

Abstract
We conducted an analysis of the Kallmann syndrome 1 (KAL-1) genotype in 17 patients with Kallmann syndrome (KS), 9 patients with normosmic idiopathic hypogonadotropic hypogonadism (nIHH) and 20 age-matched normal men in Northwestern China. To do this, we used multiplex PCR analysis with exon-flanking primers and automated sequencing techniques with peripheral blood DNA samples. Intragenic deletions were found at the KAL-1 locus in two KS patients. One case with an atrial septal defect exhibited an intragenic deletion of exon 6. Another KS patient with cryptorchidism had intragenic deletions of exons 5 and 6. For the nIHH patients, no abnormalities were observed in the exonic and flanking sequences of KAL-1. This report describes two intragenic deletions of KAL-1 in two KS patients and suggests that KAL-1 deletion might be more prevalent in KS patients with other congenital organ abnormalities than those described previously in other series from Northwestern China.
Keywords: Kallmann syndrome 1, Kallmann syndrome, normosmic idiopathic hypogonadotropic hypogonadism
Introduction
Idiopathic hypogonadotropic hypogonadism (IHH) is a failure of sexual development or reproductive function due to abnormalities in the pituitary secretion of the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH). These abnormalities can result from deficiencies in the production of gonadotropin-releasing hormone (GnRH) by the hypothalamus or by defects in GnRH function at the pituitary level. IHH can be associated with anosmia in Kallmann Syndrome (KS) or can be apparently isolated, without anosmia (normosmic IHH, nIHH). Although the vast majority of KS and nIHH cases are sporadic, recessive X-linked, autosomal dominant and autosomal recessive modes of inheritance have been described 1, 2, 3.
The Kallmann syndrome 1 (KAL-1), a candidate gene for X-linked KS, was cloned in 1991 by Legouis et al. 4. The discovery of the KAL-1 has led to a pathophysiological model correlating GnRH deficiency with defective migration of GnRH neurons due to abnormal olfactory bulb development in X-linked KS. This gene comprises 14 exons spanning approximately 210 kb on Xp22.3, escapes X-inactivation and encodes a protein (anosmin) that shares homology with molecules involved in neuronal migration and axonal pathfinding. KAL-1 encodes an extracellular matrix glycoprotein of approximately 100 kDa termed anosmin-1, and is expressed in multiple embryonic tissues and organs, including the primitive olfactory bulb and kidney 5, 6. In addition, it has a nonfunctional homolog at Yq11.2. Numerous KAL-1 gene mutations responsible for KS have been reported in X-linked familial and sporadic cases 5, 6, 7.
Here, we report a molecular analysis of the KAL-1 gene in 17 unrelated males with KS and 9 males with nIHH in Northwestern China.
Materials and methods
Patients
Between March 2006 and November 2008, 17 unrelated males with KS were diagnosed according to the clinical signs and symptoms of hypogonadism.
The olfactory bulb structures of KS patients were examined by brain magnetic resonance imaging. The other nine unrelated males with IHH were identified based on clinical signs and symptoms of hypogonadism, prepubertal testosterone (< 1.6 nmol L−1), low or inappropriately normal gonadotropin levels, normal baseline and reserve testing of other anterior pituitary hormones and normal radiological imaging of the hypothalamic–pituitary region. Anosmia/hyposmia was evaluated using the olfactory test described by Davidson and Murphy 8. All study participants came from Northwestern China, and were diagnosed at the First Hospital of Xi'an Jiaotong University (Xi'an, China). The control group comprised 20 healthy volunteers. The protocol was approved by the Ethics Committee of the First Hospital of Xi'an Jiaotong University, and all participants provided a written informed consent.
The ages of the 26 participants ranged from 13 to 37 years. All of them had had uneventful pregnancies, and anosmia or hyposmia had been present since early childhood. In adolescence, there was an absence of subnormal pubertal development. On physical examination, certain clinical characteristics were found in all individuals: normal stature, infantile genitalia and scant pubic hair. The high-resolution G-banded karyotype was 46, XY, and computed axial tomography of the hypothalamic-pituitary region did not show any disorder in any of the patients.
Blood sampling and DNA extraction
Blood samples (3 mL taken into EDTA by venipuncture) were obtained from all subjects. Immediately after collection, whole blood was stored at −80°C until use. Genomic DNA for PCR analysis was isolated from thawed whole blood using Axypre, a whole blood genomic DNA miniprep kit (Axygen biosciences, Union City, CA, USA).
Mutation analysis
KAL-1 genotypes were identified by multiplex PCR. Primer sequences corresponding to the flanking regions of the KAL-1 exons, sizes of the amplified products and amplification conditions were as reported by Hardelin et al. 9. For exons 2–13 of the KAL-1, PCR products corresponded to a segment of DNA spanning the coding region and the adjacent splice site junctions. For exons 1 and 14 of the KAL-1, which contain the 5′ and the 3′ untranslated regions, respectively, only the coding fragment was amplified using an intronic primer and a second primer annealing to the corresponding untranslated region 10, 11. PCR was performed in a 25-μL reaction buffer containing 200 μmol L−1 dNTPs, 1.5 mmol L−1 MgCl2, 10 pmol of each primer, approximately 200 ng of template DNA and 2U of thermostable Taq DNA polymerase (MBI, Vilnius, Lithuania). After a 5-min pretreatment at 95°C, the specific conditions used for all PCR amplifications (35 cycles) were denaturation for 45 seconds at 94°C; annealing for 45 seconds at 63°C for exon 1, 57°C for exon 2 and at 55°C for exons 3–14 of the KAL-1; fragment extension for 45 seconds at 72°C; and a final elongation step for 7 min at 72°C. For the amplification of exons 1, 4, 7 and 14 of the KAL-1, 10% dimethylsulfoxide was included. PCR products were electrophoresed on a 2% agarose gel stained with ethidium bromide to verify the correct size of the expected fragments. Subsequently, all PCR fragments were purified by Nucleospin Extract (Machery-Nagel, DÜren, Germany) and sequenced for both DNA strands using the BigDye terminator cycle sequencing ready reaction kit (Sangon, Shanghai, China) in an ABI 3730 Automated DNA Sequencer (Sangon). Each potential mutation or deletion was confirmed by a second independent PCR amplification and sequencing. The primers for β-actin were 5′-ACTCCCCATCCCAAGACC-3′ and 5′-CCTTAATGTCACGCACGAT-3′. A 400-bp fragment of the β-actin gene was amplified in all samples as an internal standard.
Statistical analysis
Exact 95% confidence intervals (CIs) were calculated as suggested by Agresti and Coull 12 and Brown et al. 13. This approach can be substantiated on the grounds that it is the exact algebraic counterpart to the large-sample hypothesis test and that its worth does not strongly depend on the sample size. All analyses were performed using SPSS version 13.0 statistical software (SPSS, Chicago, IL, USA).
Results
Hypogonadotropic hypogonadism was documented in all patients. Basal testosterone concentrations were consistently in the prepubertal range (1.6 nmol L−1). Meanwhile, gonadotropin levels were always below the normal adult male range (FSH, 3.0–30.0 IU/L; LH, 5.0–28.0 IU/L) and normal prepubertal male range (FSH, 1.0–9.0 IU/L; LH, 3.0–7.0IU/L).
KS patients
Genetic defects were found in only two patients out of 17 participants with KS (P = 0.118; 95% CI, 0.0353–0.2710). One KS patient with an atrial septal defect had an intragenic deletion of exon 6, whereas the remaining exons of the gene amplified normally (Figure 1). Another KS patient with cryptorchidism had intragenic deletion of exons 5 and 6, whereas the other exons of the gene amplified normally (Figure 2). In the remaining 15 KS cases, no mutations or deletion were detected in KAL-1.
Figure 1.
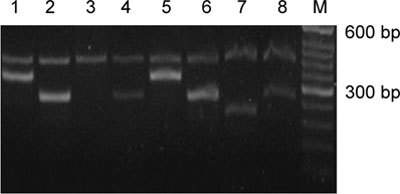
Electrophoresis pattern for exons 4–7 of KAL-1 using DNA from the KS patient with an atrial septal defect (1–4) and a normal control (5–8) based on multiplex PCR. A 400-bp fragment of the β-actin gene was used as an internal standard using primers (5′-ACTCCCCATCCCAAGACC-3′ and 5′-CCTTAATGTCACGCA CGAT-3′). M: 50 bp DNA ladder (TaKaRa Bio, Otsu, Japan); deletion of exon 6 is observed in the patient's DNA.
Figure 2.
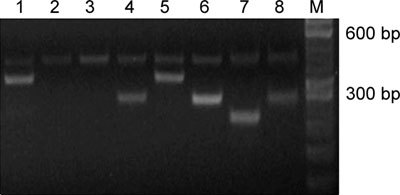
Electrophoresis pattern for exons 4–7 of KAL-1 using DNA from the KS patient with cryptorchidism (1–4) and a normal control (5–8) based on multiplex PCR. A 400-bp fragment of the β-actin gene was used as an internal standard using primers (5′-ACTCCCCATCCCAAGACC-3′ and 5′-CCTTAATGTCACGCACGAT-3′). M: 50 bp DNA ladder (TaKaRa Bio, Otsu, Japan); deletion of exons 5–6 is observed in the patient's DNA.
nIHH patients
No abnormalities were found in KAL-1 among the nIHH patients.
Discussion
Various types of KAL-1 abnormalities have been reported in patients with KS. These include missense and nonsense mutations, splice site mutations, intragenic deletions and submicroscopic chromosomal deletions involving the entire KAL-1. To date, a search for mutations in the KAL-1 has been performed in various groups of patients with X-linked KS or sporadic KS. Defects have been found to be widely distributed throughout this gene in ∼14% and 11% of patients with familial X-linked KS and males with sporadic KS, respectively 14, 15.
In our series, only two complete intragenic deletions within the coding region of the KAL-1 were identified in two KS patients. One patient, who had an atrial septal defect, showed an intragenic deletion of exon 6. The other, who had cryptorchidism, showed intragenic deletion of exons 5 and 6. No mutations or deletions were detected in the KAL-1 among the remaining KS patients from Northwestern China. In fact, Oliveira 16 concluded that autosomal genes are clearly responsible for the majority of both familial and sporadic KS cases. To date, only loss-of-function mutations in the FGFR1 gene, located at 8p11.2, have been associated with KS 17. Therefore, the possibility of the existence of a second X-linked gene causing KS or of molecular alterations located in the regulatory regions of the KAL-1 promoter, in the untranslated regions of exons 1 and 14 or within introns creating new splicing sites, cannot be excluded.
A deletion of exons in KAL-1 has been reported previously. These mutations involved entire KAL-1 deletions and interstitial deletions encoding the fibronectin III domain (FNIII)-like repeats 18. As the mutation reported by Gu et al. 19 causes a premature stop codon, it can be considered to be an extended deletion rather than a mutation that causes a defect in the functionality of the protein. This deletion comprises only part (exon 5) of the coding region of the first FNIII-like repeat of the anosmin-1 protein. The rest of the deletion comprises part of the conserved cysteine-rich N-terminal region, which corresponds to the whey acidic protein motif. In this region, several mutations have been reported to date. The patient who presented with this deletion exhibited a severe phenotype that included facial asymmetry, abnormal eye movements, bimanual synkinesis and the absence of the right kidney, in addition to other congenital malformations 20, 21.
At present, we cannot establish a correlation between the location and number of polymorphisms, and the occurrence of the mutation or the severity of the disease in our patients. Five causal genes have been identified to date for KS, namely (by chronological order of discovery), KAL-1, FGFR1, PROKR2 and PROK2, and FGF8 22. Advanced analysis has been done for the remaining KS patients in our recent work.
No mutations or deletions were detected in KAL-1 among the nIHH patients in our study. Nearly all mutations have been identified in patients with both HH and olfactory dysfunction of variable extent. In particular, there has been no report documenting a KAL-1 mutation-positive patient with normal olfactory function, although two males with apparently normal gonadal function have been identified through familial studies of their brothers with the typical KS phenotype 23, 24. This study supports this observation as well.
New promising candidate loci for human HH include genes with potential influence on the migration of GnRH neurons. GnRH neurons arise in the medial olfactory placode epithelium, migrating along the nasal septum and across the cribiform plate to reach the hypothalamus 25. Recently, two genes, nasal embryonic LH-releasing hormone factor (NELF) and early B-cell factor 2 (EBF2), were implicated in this process. NELF was first isolated in mouse, and its expression patterns in olfactory axons and GnRH cells during development are consistent with its proposed function as a migratory factor for GnRH neurons 26, 27. Therefore, these genes are good candidate genes to underlie HH, and HH patients should be studied in the future.
In conclusion, this report includes descriptions of two intragenic deletions in the KAL-1 in two KS patients and suggests that deletions in KAL-1 might be more prevalent in Chinese KS patients with other congenital organ abnormalities than that previously described in other series from Northwestern China. More candidate genes for HH and a large number of HH patients should be identified for future studies.
Acknowledgments
This work was supported by Science and Technology Project of Shannxi Province, China (No. 2008K15-02).
References
- Quinton R, Duke VM, de Zoysa PA, Platts AD, Valentine A, et al. The neuroradiology of Kallmann's syndrome: a genotypic and phenotypic analysis. J Clin Endocrinol Metab. 1996;81:3010–7. doi: 10.1210/jcem.81.8.8768867. [DOI] [PubMed] [Google Scholar]
- Fechner A, Fong S, McGovern P. A Review of Kallmann Syndrome: genetics, Pathophysiology, and Clinical Management. Obstet Gynecol Surv. 2008;123:189–94. doi: 10.1097/OGX.0b013e3181641278. [DOI] [PubMed] [Google Scholar]
- Ribeiro RS, Vieira TC, Abucham J. Reversible Kallmann syndrome: report of the first case with a KAL-1 mutation and literature review. Eur J Endocrinol. 2007;156:285–90. doi: 10.1530/eje.1.02342. [DOI] [PubMed] [Google Scholar]
- Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–35. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL-1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079–88. doi: 10.1210/jc.2003-030476. [DOI] [PubMed] [Google Scholar]
- Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;35:863–73. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- Ballabio F, Guioli S, Pragliola A, Incerti B, Bardoni B, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal pathfinding molecules. Nature. 1991;353:529–36. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- Davidson TM, Murphy C. Rapid clinical evaluation of anosmia. The alcohol sniff test. Arch Otolaryngol Head Neck Surg. 1997;123:591–94. doi: 10.1001/archotol.1997.01900060033005. [DOI] [PubMed] [Google Scholar]
- Hardelin JP, Levilliers J, Blanchard S, Carel JC, Leutenegger M, et al. Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum Mol Genet. 1993;2:373–7. doi: 10.1093/hmg/2.4.373. [DOI] [PubMed] [Google Scholar]
- del Castillo I, Cohen-Salmon M, Blanchard S, Lutfalla G, Petit C. Structure of the X-linked Kallmann syndrome gene and its homologous pseudogene on the Y chromosome. Nat Genet. 1992;2:305–10. doi: 10.1038/ng1292-305. [DOI] [PubMed] [Google Scholar]
- Georgopoulos NA, Koika V, Galli-Tsinopoulou A, Spiliotis BE, Adonakis G, et al. Renal dysgenesis and KAL-1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril. 2007;88:1311–7. doi: 10.1016/j.fertnstert.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Agresti A, Coull BA. Approximate is better than “exact” for interval estimation of binomial proportions. Am Stat. 1998;52:119–26. [Google Scholar]
- Brown LD, Cai TT, DasGupta A. Interval estimation for a binomial proportion. Stat Sci. 2001;16:101–3. [Google Scholar]
- Oliveira LM, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–8. doi: 10.1210/jcem.86.4.7420. [DOI] [PubMed] [Google Scholar]
- Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001;55:163–74. doi: 10.1046/j.1365-2265.2001.01277.x. [DOI] [PubMed] [Google Scholar]
- Oliveira LM, Seminara SB, Beranova M, Hayes FJ, Valkenburgh SB, et al. The importance of autosomal genes in Kallmann syndrome: genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab. 2001;86:1532–8. doi: 10.1210/jcem.86.4.7420. [DOI] [PubMed] [Google Scholar]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le DÛ N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–5. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Trarbach EB, Baptista MT, Garmes HM, Hackel C. Molecular analysis of KAL-1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients. J Endocrinol. 2005;187:361–8. doi: 10.1677/joe.1.06103. [DOI] [PubMed] [Google Scholar]
- Gu WX, Colquhoun-Kerr JS, Kopp P, Bode HH, Jameson L. A novel aminoterminal mutation in the KAL-1 gene in a large pedigree with X-linked Kallmann syndrome. Mol Genet Metab. 1998;6:59–61. doi: 10.1006/mgme.1998.2732. [DOI] [PubMed] [Google Scholar]
- Quinton R, Duke VM, de Zoysa PA, Platts AD, Valentine A, et al. The neuroradiology of Kallmann's syndrome: a genotypic and phenotypic analysis. J Clin Endocrinol Metab. 1996;81:3010–7. doi: 10.1210/jcem.81.8.8768867. [DOI] [PubMed] [Google Scholar]
- Dodé C, Hardelin JP. Kallmann syndrome. Eur J Hum Genet. 2009;17:139–46. doi: 10.1038/ejhg.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardelin JP, Dode C. The complex genetics of Kallmann syndrome: KAL-1, FGFR1, FGF8, ROKR2, PROK2. Sex Dev. 2008;2:181–93. doi: 10.1159/000152034. [DOI] [PubMed] [Google Scholar]
- Parenti G, Rizzolo MG, Ghezzi M, Di Maio S, Sperandeo MP, et al. Variable penetrance of hypogonadism in a sibship with Kallmann syndrome due to a deletion of the KAL gene. Am J Med Genet. 1995;57:476–8. doi: 10.1002/ajmg.1320570323. [DOI] [PubMed] [Google Scholar]
- Georgopoulos NA, Pralong FP, Seidman CE, Seidman JG, Crowley WF Jr, et al. Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab. 1997;82:213–7. doi: 10.1210/jcem.82.1.3692. [DOI] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161–4. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- Kramer PR, Wray S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000;14:1824–34. [PMC free article] [PubMed] [Google Scholar]
- Kramer PR, Wray S. Nasal embryonic LHRH factor (NELF) expression within the CNS and PNS of the rodent. Brain Res Gene Expr Patterns. 2001;1:23–6. doi: 10.1016/s1567-133x(01)00004-7. [DOI] [PubMed] [Google Scholar]