

Dear Sir,
I am Neoklis A. Georgopoulos, from Department of Obstetrics and Gynaecology, Division of Reproductive Endocrinology, University of Patras Medical School, Greece. We present an early prepubertal diagnosis of Kallmann syndrome (KS) in a child with anosmia, renal agenesis and mirror movements. KS is a congenital defect characterized by the presence of hypogona-dotropic hypogonadism and anosmia or hyposmia 1. Besides hypogonadism and anosmia, the clinical phenotype is characterized by the presence of other associated features, including, among others, mirror movements (synkinesis), cleft palate and renal agenesis/dysgenesis.
KS is a genetically heterogeneous disease showing X-linked, dominant or recessive autosomal, or oligogenic inheritance. The gene responsible for an X-linked mode of inheritance was identified in the short arm of the X chromosome (Xp22.3) and named KAL1 2. Mutations within the FGFR1 and the Prok2/ProkR2 genes have been implicated in the autosomal dominant and recessive inheritance forms, respectively. Certain non-reproductive features of KS might be linked to either KAL1 or FGFR1 gene defects. The mirror movements are present in approximately 80% of males with KAL1 mutations, but only in 10% of patients with FGFR1 gene defects. The frequency of KAL1 gene defects is estimated to be around 10% of all sporadic cases, whereas in the presence of associated renal agenesis/dysgenesis the frequency is as high as 89% 3, 4. To date, no case of KS and its associated renal agenesis caused by an FGFR1 mutation has been reported.
The diagnosis of hypogonadotropic hypogonadism can be made in a male infant during the first 6 months of life on the basis of activation of the hypothalamic–pituitary–gonadotropic axis 5. Later, in childhood, the axis is suppressed and the definitive diagnosis of KS is therefore delayed until late adolescence, when the lack of pubertal development and the absence of secondary sex characteristics become obvious. The lack of pubertal development can cause considerable psychological damage to the young patient.
We report on a child who was admitted to the Paediatric Clinic of Patras Medical School (Rio, Greece) at the age of 12 years for the evaluation of right renal agenesis. His height was 146.6 cm (25th percentile) and his weight was 38.8 kg (50th percentile). On clinical examination by a paediatric endocrinologist, his penis size was found to be normal for a prepubertal boy (length 5 cm, diameter 3.5 cm); his right testis was palpable in the scrotum, which was 2 cm in diameter, but his left testis was not palpable. The associated clinical features included an arched palate, mirror movements and anosmia. He was the only child in the family. As a detailed family history showed no evidence of hypogonadism, anosmia/hyposmia and/or delayed puberty in any relatives, the case was considered sporadic.
Serum testosterone level was 8.46 ng dL-1, and there was no response of luteinizing hormone (LH) or follicle-stimulating hormone (FSH) to a gonadotropin-releasing hormone (GnRH) (100 μg) challenge (LH: < 0.01 mIU mL-1, FSH: 0.2 mIU mL-1). A detailed magnetic resonance imaging of the brain showed the absence of the olfactory bulbs and tracts and atrophy of the olfactory gyri.
On the basis of the knowledge that KAL1 gene defects are highly probable in the presence of renal agenesis, we decided to sequence the KAL1 gene for a possible early clinical and genetic diagnosis of KS in our patient. We believed that the result of the genetic test would be worth considering, along with the fact that the patient had no clinical signs of hypogonadism.
Sequence analysis of the KAL1 gene identified two genetic defects in the patient. The first mutation was a 259G to A transition in exon 1, turning codon 37 encoding glutamic acid into lysine (E37K), and the second an 835A to T transversion in exon 5, turning codon 235 encoding threonine into serine (T235S) (Figure 1). Neither mutation has been described earlier, and they are not identified in the sequence analysis of KAL1 gene in 53 normal men or in the two major public Single Nudeotide Polymorphism (SNP) databases, GenBank and Ensembl. Sequence analysis was carried out for the mother, because the KAL1 gene is located on the X chromosome that is passed on from the mother to the male proband. The mother was a heterozygote for exon 1 with a normal and mutant allele, but she was a wild-type homozygote for exon 5. Therefore, the mother transmitted the mutation in exon 1 to her son. The fact that the second mutation in her genome was absent suggested that the son had a de novo mutation in exon 5 (Figure 1).
Figure 1.
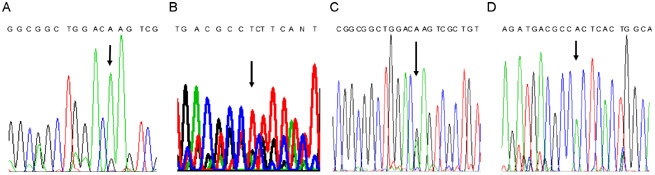
Electrochromatograms showing the two point mutations. (A) at 259 bp of exon 1 and (B) at 835 bp of exon 5 of KAL1 gene identified in the patient with Kallmann syndrome and renal aplasia and the heterozygotous status of his mother for (C) exon 1 and homozygotous status of the wild type allele of his mother for (D) exon 5.
The protein encoded by the KAL1 gene, named anosmin-1, is a glycoprotein of the extracellular matrix of unknown function. It has an amino terminus with a cysteine-rich domain, including a whey acidic protein-like four-disulphide core motif (WAP), four fibronectin type III domains (FnIII R1–R4) and a carboxyl-terminal histidine-rich domain. The E37K mutation is localized to the amino-terminal cysteine-rich domain of anosmin-1 and involves the non-conservative replacement of an acidic amino acid by a basic amino acid. The T235S mutation lies within the first R1 FNIII of the protein near a major cell adhesion site, which contains one of the predicted heparin sulphate binding sites of anosmin. A sequence-based prediction of the effects of these amino acid substitutions using the SIFT (Sorting Intolerant From Tolerant) software (http://blocks.fhcrc.org/sift/SIFT.html) revealed that the 259G to A transition (E37K) could affect protein function (with a score of 0.04), whereas the 835A to T transversion (T235S) was predicted to be tolerable. Indeed, with the latter mutation, at the protein level, the amino acid threonine was replaced by serine at position 235. Both amino acids are polar and they differ by only one methyl group (–CH3); therefore, the change was predicted to be tolerable. In contrast, the change from glutamic acid to lysine at position 37 replaces an acidic amino acid with a basic one, which might seriously affect protein function.
To our knowledge, a genetic diagnosis of KS has never been reported during childhood. As our case was sporadic, any attempt to identify the genotype would have been extremely costly and time consuming. The KAL1 gene consists of 14 exons, and all reported causative mutations are scattered within the gene without any specific clustering. The finding of a right renal agenesis in a child with coexistent anosmia made sequencing the entire coding region of the KAL1 gene worthwhile, because the possibility of the identification of a causative mutation increased from 9% to 89%. It is therefore worthwhile to apply a genetic test only in the presence of associated non-reproductive clinical phenotypes in KS.
Although a normal sequence in the KAL1 gene would not exclude KS, the finding of a causative mutation would further strengthen the diagnosis. Previously, an X-linked mode of inheritance dissociation of anosmia, delayed puberty and hypogonadotropic hypogonadism was reported in members of the same family 4, 6. In this study, the association of the right renal agenesis with anosmia strengthened the clinical diagnosis of KS, and it was further supported by the identification of the causative molecular defect. A normal clinical phenotype has not been reported in a case with KAL1 gene mutation.
Through this letter, we wish to emphasise the im- portance of the presence of renal dysgenesis/agenesis and mirror movements as strong indicators for the presence of KAL1 gene mutations in patients with anosmia, even in childhood.
Upon a prepubertal genetic diagnosis of the syndrome, the clinician can undertake therapeutic intervention for puberty induction at the age of 14 years, offering the patient a pubertal maturation that is similar to that of his teenage peers. As the correlations between genotypes and phenotypes cannot be predicted with certainty, treatment can be discontinued and the gonadotropic axis can be re-evaluated after puberty is reached. The documentation of the X-linked mode of inheritance could reassure these patients that the disease can only be transmitted as a recessive trait in their female offspring, and prenatal diagnosis could identify the presence of the causative mutation in the KAL1 gene in the male offspring of the next generation.
Acknowledgments
This work has been sponsored by a grant from the Hellenic Endocrine Society (Athens, Greece).
References
- Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Ment Defic. 1944;48:203–36. [Google Scholar]
- del Castillo I, Cohen-Salmon M, Blanchard S, Lutfalla G, Petit C. Structure of the X-linked Kallmann syndrome gene and its homologous pseudogene on the Y chromosome. Nat Genet. 1992;2:305–10. doi: 10.1038/ng1292-305. [DOI] [PubMed] [Google Scholar]
- Georgopoulos NA, Koika V, Galli-Tsinopoulou A, Spiliotis BE, Adonakis G, et al. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril. 2007;88:1311–7. doi: 10.1016/j.fertnstert.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Georgopoulos NA, Pralong FP, Seidman CE, Seidman JG, Crowley WF, Jr, et al. Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab. 1997;82:213–7. doi: 10.1210/jcem.82.1.3692. [DOI] [PubMed] [Google Scholar]
- Forest MG, Cathiard AM, Bertrand J. Evidence of testicular activity in early infancy. J Clin Endocrinol Metab. 1973;37:148–51. doi: 10.1210/jcem-37-1-148. [DOI] [PubMed] [Google Scholar]
- Waldstreicher J, Seminara SB, Jameson JL, Geyer A, Nachtigall LB, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388–95. doi: 10.1210/jcem.81.12.8954047. [DOI] [PubMed] [Google Scholar]