higher in the FL group than in the F group. Bar labels indicate the (‘study and reference number for the transition’) and the genes. Studies: W[19], Sn[20], M[21], S[22], C[23], P[24], Wi[5], Wh[26], D[25], Po[27], H[28], Pe[29], Mi[30], Z[31], Mu[33]. Transition reference numbers correspond to clade labels shown in the electronic supplementary material, figures S1–S8. (Online version in colour.)
(b). Overall substitution rates
(i). Whole-tree results
Mitochondrial whole-tree OSRs (figure 1b, darker bars) were generally positive but not significantly so across the six studies (results in five of six studies were positive, p = 0.2188). Nuclear whole-tree OSR patterns (figure 1b, lighter bars) were also not consistently positive (four of six studies were positive, one negative and one majority negative).
(ii). Sister-clade relative rates test results
Thirty-nine independent transitions were analysed by relative rates tests. The 30 mitochondrial results showed a significant pattern in which FL lineages had higher relative rates (21 of 30 positive, p = 0.0428), whereas the 32 nuclear results did not (19 of 32 positive, p = 0.3771) (relative-rate results in the electronic supplementary material, table S6).
(c). Sensitivity testing
No significant negative relationship was observed between the square roots of the sum of the branch lengths and the absolute differences in dN/dS rates for the sister pairs. Therefore we did not remove any datasets from dN/dS sister-comparison binomial pattern testing.
We do not expect that the node-density effect has produced the observed dN/dS sister-clade results. Out of the gene datasets in dN/dS sister-clade analysis (82 sets) (see the electronic supplementary material, table S4), almost half (40) were balanced comparisons in which the FL and F groups have an equal number of tips. The remainder of datasets were divided between the FL category having a higher number of individuals per clade (20) and the F category having a higher number of individuals per clade (22).
FFL and both-sexes-flightless (bsFL) transitions were equally represented in the number of mitochondrial and nuclear sister-clade dN/dS comparisons. These flight loss types did not differ in mitochondrial dN/dS patterns (for FFL 11 of 15 transitions were positive versus bsFL 12 of 15 positive (p = 1.000)), dN results (FFL 12 of 15 versus bsFL 12 of 15 (p = 1.000)) or dS results (FFL 7 of 15 versus bsFL 10 of 15 (p = 0.4621)). Nuclear sister-clade patterns also did not differ significantly by flight loss type for dN/dS results (FFL 8 of 13 positive versus bsFL 4 of 13 positive (p = 0.2377)), dN results (FFL 7 of 13 versus bsFL 5 of 13 (p = 0.6951)) or dS results (FFL 7 of 13 versus bsFL 9 of 13 (p = 0.6882)).
4. Discussion
FL insect groups displayed higher mitochondrial dN/dS ratios significantly more often than their F relatives. This result was consistent across both whole-tree studies and across sister comparisons, with FL dN/dS rates approximately double those in F lineages. Additionally, there was a significant pattern of mitochondrial OSRs being higher in FL lineages across the sister comparisons. The nuclear patterns for both dN/dS ratios and OSRs were not consistent across whole-tree studies or across the sister-clade comparisons.
Effective population size (Ne) differences are expected to act genome-wide; therefore, the difference in mitochondrial and nuclear patterns suggests that Ne differences may not be the sole or main cause of the patterns. Ne may still play a role in the mitochondrial patterns observed. The Ne of mitochondrial genes is inherently fourfold smaller than autosomal genes. If flight loss has caused a further reduction in Ne, more deleterious mutations may behave as neutral in mitochondrial genes and be fixed due to drift. Reduced Ne, for example owing to island living, may affect mitochondrial and nuclear genes differently and only cause accelerated rates in mitochondrial genes [46]. Overall however, it does not appear that there are strong Ne differences consistent across the majority of cases of flight loss tested. The various reasons for expecting Ne differences described in the introduction may in fact be counteracted by the increase in fecundity observed for FL groups [47].
The strong pattern across mitochondrial genes is likely at least partially due to a relaxation of selective constraints associated with flight loss, as was observed for bird flight loss [16]. Flight is an energetically costly activity; insect flying metabolic rate is estimated at 50 times higher than that at rest [17,18]. Therefore, the loss of flight may result in decreased selection pressure to maintain efficient energy production. For example, the winged morph of a grasshopper species consumes significantly more energy than the wingless morph [48], and winged individuals of pea aphids show increased transcription levels of genes related to energy production relative to unwinged morphs [49]. Although these studies examined intraspecific differences in flight ability, energy consumption or expression differences might be expected between FL and F species as well. These expectations of differences in energy-gene constraints are not limited to flight loss itself, as patterns among mitochondrial genes are also observed for mammals that are less locomotive compared with their more highly locomotive relatives [16]. The relaxation of purifying selection in those relevant genes could allow more non-synonymous mutations to accumulate. This would be expected in both mitochondrial and nuclear genes involved with energy production. Since the OXPHOS pathway produces 95% of the ATP needed for locomotion [50,51], these genes are expected to show the strongest effects. The OXPHOS genes included in this study are the mitochondrial genes COI, COII and cytB. Shen et al. [16] did not observe a significant difference between weakly and strongly locomotive birds for COI specifically, as was found in this study of insects. Insects are ectotherms and do not have metabolic rates that are as high as endotherms such as birds [52]. Thus, it could be that insects may expend a higher proportion of their total energy budget on flight than birds, and a stronger pattern in OXPHOS genes might therefore be expected in insects relating to locomotion.
Most of the energy-linked genes that were available for this study are mitochondrial. The nuclear gene IDH is also involved in energy metabolism; it has been observed in a species of cricket that the long-winged morph had greater transcript abundance and protein concentration of the lipogenic enzyme NADP ± IDH than the short-winged (FL) morph [53]. However, we expect that mitochondrial and nuclear genes in this study generally represent different functional categories, and that mitochondrial genes would show the strongest patterns relating to energy. If indeed the patterns are related to energy metabolism, the results of this study do not give support to the hypothesis by Martin & Palumbi [54], where higher mutation rates are expected with increased metabolic rates. By that hypothesis, we would expect FL lineages also to have decreased dS rates compared with F lineages due to reduced metabolic rates. Although this may be true within certain groups included in the study, we did not observe such patterns in dS rates overall in our analyses.
Interestingly, there was no difference in patterns between FFL and bsFL comparisons, with mitochondrial patterns being strong in each category. Also interesting is the observation of a strong pattern in mitochondrial genes at all, given the major hypothesized cause of flight loss (specifically in females) as an energy trade-off [18,47]. If the energy saved by being FL is being used elsewhere by the organism, we might not expect the genetic energy pathways to be under less intense purifying selection. In addition, half of the cases of flight loss used in this study are transitions to female flightlessness, where relaxed purifying selection would allow deleterious mutations in OXPHOS genes to be passed onto F male offspring. Further work could examine whether the F male counterparts experience any reduction in flight ability, either due to genetic correlation of wing-related genes with females that have wing reduction, as suggested by [18], or due to sharing of those energy-relevant genes with the females.
Complete agreement across all datasets was not an original expectation of this study owing to expected differences in positive or purifying selection across taxa and genes. While there was variability among datasets, our overall consistent patterns are quite remarkable in the light of the possible confounding influences upon molecular rates. In using flight loss as a predictor of molecular rates, if co-occurring factors that were excluded here (parasitism and island living) were instead included, these might enhance the pattern of higher dN/dS ratios in FL groups. More work is needed to delve deeper into the molecular patterns to elucidate additional influences, for example, of positive or purifying selection. Future studies using larger amounts of genetic data for individual transitions could further test for genome-wide patterns and perform genome-wide scans for positive selection in order to identify the types of genes involved in and affected by flight loss.
In conclusion, we have shown that FL insect lineages have higher ratios of non-synonymous-to-synonymous substitutions and higher OSRs than related F lineages for mitochondrial genes but not nuclear genes. While differences in effective population size between FL and F lineages may exist, our results are more favourable to the hypothesis of relaxed selective constraints relating to energy metabolism in FL groups. This study has brought to light an interesting wide-spread pattern in insect molecular evolution.
Acknowledgements
We thank T. Crease, J. Fu, R. Gregory and two anonymous reviewers for helpful suggestions. We also thank the researchers who produced the sequence data that we used in the analysis and authors who provided alignments directly.
Funding statement
This research was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant to S.J.A., Canada Graduate Scholarship to T.F.M.), the Government of Ontario (Ontario Graduate Scholarship to T.F.M.) and the University of Guelph (Integrative Biology PhD Award to T.F.M.).
References
- 1.Roff DA. 1990. The evolution of flightlessness in insects. Ecol. Monogr. 60, 389–421 (doi:10.2307/1943013) [Google Scholar]
- 2.Kingsolver JG, Koehl MAR. 1994. Selective factors in the evolution of insect wings. Annu. Rev. Entomol. 39, 425–451 (doi:10.1146/annurev.en.39.010194.002233) [Google Scholar]
- 3.Mayhew PJ. 2007. Why are there so many insect species? Perspectives from fossils and phylogenies. Biol. Rev. 82, 425–454 (doi:10.1111/j.1469-185X.2007.00018.x) [DOI] [PubMed] [Google Scholar]
- 4.Wagner DL, Liebherr JK. 1992. Flightlessness in insects. Trends. Ecol. Evol. 7, 216–220 (doi:10.1016/0169-5347(92)90025-7) [DOI] [PubMed] [Google Scholar]
- 5.Whiting MF, Bradler S, Maxwell T. 2003. Loss and recovery of wings in stick insects. Nature 421, 264–267 (doi:10.1038/nature01313) [DOI] [PubMed] [Google Scholar]
- 6.McCulloch GA, Wallis GP, Waters JM. 2009. Do insects lose flight before they lose their wings? Population genetic structure in subalpine stoneflies. Mol. Ecol. 18, 4073–4087 (doi:10.1111/j.1365-294X.2009.04337.x) [DOI] [PubMed] [Google Scholar]
- 7.Ikeda H, Nishikawa M, Sota T. 2012. Loss of flight promotes beetle diversification. Nat. Commun. 3, 648–654 (doi:10.1038/ncomms1659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Templeton AR. 2006. Population genetics and microevolutionary theory. Hoboken, NJ: John Wiley & Sons, Inc [Google Scholar]
- 9.Lester SE, Ruttenberg BI, Gaines SD, Kinlan BP. 2007. The relationship between dispersal ability and geographic range size. Ecol. Lett. 10, 745–758 (doi:10.1111/j.1461-0248.2007.01070.x) [DOI] [PubMed] [Google Scholar]
- 10.Woolfit M, Bromham L. 2005. Population size and molecular evolution on islands. Proc. R. Soc. B 272, 2277–2282 (doi:10.1098/rspb.2005.3217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohta T. 1972. Population size and rate of evolution. J. Mol. Evol. 1, 305–314 (doi:10.1007/BF01653959) [PubMed] [Google Scholar]
- 12.Ohta T. 1992. The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286 (doi:10.1146/annurev.es.23.110192.001403) [Google Scholar]
- 13.Eyre-Walker A, Keightley PD. 2007. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (doi:10.1038/nrg2146) [DOI] [PubMed] [Google Scholar]
- 14.Foltz DW. 2003. Invertebrate species with nonpelagic larvae have elevated levels of nonsynonymous substitutions and reduced nucleotide diversities. J. Mol. Evol. 57, 607–612 (doi:10.1007/s00239-003-2495-5) [DOI] [PubMed] [Google Scholar]
- 15.Foltz DW, Mah CL. 2010. Difference in larval type explains patterns of nonsynonymous substitutions in two ancient paralogs of the histone H3 gene in sea stars. Evol. Dev. 12, 222–230 (doi:10.1111/j.1525-142X.2010.00406.x) [DOI] [PubMed] [Google Scholar]
- 16.Shen Y-Y, Shi P, Sun Y-B, Zhang Y-P. 2009. Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Res. 19, 1760–1765 (doi:10.1101/gr.093138.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roff DA. 1991. Life history consequences of bioenergetic and biomechanical constraints on migration. Am. Zool. 31, 205–216 (doi:10.1093/icb/31.1.205) [Google Scholar]
- 18.Roff DA, Fairbairn DJ. 1991. Wing dimorphisms and the evolution of migratory polymorphisms among the Insecta. Am. Zool. 31, 243–251 (doi:10.1093/icb/31.1.243) [Google Scholar]
- 19.Wahlberg N, Snäll N, Viidalepp J, Ruohomäki K, Tammaru T. 2010. The evolution of female flightlessness among Ennominae of the Holarctic forest zone (Lepidoptera, Geometridae). Mol. Phylogenet. Evol. 55, 929–938 (doi:10.1016/j.ympev.2010.01.025) [DOI] [PubMed] [Google Scholar]
- 20.Snäll N, Tammaru T, Wahlberg N, Viidalepp J, Ruohomäki K, Savontaus M-L, Huoponen K. 2007. Phylogenetic relationships of the tribe Operophterini (Lepidoptera, Geometridae): a case study of the evolution of female flightlessness. Biol. J. Linn. Soc. 92, 241–252 (doi:10.1111/j.1095-8312.2007.00834.x) [Google Scholar]
- 21.Medeiros MJ, Gillespie RG. 2010. Biogeography and the evolution of flightlessness in a radiation of Hawaiian moths (Xyloryctidae: Thyrocopa). J. Biogeogr. 38, 1–11 (doi:10.1111/j.1365-2699.2010.02402.x) [Google Scholar]
- 22.South A, Stanger-Hall K, Jeng M-L, Lewis SM. 2010. Correlated evolution of female neoteny and flightlessness with male spermatophore production in fireflies (Coleoptera: Lampyridae). Evolution 65, 1099–1113 (doi:10.1111/j.1558-5646.2010.01199.x) [DOI] [PubMed] [Google Scholar]
- 23.Cunha RL, Verdú JR, Lobo JM, Zardoya R. 2011. Ancient origin of endemic Iberian earth-boring dung beetles (Geotrupidae). Mol. Phylogenet. Evol. 59, 578–586 (doi:10.1016/j.ympev.2011.03.028) [DOI] [PubMed] [Google Scholar]
- 24.Petersen FT, Meier R, Kutty SN, Wiegmann BM. 2007. The phylogeny and evolution of host choice in the Hippoboscoidea (Diptera) as reconstructed using four molecular markers. Mol. Phylogenet. Evol. 45, 111–122 (doi:10.1016/j.ympev.2007.04.023) [DOI] [PubMed] [Google Scholar]
- 25.Dittmar K, Porter ML, Murray S, Whiting MF. 2006. Molecular phylogenetic analysis of nycteribiid and streblid bat flies (Diptera: Brachycera, Calyptratae): implications for host associations and phylogeographic origins. Mol. Phylogenet. Evol. 38, 155–170 (doi:10.1016/j.ympev.2005.06.008) [DOI] [PubMed] [Google Scholar]
- 26.Whiting MF. 2002. Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zool. Scr. 31, 93–104 (doi:10.1046/j.0300-3256.2001.00095.x) [Google Scholar]
- 27.Pollmann C, Misof B, Sauer KP. 2008. Molecular phylogeny of panorpodid scorpionflies: an enigmatic, species-poor family of Mecoptera (Insecta). Org. Divers. Evol. 8, 77–83 (doi:10.1016/j.ode.2006.12.001) [Google Scholar]
- 28.Hayashi F, Kamimura Y, Nozaki T. 2008. Origin of the transition from aquatic to terrestrial habits in Nothopsyche caddisflies (Trichoptera: Limnephilidae) based on molecular phylogeny. Zool. Sci. 25, 255–260 (doi:10.2108/zsj.25.255) [DOI] [PubMed] [Google Scholar]
- 29.Petersen MJ, Bertone MA, Wiegmann BM, Courtney GW. 2010. Phylogenetic synthesis of morphological and molecular data reveals new insights into the higher-level classification of Tipuloidea (Diptera). Syst. Entomol. 35, 526–545 (doi:10.1111/j.1365-3113.2010.00524.x) [Google Scholar]
- 30.Mitchell A, Mitter C, Regier JC. 2006. Systematics and evolution of the cutworm moths (Lepidoptera: Noctuidae): evidence from two protein-coding nuclear genes. Syst. Entomol. 31, 21–46 (doi:10.1111/j.1365-3113.2005.00306.x) [Google Scholar]
- 31.Zahiri R, Kitching IJ, Lafontaine JD, Mutanen M, Kaila L, Holloway JD, Wahlberg N. 2010. A new molecular phylogeny offers hope for a stable family level classification of the Noctuoidea (Lepidoptera). Zool. Scr. 40, 158–173 (doi:10.1111/j.1463-6409.2010.00459.x) [Google Scholar]
- 32.Hunter AF. 1995. The ecology and evolution of reduced wings in forest macrolepidoptera. Evol. Ecol. 9, 275–287 (doi:10.1007/BF01237773) [Google Scholar]
- 33.Mutanen M, Wahlberg N, Kaila L. 2010. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc. R. Soc. B 277, 2839–2848 (doi:10.1098/rspb.2010.0392) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda H, Kagaya T, Kubota K, Abe T. 2008. Evolutionary relationships among food habit, loss of flight, and reproductive traits: life-history evolution in the Silphinae (Coleoptera: Silphidae). Evolution 62, 2065–2079 (doi:10.1111/j.1558-5646.2008.00432.x) [DOI] [PubMed] [Google Scholar]
- 35.Ogden TH, Gattolliat JL, Sartori M, Staniczek AH, Soldán T, Whiting MF. 2009. Towards a new paradigm in mayfly phylogeny (Ephemeroptera): combined analysis of morphological and molecular data. Syst. Entomol. 34, 616–634 (doi:10.1111/j.1365-3113.2009.00488.x) [Google Scholar]
- 36.Ruffieux L, Elouard J-M. 1998. Flightlessness in mayflies and its relevance to hypotheses on the origin of insect flight. Proc. R. Soc. Lond. B 265, 2135–2140 (doi:10.1098/rspb.1998.0550) [Google Scholar]
- 37.Wright S, Keeling J, Gillman L. 2006. The road from Santa Rosalia: a faster tempo of evolution in tropical climates. Proc. Natl Acad. Sci. USA 103, 7718–7722 (doi:10.1073/pnas.0510383103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stone G, French V. 2003. Evolution: Have wings come, gone and come again? Curr. Biol. 13, R436–R438 (doi:10.1016/S0960-9822(03)00364-6) [DOI] [PubMed] [Google Scholar]
- 39.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (doi:10.1093/molbev/msm088) [DOI] [PubMed] [Google Scholar]
- 41.Ho SYW, Shapiro B, Phillips MJ, Cooper A, Drummond AJ. 2007. Evidence for time dependency of molecular rate estimates. Syst. Biol. 56, 515–522 (doi:10.1080/10635150701435401) [DOI] [PubMed] [Google Scholar]
- 42.Hovmoller R, Pape T, Kallersjo M. 2002. The Palaeoptera problem: basal Pterygote phylogeny inferred from 18S and 28S rDNA sequences. Cladistics 18, 313–323 (doi:10.1006/clad.2002.0199) [DOI] [PubMed] [Google Scholar]
- 43.Robinson M, Gouy M, Gautier C, Mouchiroud D. 1998. Sensitivity of the relative-rate test to taxonomic sampling. Mol. Biol. Evol. 15, 1091–1098 (doi:10.1093/oxfordjournals.molbev.a026016) [DOI] [PubMed] [Google Scholar]
- 44.Welch JJ, Waxman D. 2008. Calculating independent contrasts for the comparative study of substitution rates. J. Theor. Biol. 251, 667–678 (doi:10.1016/j.jtbi.2007.12.015) [DOI] [PubMed] [Google Scholar]
- 45.Garland T, Harvey PH, Ives AR. 1992. Procedure for the analysis of comparative data using phylogenetically independent contrasts. Syst. Biol. 41, 18–32 (doi:10.1093/sysbio/syp096) [Google Scholar]
- 46.Smith BT, Klicka J. 2013. Examining the role of effective population size on mitochondrial and multilocus divergence time discordance in a songbird. PLoS ONE 8, e55161 (doi:10.1371/journal.pone.0055161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guerra PA. 2011. Evaluating the life-history trade-off between dispersal capability and reproduction in wing dimorphic insects: a meta-analysis. Biol. Rev. 86, 813–35 (doi:10.1111/j.1469-185X.2010.00172.x) [DOI] [PubMed] [Google Scholar]
- 48.Lock K, Verslycke T, Janssen CR. 2006. Energy allocation in brachypterous versus macropterous morphs of the pygmy grasshopper Tetrix subulata (Orthoptera: Tetrigidae). Entomol. Gener. 28, 269–274 (doi:10.1127/entom.gen/28/2006/269) [Google Scholar]
- 49.Brisson JA, GK Davis, Stern DL. 2007. Common genome-wide patterns of transcript accumulation underlying the wing polyphenism and polymorphism in the pea aphid (Acyrthosiphon pisum). Evol. Dev. 9, 338–346 (doi:10.1111/j.1525-142X.2007.00170.x) [DOI] [PubMed] [Google Scholar]
- 50.Erecinska M, Wilson DF. 1982. Regulation of cellular energy metabolism. J. Membr. Biol. 70, 1–14 (doi:10.1007/BF01871584) [DOI] [PubMed] [Google Scholar]
- 51.Shen Y-Y, Liang L, Zhu Z-H, Zhou W-P, Irwin DM, Zhang Y-P. 2010. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl Acad. Sci. USA 107, 8666–8671 (doi:10.1073/pnas.0912613107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gillooly JF, Brown JH, West GB, Savage VM, Charnov EL. 2001. Effects of size and temperature on metabolic rate. Science 293, 2248–2251 (doi:10.1126/science.1061967) [DOI] [PubMed] [Google Scholar]
- 53.Schilder RJ, Zera AJ, Black C, Hoidal M, Wehrkamp C. 2011. The biochemical basis of life history adaptation: molecular and enzymological causes of NADP 1-isocitrate dehydrogenase activity differences between morphs of Gryllus firmus that differ in lipid biosynthesis and life history. Mol. Biol. Evol. 28, 3381–3393 (doi:10.1093/molbev/msr171) [DOI] [PubMed] [Google Scholar]
- 54.Martin AP, Palumbi SR. 1993. Body size, metabolic rate, generation time, and the molecular clock. Proc. Natl Acad. Sci. USA 90, 4087–4091 (doi:10.1073/pnas.90.9.4087) [DOI] [PMC free article] [PubMed] [Google Scholar]