

Abstract
Autoimmune cytopenias are a recognized complication of hematopoietic stem cell transplant (HSCT), and are considered to be a feature of chronic graft-versus-host disease (cGVHD). We report on a cohort of very young infants (≤3 months of age) receiving HSCT from unrelated donor umbilical cord blood for genetic disorders who developed posttransplant autoimmune cytopenias at an increased rate compared to older aged controls. These infants received a conditioning regimen consisting of busulfan, cyclophosphamide, and antithymocyte globulin (ATG). All infants received HLA mismatched unrelated umbilical cord blood as graft source. GVHD prophylaxis was either cyclosporine + methylprednisolone (n = 16) or cyclosporine + mycophenolate mofetil (n =3). Engraftment, acute GVHD (aGVHD) and cGVHD, survival, treatment-related mortality (TRM), and deaths were evaluated. Ten patients developed cGVHD manifesting as autoimmune cytopenias at a median 247 days posttransplant with a cumulative incidence of 44% (95% confidence interval [CI] 21%–68%) and 56% (95% CI 32%–80%) at 1 and 2 years, respectively. In 6 of 10 patients developing autoimmune cytopenias, cGVHD presented as autoimmune cytopenia de novo. The cytopenias observed included anemia (n =4), thrombocytopenia (n =1), anemia with thrombocytopenia (n =3), and pancytopenia (n =2). No graft factors were identified as being significant to development of cGVHD. All patients responded to treatment with methylprednisolone, azithioprine ± rituximab. One patient required splenectomy. We hypothesize that posttransplant immunosuppression interferes with normal immune ontogeny creating immune dysregulation and graft directed cell destruction. Alternative strategies to prevent GVHD should be considered for this unique patient population.
Keywords: Autoimmune hemolytic anemia, Unrelated umbilical cord blood transplantation, Chronic graft versus host disease
INTRODUCTION
Autoimmune cytopenias are a recognized complication after allogeneic hematopoietic stem cell transplantation (HSCT), and are considered to be a feature of chronic graft-versus-host disease (cGVHD) when another symptom of cGVHD is present [1,2]. In adults, the incidence of autoimmune hemolytic anemia (AIHA) is 3%–4% in patients receiving either conventional or T cell-depleted grafts [3–5]. A slightly higher rate of ~6% has been the reported in the pediatric literature [6,7]. There are limited reports about autoimmune cytopenias and cGVHD in very young babies undergoing HSCT. Over the past decade, we have transplanted a cohort of very young babies with early infantile lysosomal storage diseases and other genetic disorders. We have observed a higher than expected rate of autoimmune cytopenias in these infants and describe the cases in this report. We propose that autoimmune cytopenias can be a primary manifestation of cGVHD in young pediatric patients undergoing allogeneic HSCT.
MATERIALS AND METHODS
Patients
Between September 1998 and April 2007, 19 consecutive infants (≤3 months of age) diagnosed with early infantile lysosomal storage diseases (n = 18) or hemoglobinopathies (n = 1) who were referred to our center for transplantation and who lacked appropriate HLA matched related donors received cord blood transplants from unrelated donors. Fourteen were diagnosed prenatally and 5 were diagnosed at birth. All diagnoses were confirmed prior to transplant. Eight patients were enrolled in an institutional-based phase I/II study of cord blood transplantation for the treatment of nonmalignant diseases. Ten patients were enrolled on the Cord Blood Transplantation Study (COBLT) [8], and 1 patient was enrolled in a phase I trial using ex vivo expansion of ALDH cells [9]. All studies were approved by the institutional review board of Duke University Medical Center. Written informed consent was obtained from the infant’s parents before enrollment.
Selection of Donors
Intermediate-resolution typing for HLA class I (A and B) and high resolution for HLA-DRB1 were used for donor selection. The cord blood unit with the highest number of nucleated cells (minimum, 3 × 107/kg of recipient body weight) and closest HLA match (minimum 4 of 6 HLA loci match) was selected. Units were tested to ensure that the donor was neither affected with nor a carrier of the condition for which the infant was being transplanted.
Transplantation Procedure
Cryopreserved units of cord blood were thawed and washed per the procedure of Rubinstein et al. [10]. For the single patient on the ALDH trial, the 80% fraction was transplanted using conventional methods, whereas the 20% fraction was sorted to isolate ALDH bright cells that were subsequently primed ex vivo for 5 days using cytokines before infusion [9]. All thawed cord blood units were tested for total number of nucleated cells (TNC), clonal hematopoietic progenitor cells, CD34+and CD3+cells, cell viability, ABO and Rh typing, and microbial cultures (bacteria and fungal).
Conditioning Regimen
The conditioning regimen for all patients included 16 doses of busulfan (Bu; 20 mg/m2 orally) from days –9 through –6; 4 doses of cyclophosphamide (Cy; 50 mg/m2 i.v.) from days –5 through –2; and 3 doses of equine antithymocyte globulin (ATG; 30 mg/kg i.v.) from days –3 through –1. Bu pharmacokinetics were monitored with the first dose to target steady-state concentrations between 600–900 ng/mL. Patients received phenytoin during Bu as seizure prophylaxis and mesna during Cy for prophylaxis against hemorrhagic cystitis.
Posttransplant Regimen and Supportive Care
All children received prophylaxis against GVHD with cyclosporine (CSA) + methylprednisone (n = 16) or CSA + mycophenolate mofetil (MMF; n = 3). CSA was dosed to target levels between 200 and 300 ng/mL, and MMF was dosed at 15 mg/kg/dose i.v. every 8 hours. GVHD prophylaxis was continued at full dose for 6–9 months, then weaned as tolerated. Treatment options for GVHD were based on severity and included topical steroids, dose escalation of methylprednisone, change to tacrolimus, or addition of daclizumab (Hoffmann-LaRoche, Nutley, NJ).
All patients were nursed in reverse isolation rooms under positive pressure and HEPA filtration. Standard prophylaxis was used against pneumocystis carinii, acyclovir for viral, and voriconazole for fungal infections. Empiric antibiotic treatment was started with the first febrile episode and continued through engraftment. Intravenous immune globulin (500 mg/ kg/dose) was administered weekly through day 100 then monthly through 1 year, then weaned as tolerated. As prophylaxis against veno-occlusive disease (VOD), a continuous infusion of low-dose heparin was used through day 28. Patients received transfusions of leukocyte-depleted, irradiated-packed red blood cells and platelets. Granulocytecolony stimulating factor (G-CSF) was administered from day 0 until hematopoietic recovery and then was weaned. Confirmation of engraftment was performed using fluorescein in situ hybridization (FISH) or restricted fragment length polymorphism (RFLP).
Study Subjects
Potential study subjects were identified by retrospective review of the transplant program research database using age at transplant as initial criteria. To be eligible for the study, patients had to be ≤3 months of age at transplant, recipients of unrelated UCBT with myeloablative conditioning, and have a diagnosis of a nonmalignant disorder. Data was collected via chart review for demographics, information regarding cord blood unit, complications following transplant including diagnosis, treatment, and response to treatment of an autoimmune cytopenia. Data were compared to 141 older aged historic and contemporary controls at our center.
Laboratory Evaluation
Autoimmune hemolytic anemia was defined as development of anemia in a patient with stable engraftment where there was a hemoglobin drop of >2 g/dL, transfusions needed in prior transfusion-independent patients, and evidence of hemolysis by positive direct antiglobulin test (DAT) and/or indirect Coomb’s test, peripheral blood cell morphology, reticulocyte count, and bilirubin level. The DAT was performed to detect the presence of IgG or IgM antibodies and complement on the patient’s red blood cells (Immucor, Norcross, GA). If initial testing was positive, further testing was conducted to determine if the reaction was the result of IgG and/or complement specifically C3. If IgG antibodies were present, an eluate was prepared and tested to determine antibody specificity. Thrombocytopenia was defined as platelet count <50,000 cells/μL. Neutropenia was present if the absolute neutrophil count was <500/mm3. Direct and indirect antiplatelet antibody testing was performed if thrombocytopenia was present. Neutrophil cytoplasmic antibodies were enumerated when possible.
Acute and Chronic GVHD (aGVHD, cGHVD) Scoring
The severity of aGVHD was scored using standard criteria [11]. cGVHD was scored using National Institute of Health (NIH) consensus criteria [1]. However, patients with isolated immune cytopenias were also scored as having cGVHD.
Statistical Analysis
Neutrophil engraftment was defined as the first day of 3 consecutive days of an absolute neutrophil count (ANC) of ≥500 donor cells/mm3; platelet engraftment was defined as the day of achieving an un-transfused count of ≥50,000 platelets/mm3 for 7 days. aGVHD was scored as the maximum grade in all patients within 100 days [11] and the cGVHD at the highest level per consensus criteria [1]. The probabilities of neutrophil and platelet engraftment, aGVHD, and cGVHD and cytopenia were estimated using the cumulative-incidence-function method [12]. Neutrophil engraftment, platelet engraftment, aGVHD, and cGVHD and cytopenia were assessed in patients surviving past day 15 and treating death as a competing risk. Differences between subgroups were compared using Grays K-Sample Test [13]. The probability of overall survival (OS) was calculated with the use of the Kaplan-Meier estimator [14] and differences between groups were compared using the log-rank statistics [15]. Analyses were completed using the SAS system, version 8.2, and R, version 2.1.1.
RESULTS
Patients Characteristics
From September 1998 through April 2007, 19 very young infants (11 males, 8 females) received cord blood transplants from unrelated donors. The median weight and age at transplantation were 4.20 kg (range: 2.74 to 6.75) and 36 days (range: 18 to 117 days), respectively (Table 1). Diagnoses included Krabbe disease (n = 12), Hurler’s syndrome (MPS I; n = 2), Hunter’s syndrome (MPS II; n = 2), Metachromatic Leukodystrophy (n = 1), Tay Sachs disease (n = 1) and Beta-Thalassemia Major (n = 1).
Table 1.
Characteristics of the Study Population and the Unrelated Cord Blood Donors
| Pt No. | Diagnosis | Days of Life at Time of Transplant | HLA Match (No./Total No.) | HLA Mismatch | Blood Type Pt | Blood Type Donor | Pt Sex | Unit Sex |
|---|---|---|---|---|---|---|---|---|
| Cytopenias without other GVHD manifestations | ||||||||
| 2 | Thalassemia | 91 | 4/6 | A31/30;Bbl/7 | A+ | A+ | M | M |
| 6 | Krabbe | 29 | 4/6 | B57/8;DR0402/1302 | B+ | B+ | M | M |
| 12 | Krabbe | 117 | 5/6 | A11/29 | O+ | A+ | M | F |
| 13 | Krabbe | 26 | 4/6 | Abl/11;B39/18 | O+ | O+ | F | F |
| 15 | Krabbe | 22 | 5/6 | DR9/8 | O+ | O+ | F | F |
| 19 | Krabbe | 33 | 5/6 | A31/2 | A+ | A+ | M | F |
| Cytopenias with cGVHD | ||||||||
| 9 | MPS II | 44 | 5/6 | DR11/7 | O+ | A− | M | F |
| 10 | MPS I | 113 | 4/6 | A33/68;DR16/13 | B+ | A+ | M | M |
| 16 | MLD | 62 | 5/6 | A1/3 | O+ | O+ | M | F |
| 17 | Krabbe | 29 | 4/6 | B55/51;DR15/4 | A+ | O+ | F | M |
| No cytopenias or cGVHD | ||||||||
| 1 | MPS I | 88 | 5/6 | A28/29 | A+ | O+ | F | F |
| 3 | Krabbe | 55 | 4/6 | A30/25;B44/53 | O− | A+ | M | M |
| 4 | Krabbe | 29 | 4/6 | A2/33;B39/bl | AB+ | B+ | F | M |
| 5 | Tay-Sachs | 22 | 4/6 | B50/41;DRB1 1302/1303 | B+ | A+ | M | M |
| 7 | Krabbe | 37 | 5/6 | B40/58 | O+ | A− | F | M |
| 8 | MPS I | 62 | 5/6 | A68/2 | O+ | O+ | F | F |
| 11 | Krabbe | 26 | 5/6 | B18/44 | A− | A+ | M | M |
| 14 | Krabbe | 22 | 5/6 | B44/14 | A− | A+ | M | M |
| 18 | Krabbe | 18 | 4/6 | A25/11;B18/44 | B+ | A+ | F | M |
| Mean ± SD | 48 ± 31 | |||||||
| Median | 36 | |||||||
GVHD indicates graft-versus-host disease; cGVHD, chronic graft-versus-host disease; Pt, patient; bl, blank.
Graft Characteristics
All infants were transplanted with a single unrelated cord blood unit selected by cell dose and HLA matching. The units contained a median TNC before cryopreservation of 18.78 × 107/kg of recipient body weight (range: 12.34 × 107/kg to 50.37 × 107/kg (Table 2). The median number of total nucleated infused cells were 15.70 × 107/kg (range: 8.42 × 107/ kg to 32.40 × 107/kg). The median numbers of CD34+ cells and CD3+ infused were 3.18 × 105/kg (range: 1.27 × 105/kg to 11.02 × 105/kg) and 26.50 × 106/kg (range: 15.23 106/kg to 90.31 × 106/kg), respectively. Units were matched at 4 of 6 (n = 9), or 5 of 6 (n = 10) HLA loci using intermediate resolution DNA typing for Class I A + B and high-resolution DNA typing for DRB1.
Table 2.
Characteristics of Transplanted Umbilical Cord Blood Units
| Pt No. | Total Nucleated Cells Cryopreserved (×107/kg) | Total Nucleated Cells Reinfused (×107/kg) | Colony-Forming Units Infused (×104/kg) | CD34+ Cells Infused (×105/kg) |
|---|---|---|---|---|
| Cytopenias without other GVHD manifestations | ||||
| 2 | 31.82 | 19.10 | 7.64 | 6.11 |
| 6 | 39.20 | 25.00 | 23.75 | 5.65 |
| 12 | 14.74 | 19.90 | 15.92 | 3.18 |
| 13 | 13.48 | 9.92 | 4.46 | 1.69 |
| 15 | 18.78 | 13.20 | 10.56 | 8.45 |
| 19 | 42.75 | 31.60 | 0 | 2.84 |
| Cytopenias with cGVHD | ||||
| 9 | 14.85 | 10.00 | 4.50 | 1.48 |
| 10 | 24.63 | 20.73 | 14.51 | 4.68 |
| 16 | 16.42 | 12.60 | 3.15 | 1.51 |
| 17 | 38.23 | 27.40 | 20.55 | 5.48 |
| No cytopenias or cGVHD | ||||
| 1 | 18.63 | 15.70 | 51.81 | 8.01 |
| 3 | 21.93 | 14.20 | 6.39 | 1.80 |
| 4 | 19.79 | 11.50 | 10.93 | 1.27 |
| 5 | 18.49 | 12.41 | 13.03 | 1.81 |
| 7 | 17.36 | 15.84 | 11.09 | 3.72 |
| 8 | 12.34 | 8.42 | 8.00 | 1.94 |
| 11 | 17.76 | 15.40 | 23.10 | 2.60 |
| 14 | 50.37 | 32.40 | 105.30 | 11.02 |
| 18 | 33.03 | 18.60 | 13.02 | 7.25 |
| Mean ± SD | 24.45 ± 11.27 | 17.57 ± 7.14 | 18.30 ± 23.92 | 4.24 ± 2.86 |
| Median | 18.78 | 15.70 | 11.08 | 3.18 |
GVHD indicates graft-versus-host disease; cGVHD, chronic graft-versus-host disease; Pt, patient.
Engraftment and Chimerism
Neutrophil (ANC >500/mm3) and platelet engraftment (platelets >50,000 cells/μL) occurred in a median of 19 days (range: 10 to 40) and 60 days (range: 30 to 164), respectively (Table 3). The cumulative incidence of neutrophil engraftment at 42 days was 100% (95% confidence interval [CI] 89%–100%) and platelet engraftment at 180 days was 100% (95% CI 89%–100%). Red cell and platelet transfusions were no longer required at a median 59 days (range: 17 to 385) and 50 days (range: 25 to 164), respectively. Seventeen of the 18 evaluable infants obtained full donor chimerism (defined as >98% donor). One infant diagnosed with MPS II remained a mixed chimera throughout his life. An additional infant with meta-chromatic leukodystrophy experienced secondary graft failure 2.5 years posttransplant.
Table 3.
Engraftment, Chimerism, Incidence of GVHD and Event-Free Survival
| Pt No. | Diagnosis | Engraftment (Days)*
|
RBC Transfusion Independence (Days)† | Chimerism (% Donor)
|
Acute GVHD Grade‡ | Chronic GVHD Grade | Event-Free Survival (Days)§ | Cause of Death | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Neutrophil | Platelet | At 100 Days | At 1 Year | |||||||
| Cytopenias without other GVHD manifestations | ||||||||||
| 2 | Thalassemia | 14 | 33 | 17 | >99 | >99 | 1 | Cytopenia | 3440 | |
| 6 | Krabbe | 20 | 101 | 115 | >99 | >99 | 1 | Cytopenia | 2684 | |
| 12 | Krabbe | 31 | 91 | 77 | >98 | >99 | 3 | Cytopenia | 1480 | |
| 13 | Krabbe | 14 | 48 | 55 | >98 | >98 | 1 | Cytopenia | 1368 | |
| 15 | Krabbe | 25 | 64 | 62 | 94 | 98 | 1 | Cytopenia | 1321 | |
| 19 | Krabbe | 31 | 141 | 385 | 97 | NE | 2 | Cytopenia | 353 | |
| Cytopenias with cGVHD | ||||||||||
| 9 | MPS II | 20 | 56 | 52 | 50 | 22 | 1 | Cytopenia, Extensive | Died | Multisystem organ failure (day 653) |
| 10 | MPS II | 14 | 89 | 80 | >99 | >98 | 2 | Cytopenia, Limited (Skin) | 1879 | |
| 16 | MLD | 40 | 164 | 190 | 63 | 100 | 1 | Cytopenia, Limited (Skin) | 1293 | |
| 17 | Krabbe | 17 | 49 | 70 | 97 | 100 | 1 | Cytopenia, Limited (Skin) | 955 | |
| No cytopenias or cGVHD | ||||||||||
| 1 | MPS I | 10 | 33 | 25 | >99 | >99 | 0 | None | 3468 | |
| 3 | Krabbe | 14 | 53 | 49 | >99 | >99 | 1 | None | 3391 | |
| 4 | Krabbe | 13 | 49 | 39 | 100 | 100 | 1 | None | 2977 | |
| 5 | Tay-Sachs | 20 | 90 | 103 | >99 | >99 | 2 | None | Died | Probable infection (day 1686) |
| 7 | Krabbe | 20 | 67 | 53 | 99 | 100 | 1 | None | 2264 | |
| 8 | MPS I | 19 | 47 | 47 | >99 | >99 | 2 | None | 1986 | |
| 11 | Krabbe | 19 | 74 | 70 | >99 | >98 | 2 | None | 1725 | |
| 14 | Krabbe | 11 | 30 | 51 | >98 | >98 | 1 | None | 1327 | |
| 18 | Krabbe | NE | NE | NE | NE | NE | NE | NE | Died | Pulmonary Hypertension (day 15) |
| Mean ± SE | 20 ± 2 | 71 ± 9 | 86 ± 20 | 1994 ± 238 | ||||||
| Median (range) | 19 (10–40) | 60 (30–164) | 59 (17–385) | 1802 (353–3468) | ||||||
MPS I indicates mucopolysaccharidosis type I (Hurler’s Syndrome); Krabbe, globoid cell leukodystrophy, Krabbe disease; MPS II, mucopolysaccharidosis type II (Hunter’s Syndrome); MLD, metachromatic leukodystrophy; EFS, event-free survival; Pt, patient.
Neutrophil engraftment was defined as an absolute neutrophil count of >500 per cubic millimeter on at least 3 consecutive days, and platelet engraftment was defined as a count >50,000 per cubic millimeter without any transfusion support for at least 7 days.
RBC transfusion independence was defined as 7 days from last transfusion with no further transfusion support needed.
The 15 infants with acute graft-versus-host disease (aGVHD) had skin manifestations.
EFS as of April 1, 2008.
aGVHD and cGVHD
In the 18 infants evaluable for aGVHD, 12 had either grade 0 (n = 1) or grade I (n = 11) (Table 3). Grades II–IV aGVHD occurred in 6 infants (grade II n = 5; grade III n = 1; grade IV n = 0). The median time to onset of among patients with grades II–IV was 12 days (range: 7–35) with a cumulative incidence of grades II–IV at 100 days of 33% (95% CI 11%–56%) (Figure 1). One patient experienced grade III aGVHD on day 35 posttransplant (Figure 1). Treatment included topical steroids (n = 10) and methyl-prednisolone pulse (n = 7), tacrolimus (n = 4), and daclizimab (n = 4).
Figure 1.

Cumulative incidence of aGVHD after unrelated cord blood transplantation in very young infants. Cumulative incidence of grades II–IV and grades III–IV aGVHD in the study patients is displayed.
The cumulative incidence of cGVHD defined according to conventional diagnostic criteria [1] was 17% (95% CI 0%–35%) and 23% (95% CI 2%–43%), at 1 and 2 years, respectively (Figure 2A). One patient had extensive cGVHD involving skin, gastrointestinal track, and the lung cGVHD, whereas 3 other infants had disease limited disease of the skin (Table 3). We examined possible factors contributing to GVHD, including TNC cell dose, HLA-matching, CD34+ cell dose, CD3+ cell dose, colony-forming units, ABO/Rh mismatch, and unit sex. None of these factors influenced the incidence or severity of aGVHD or cGVHD in this small patient cohort (data not shown).
Figure 2.
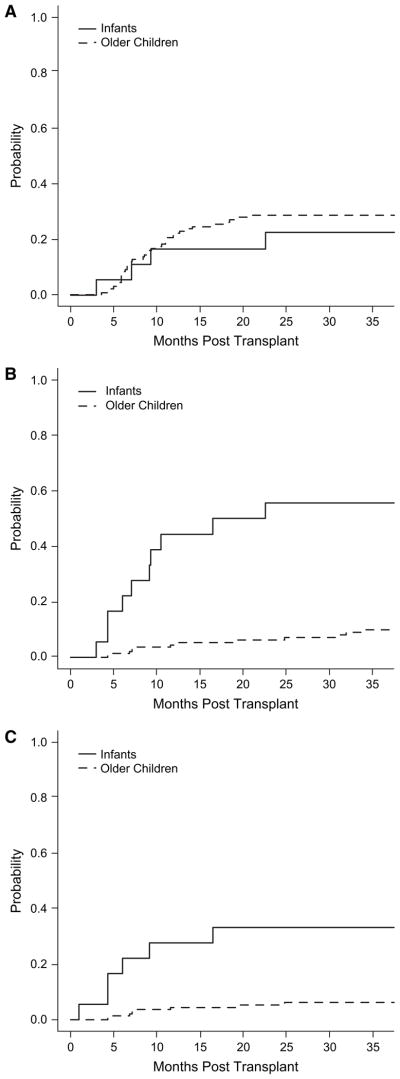
Cumulative incidence of cGVHD (A) and autoimmune cytopenias (B, C) in very young infants compared to older children undergoing UCBT. (A) The cumulative incidence of cGVHD in the infant population (solid line) compared to a historic control group of all other metabolic patients treated at Duke (dotted line). (B) The cumulative incidence of autoimmune cytopenias in the study patients (solid line) compared to the older control group of all other metabolic patients treated at Duke (dotted line). (C) The cumulative incidence of cytopenias without other manifestations of cGVHD between the infants (solid line) and older children (dotted line).
Autoimmune Diseases
Ten infants were diagnosed with an autoimmune cytopenia (Table 4). The overall cumulative incidence of autoimmune cytopenias, with or without other manifestations of cGVHD, was 44% (95% CI 21%–68%) and 56% (95% CI 32%–80%) at 1 and 2 years, respectively (Figure 2B and C). The median onset was 247 days posttransplant (range: 92 to 687). The cytopenias observed included anemia (n = 4), thrombocytopenia (n = 1), anemia with thrombocytopenia (n = 3), and pancytopenia (n = 2).
Table 4.
Characteristics of Patients Transplanted in Neonatal Period that Later Developed an Autoimmune Cytopenia
| Pt No. | Pretranplant Diagnosis | Initial GVHD Prophylaxis | Type of Cytopenia | Posttransplant Onset (Days) | RBC Antibody | Plt Antibody | Tacrolimus Exposure | Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Cytopenias without other GVHD manifestations | |||||||||
| 2 | Beta-thalassemia | CsA, Methypred | AIHA, thrombocytopenia | 131 | IgG+ panreactive | No | Methylpred, IVIG, Azathioprine | Alive. Resolved. | |
| 6 | Krabbe | CsA, Methypred | Thrombocytopenia | 279 | No | Methylpred, IVIG, d/c CSA | Alive. Resolved. | ||
| 12 | Krabbe | CsA, Methypred | AIHA | 503 | IgG+, panreactive, C3+ | No | Methylpred, IVIG, Azathioprine, Rituximab | Alive. On treatment with intermittent hemolysis. | |
| 13 | Krabbe | CsA, Methypred | AIHA | 183 | IgG+ panreactive, C3+ | Yes | Methylpred, Azathioprine, Rituximab, d/c CSA | Alive. Resolved. | |
| 15 | Krabbe | CsA, Methypred | AIHA, thrombocytopenia, neutropenia | 132 | IgG+, panreactive, C3+ | Yes | Yes | Methylpred, Azathioprine, Rituximab, EPO, GCSF, d/c CSA | Alive. Continues on azathioprine (weaning). |
| 19 | Krabbe | CsA, MMF | AIHA | 320 | IgG+, panreactive (Anti-e, C) | No | Methylpred, Azathioprine, Rituximab | Alive. On treatment. | |
| Cytopenias with cGVHD | |||||||||
| 9 | MPS II | CsA, Methypred | AIHA, thrombocytopenia | 92 | IgG+ panreactive, C3+, (Anti-E) | Yes | Yes | Methylpred, IVIG, Azathioprine, Rituximab, Splenectomy, d/c CSA | Died of multisystem organ failure. Had secondary graft failure and underwent second transplant. |
| 10 | MPS II | CsA, Methypred | AIHA, thrombocytopenia | 283 | IgG+, C3+, (Anti-A) | Yes | Yes | Methylpred, Rituximab (daclizimab for skin GVHD) | Alive. Resolved. |
| 16 | MLD | CsA, MMF | AIHA, thrombocytopenia, neutropenia | 687 | Yes | No | Methylpred, Rituximab | Alive. Ongoing pancytopenia. Secondary graft failure requiring second transplant. | |
| 17 | Krabbe | CsA, MMF | AIHA | 215 | IgG+ panreactive, (Anti-E) | Yes | Methylpred, Azathioprine, Rituximab, d/c CSA | Alive. On azathioprine and steroids for skin cGVHD. | |
Krabbe indicates globoid cell leukodystrophy (Krabbe disease); MPS II, mucopolysaccaridosis type II (Hunter’s Syndrome); MLD, metachromatic leukodystrophy; DAT, direct antibody testing; Methylpred, methyl-prednisolone; CSA, cyclosporine; MMF, mycophenolate; EPO, erythropoietin; G-CSF, granulocyte-colony stimulating factor; GVHD, graft-versus-host disease; Pt, patient.
Of the 9 patients with anemia, 8 had a positive Coombs with IgG polyspecific autoantibodies. Four patients also had complement fixation noted on red cells by direct antiglobulin testing. On eluate testing, the specificity of the antibody was identified as antiE in 2 patients with 1 patient having anti-A. Four of the 6 infants with thrombocytopenia had elevated levels of direct antiplatelet antibodies.
Nine of the 10 infants developed an autoimmune cytopenia while on CSA (n = 6) or tacrolimus (n = 3) with methylprednisolone for GVHD treatment or prophylaxis. One infant was on physiologic doses of hydrocortisone secondary to adrenal suppression.
Treatment of Autoimmune Cytopenias
All 10 infants received methylprednisolone for treatment of the cytopenias. Eight infants also received Rituximab therapy (2 infants were treated prior to widespread rituximab use). CSA was withdrawn in 5 infants. Other treatments included intravenous immu-noglobulins (n = 4), Azathioprine (n = 7), splenectomy (n = 1) (Table 4). Erythropoietin was administered to 1 infant without response. G-CSF was used in the 2 infants with neutropenia. The median treatment length was 21 months (range: 8–35).
As of April 2008, 3 of 10 infants are continuing to receive immunosuppressive therapies for autoimmune cytopenias. The DAT remains positive in 1 with active AIHA and 1 other infant who continues with intermittent hemolysis. One infant remains on immunosuppression for skin cGVHD.
Comparison with Older Children
We compared the incidence of cGVHD and auto-immune cytopenias with or without other manifestations of cGVHD in this group of very young infants to a cohort of 141 older children (median age = 1.50 years) with inherited metabolic disease who underwent cord blood transplantation between August 1995 and April 2007 at our center [16]. Although the overall incidence of cGVHD in the infants was equivalent to that of older children (Figure 2A), the cumulative incidence of autoimmune cytopenia with or without other manifestations of cGVHD for the infants was significantly higher at 1 and 2 years (44% [95% CI 21%–68%] and 56% [95% CI 32%–80%]) compared to the older children (1 year: 5% [95% CI 1%–8%], 2 years: 6% [95% CI 2%–10%], P < .01) (Figure 2B). The cumulative incidence of isolated autoimmune cytopenias was also significantly higher in the infants compared to the older children (28% [95% CI 6%–49%] versus 5% [95% CI 2%–9%], P < .01) (Figure 2C).
Survival
As of April 2008, 16 of 19 infants were alive (median: 1802 days; range: 353 to 3468 days) for an OS in this cohort of 95% (95% CI 85%–100%),89% (95% CI 75%–100%), and 80% (95% CI 59%–100%) at 1, 2, and 5 years, respectively (Table 3). One infant with Krabbe died on posttransplant day 15 from pulmonary hypertension. An infant with Hunter’s (MPS II) died 653 days posttransplant from multisystem organ failure. An infant with Tay-Sachs died abruptly on day 1686 from unknown causes.
DISCUSSION
In this report, we describe a cohort of younger infants undergoing unrelated, single-unit UCBT for nonmalignant disorders after myeloablative chemotherapy-based conditioning who developed an unexpectedly high incidence of posttransplant immune mediated hematologic disorders. Rates of aGVHD and cGVHD were similar to those in previous reports. No patient had Epstein-Barr Virus lymphoproliferative disease and none of the immune diseases were triggered by a known viral illness.
Because of their small size, all infants received very high cell doses from their donor grafts. Engraftment and survival occurred at higher rates than those experienced by older children and adults. In this group, overall survival was 95%, 89%, and 80% at 1, 2, and 5 years, respectively. Neutrophil and platelet engraftment occurred (with a cumulative incidence of 100%) in a median of 19 and 60 days, respectively. Infants were treated with therapy first to remove T cell suppression and then to target B cells. We found that combination treatment with steroids, rituximab, and/ or azithioprine was most effective. In contrast to other reports [3–5,7], there was increase in mortality associated with the development of an autoimmune cytopenia. Rather, this cohort exhibited an excellent response to therapy and an excellent OS.
AIHA and other immune cytopenias have been reported in both adults and children undergoing allogeneic HSCT. In adults, Drobyski et al. [3] reported an overall incidence of AIHA of 3% in patients receiving T cell-depleted grafts with an incidence of 5% in patients who survived >6 months (3). Chen et al. [4] reviewed data from 293 patients and noted that the patients fell into 2 distinct groups: an early onset (2–8 months) with a cold antibody and a late onset (6–18 months) associated with a warm antibody. Sanz et al. [5] described 272 patients with an overall incidence of AIHA of 4.44%. Independent risk factors identified for AIHA included HSCT from an unrelated donor and the presence of extensive cGVHD. In contrast to the young patients in this report, AIHA in adults was associated with a poor overall prognosis, although often not directly because of the process [3–5,7]. O’Brien et al. [7] described the largest series of pediatric patients who received unrelated donor HSCT with 19 (6%) of 303 patients developing AIHA. None of the patients who received a related donor transplant in their series developed AIHA (n = 136). In their series, children<10 years of age and those undergoing transplant for nonmalignant diagnosis (specifically metabolic diseases) were significantly more likely to develop AIHA as a posttransplant complication. Both stem cell source and graft manipulation to deplete T cells were not found to be risk factors, which are in contrast to other reports [3,6]. There was no difference in the overall rate of cGVHD in children who developed AIHA and those who did not. A high rate of mortality (53%) was reported in children developing AIHA in this series.
Horn et al. [6] described a high incidence (19.5%) of AIHA in a series of 41 children with SCID after T cell-depleted haploidentical bone marrow or PBSC transplantation. The incidence was 6% in patients who survived >6 months, which is comparable to other reports. All infants were found to have decreased CD4 and/or CD8 cells posttransplant. They hypothesized that the paucity of regulatory T cells allowed for B cell clonal expansion resulting in the autoimmune phenomena. Godder et al. [17] described 40 patients with hematologic malignancies or marrow failure who survived >120 days. Five of the 40 patients developed AIHA as the principal manifestation of cGVHD after receiving T cell-depleted partially mismatched related donor BMT. Immune function recovery was not different in the affected patients. aGVHD was also not noted to be a risk factor. It was proposed that HLA mismatch may have been a risk factor in addition to the T cell depletion process. Isolated case reports also exist, which describe AIHA and other cytopenias in children after all types of HSCT [18–26].
There are also rare case reports of patients developing autoimmune cytopenias from 6–9 years after cardiac transplantation (age: 7 months 4 years). Tubman et al. [27] described 3 pediatric patients with autoimmune cytopenias and hypothesized that the significant B cell immunosuppression by calcineurin inhibitions was a risk factor for development of the cytopenia. All were treated with and responded to rituximab therapy. Rawal et al. [28] described a pediatric patient transplanted at 15 months of age who developed at age 11 years multiple episodes of autoimmune cytopenias (anemia, thrombocytopenia, and neutropenia) and acquired glanzmann thrombasthenia several years after cardiac transplantation. This patient initially presented with a presumed viral myocarditis that may have recapitulated robust antibody formation.
The pathogenesis of autoimmune cytopenias in these young patients is not clear at this time. It may be because of an effect of treatment with calcineurin inhibitors or ATG on the developing thymus. All patients in this series received GVHD prophylaxis with cyclosporine, which is known to inhibit thymic-dependent clonal deletion, which disrupts the reconstitution of the immune system [29]. CSA also suppresses IL-2-dependent proliferation and function of regulatory T cells, which could then lead to the development of autoimmune disease [30]. However, this does not fully explain the increased incidence of autoimmune disease in these very young patients compared to our older patient cohort, who were also receiving cyclosporine for prophylaxis against GVHD. It is possible that the relatively higher T cell dose given to these patients from the cord blood unit graft may have accentuated the risk of both GVHD and immune dysregulation in the setting of CSA therapy. Future studies, characterizing the composition of the T cell compartment at the time of onset of autoimmune disease, may elucidate the mechanism underlying this problem.
In this report, we present the outcomes of a cohort of very young infants undergoing UCBT for genetic disorders in the first few months of life. We observed a markedly increased rate of posttransplant autoimmune cytopenias. In these patients, we suspect this was because of immune dysregulation associated with GVHD prophylaxis during the first year of life, leading to aberrant immune ontogeny. We are planning a prospective study to characterize the early phases of immune reconstitution in this unique group of patients.
Acknowledgments
The authors are indebted to the inpatient and out-patient nursing staffs, physicians, nurse coordinators, nurse practitioners, social workers, and other allied staff of the Pediatric Blood and Marrow Transplant Program at Duke University Medical Center for caring for these patients, to the staff of the Stem Cell Laboratory for processing the units of umbilical cord blood and to the referring physicians and parents of these children for allowing us to treat them and study and report the consequences of this therapy.
References
- 1.Filipovich A, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11:945–955. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Couriel D, Carpenter P, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: V. Ancillary therapy and supportive care working group report. Biol Blood Marrow Transplant. 2006;12:375–396. doi: 10.1016/j.bbmt.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Drobyski W, Potluri J, Sauer D, Gottschall J. Autoimmune hemolytic anemia followingTcell-depleted allogeneic bonemarrowtransplantation. BoneMarrow Transplant. 1996;17:1093–1099. [PubMed] [Google Scholar]
- 4.Chen F, Owen I, Savage D, et al. Late onset haemolysis and red cell autoimmunisation after allogeneic bone marrow transplant. Bone Marrow Transplant. 1997;19:491–495. doi: 10.1038/sj.bmt.1700677. [DOI] [PubMed] [Google Scholar]
- 5.Sanz J, Arriaga F, Montesinos P, et al. Autoimmune hemolytic anemia following allogeneic hematopoietic stemcell transplantation in adult patients. BoneMarrow Transplant. 2007;39:555–561. doi: 10.1038/sj.bmt.1705641. [DOI] [PubMed] [Google Scholar]
- 6.Horn B, Viele M, Mentzer W, Mogck N, DeSantes K, Cowan M. Autoimmune hemolytic anemia in patients with SCID after T-cell depleted BM and PBSC transplantation. Bone Marrow Transplant. 1999;24:1009–1013. doi: 10.1038/sj.bmt.1702011. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien T, Eastlund T, Peters C, et al. Autoimmune haemolytic anaemia complicating haematopoietic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated transplants for non-malignant diseases. Br J Haematol. 2004;127:67–75. doi: 10.1111/j.1365-2141.2004.05138.x. [DOI] [PubMed] [Google Scholar]
- 8.Martin PL, Carter SL, Kernan NA, et al. Results of the Cord Blood Transplantation Study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12:184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]; Page KM, et al. Biol Blood Marrow Transplant. 2008;14:1108–1117. doi: 10.1016/j.bbmt.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurtzberg J, Balber A, Mendizabal A, et al. Preliminary results of a pilot trial of unrelated umbilical cord blood transplantation (UCBT) augmented with cytokine-primed aldehyde dehydrogenase- bright (ALDHbr) cells. Blood. 2006;108:abst 3641. [Google Scholar]
- 10.Rubinstein P, Dobrila L, Rosenfield R, et al. Processing and cryopreservation of placental/umbilical cord blood for unrelated bone marrow reconstitution. Proc Natl Acad Sci USA. 1995;92:10119–10122. doi: 10.1073/pnas.92.22.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus conference on acute gvhd grading. Bone Marrow Transplant. 1995;15:825–828. [PubMed] [Google Scholar]
- 12.Gooley T, Leisenring W, Crowley J, Storer B. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18:695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 13.Gray R. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 14.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 15.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–748. [PubMed] [Google Scholar]
- 16.Prasad VK, Kurtzberg J. Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 2008;41:99–108. doi: 10.1038/sj.bmt.1705970. [DOI] [PubMed] [Google Scholar]
- 17.Godder K, Pati A, Abhyankar S, Lamb L, Armstrong W, Henslee-Downey P. De novo chronic graft-versus-host disease presenting as hemolytic anemia following partially mismatched related donor bone marrow transplant. Bone Marrow Transplant. 1997;19:813–817. doi: 10.1038/sj.bmt.1700746. [DOI] [PubMed] [Google Scholar]
- 18.Raj A, Bertolone S, Cheerva A. Successful treatment of refractory autoimmune hemolytic anemia with monthly rituximab following nonmyeloablative stem cell transplantation for sickle cell disease. J Pediatr Hematol Oncol. 2004;26:312–314. doi: 10.1097/00043426-200405000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Corti P, Bonanomi S, Vallinoto C, et al. Rituximab for immune hemolytic anemia following T- and B-cell-depleted hematopoietic stem cell transplantation. Acta Haematol. 2003;109:43–45. doi: 10.1159/000067271. [DOI] [PubMed] [Google Scholar]
- 20.Hall J, Martin P, Wood S, Kurtzberg J. Unrelated umbilical cord blood transplantation for an infant with beta-thalassemia major. J Pediatr Hematol Oncol. 2004;26:382–385. doi: 10.1097/00043426-200406000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Urban C, Benesch M, Sovinz P, Schwinger W, Lackner H. Fatal Evans’ syndrome after matched unrelated donor transplantation for hyper-IgM syndrome. Eur J Haematol. 2004;72:444–447. doi: 10.1111/j.1600-0609.2004.00256.x. [DOI] [PubMed] [Google Scholar]
- 22.Dovat S, Roberts R, Wakim M, Stiehm E, Feig S. Immune thrombocytopenia after umbilical cord progenitor cell transplant: response to vincristine. Bone Marrow Transplant. 1999;24:321–323. doi: 10.1038/sj.bmt.1701888. [DOI] [PubMed] [Google Scholar]
- 23.Pratt G, Kinsey S. Remission of severe, intractable, autoimmune haemolytic anaemia following matched unrelated donor transplantation. Bone Marrow Transplant. 2001;28:791–793. doi: 10.1038/sj.bmt.1703232. [DOI] [PubMed] [Google Scholar]
- 24.Mullen C, Thompson J, Richard L, Chan K. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia. Bone Marrow Transplant. 2000;25:1093–1099. doi: 10.1038/sj.bmt.1702397. [DOI] [PubMed] [Google Scholar]
- 25.Sevilla J, Gonzalez-Vicent M, Madero L, Diaz M. Acute autoimmune hemolytic anemia following unrelated cord blood transplantation as an early manifestation of chronic graftversus- host disease. Bone Marrow Transplant. 2001;28:89–92. doi: 10.1038/sj.bmt.1703087. [DOI] [PubMed] [Google Scholar]
- 26.Hongeng S, Tardtong P, Worapongpaiboon S, Ungkanont A, Jootar S. Successful treatment of refractory autoimmune haemolytic anaemia in a post-unrelated bone marrow transplant paediatric patient with rituximab. Bone Marrow Transplant. 2002;29:871–872. doi: 10.1038/sj.bmt.1703551. [DOI] [PubMed] [Google Scholar]
- 27.Tubman VN, Smoot L, Heeney MM. Acquired immune cytopenias post-cardiac transplantation respond to rituximab. Pediatric Blood Cancer. 2006;48:339–344. doi: 10.1002/pbc.20761. [DOI] [PubMed] [Google Scholar]
- 28.Rawal A, Sarode R, Curtis BR, Karandikar NJ, Friedman K, Rogers ZR. Acquired Glanzmann’s Thrombasthenia as part of multiple-autoantibody syndrome in a pediatric heart transplant patient. J Pediatr. 2004:672–674. doi: 10.1016/j.jpeds.2003.12.040. [DOI] [PubMed] [Google Scholar]
- 29.Hess A, Thoburn C. Immune Tolerance to Self-Major Histocompatability Complex Class II Antigens after Bone Marrow Transplantation: Role of Regulatory T Cells. Biol Blood Marrow Transplant. 2006;12:518–529. doi: 10.1016/j.bbmt.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Zeiser R, Nguyen V, Beilhack A, et al. Inhibition of CD4+ CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood. 2006;108:390–399. doi: 10.1182/blood-2006-01-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]; Biol Blood Marrow Transplant. 2008;14:1108–1117. doi: 10.1016/j.bbmt.2008.07.006. Autoimmune Cytopenias in Young Infants after UCBT. [DOI] [PMC free article] [PubMed] [Google Scholar]