

Summary
Lipid droplets (LDs) are cellular storage organelles for neutral lipids that vary in size and abundance according to cellular needs. Physiological conditions that promote lipid storage rapidly and markedly increase LD volume and surface. How the need for surface phospholipids is sensed and balanced during this process is unknown. Here, we show that phosphatidylcholine (PC) acts as a surfactant to prevent LD coalescence, which otherwise yields large, lipolysis resistant LDs and triglyceride (TG) accumulation. The need for additional PC to coat the enlarging surface during LD expansion is provided by the Kennedy pathway, which is activated by reversible targeting of the rate-limiting enzyme, CTP:phospho-cholin cytidylyltransferase (CCT), to growing LD surfaces. The requirement, targeting, and activation of CCT to growing LDs were similar in cells of Drosophila and mice. Our results reveal a mechanism to maintain PC homeostasis at the expanding LD monolayer through targeted activation of a key PC synthesis enzyme.
Introduction
LDs store lipids for metabolic energy and membrane precursors (Walther and Farese, 2009). A phospholipid monolayer surrounds a core of neutral lipids, such as sterol esters or TG, and is decorated by LD-specific proteins. Changes in LD size directly reflect lipid synthesis and degradation. During conditions that promote TG storage, such as an excess of fatty acids, LDs increase in abundance and size to sequester fatty acids and protect cells from toxicity (Listenberger et al., 2003). As a consequence, LD volumes increase dramatically within hours. Massive amounts of neutral lipids are deposited in their cores, and large amounts of phospholipids are added to their surfaces. Conversely, lipids may be rapidly consumed during times of need. Hence, LDs provide a fascinating example of rapid and profound changes in organelle size.
The mechanisms of LD formation and expansion are mostly unknown. Synthesis of neutral lipids for LD cores is catalyzed by diacylglycerol O-acyltransferases, sterol O-acyltransferases, and other enzymes (Buhman et al., 2001) and is regulated in part by substrate availability and flux through the reactions. Little is known concerning phospholipid addition to the expanding LD-delimiting monolayer. Phospholipid remodeling enzymes localize to LDs (Moessinger et al., 2011) but by themselves cannot provide additional phospholipids for expanding LDs. Previously, in a systematic screen, we found that knockdown of enzymes important for phospholipid biosynthesis yielded cells with very large LDs by promoting LD fusion (Guo et al., 2008). Among those enzymes giving a strong phenotype were both iso-forms of CCT, key enzymes of PC synthesis, and both enzymes were found to localize to LDs (Guo et al., 2008). These findings provided clues to the importance of phospholipids in regulating LD morphology but did not address the mechanism of phospholipid homeostasis during LD expansion. Here, we investigated this mechanism by examining the function of CCT targeting to LDs and its role in maintaining PC levels during LD expansion.
Results
CCT Regulates Phospholipid Homeostasis during LD Expansion
In mammalian cells, more than 90% of LD surface phospholipids are phosphatidylethanolamine (PE) and PC (Tauchi-Sato et al., 2002). In comparison with mammalian cells (PE, 20%–30%, and PC, 50%–60%; Esko and Raetz, 1980) Drosophila S2 cells and LDs contain more PE (50%–60%) than PC (20%–25%) (Jones et al., 1992; Figure 1A). To identify phospholipid synthesis enzymes regulating LD size, we determined the effect of their depletion on LD morphology by RNAi. We included enzymes of the Kennedy pathways of PE and PC synthesis (Figures 1B and 1C) and enzymes of the PS decarboxylase pathway that yield PE (see Figure S1A available online). For each experiment, we confirmed the knockdown efficiency by qPCR or western blot (Figures S1B-S1D and data not shown). Only knockdowns of enzymes catalyzing PC synthesis resulted in LD phenotypes, indicating a key role for PC in LD expansion. The knockdown of PE synthesis enzymes modestly depleted PE levels in LDs but did not change morphology (Figure 1B, data not shown). CCT1 depletion yielded the greatest decrease in PC levels and the strongest phenotype—few very large or “giant” LDs (Figures 1A and 1C). This strong effect likely reflects that CCT1 catalyzes the rate-limiting step of PC synthesis (Vance and Vance, 2008). PC is also synthesized by sequential methylation of PE (e.g., in mammalian hepatocytes; Vance et al., 1997), but this pathway is not present in Drosophila (based on sequence similarity searches; data not shown).
Figure 1. CCT Is a Major Phospholipid Synthesis Enzyme Required during LD Expansion.
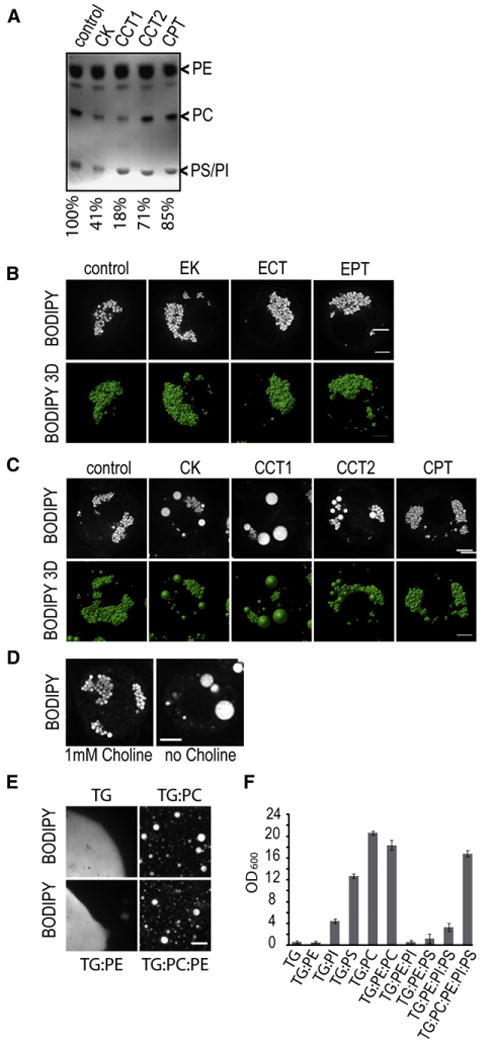
(A) RNAi-mediated depletion of several PC synthesis enzymes reduces PC levels in LDs as analyzed by TLC.
(B) RNAi depletion of PE synthetic enzymes does not affect LD morphology in Drosophila S2 cells after loading with 1 mM oleate for 12 hr. Confocal midsections (upper panels) and 3D reconstructions (lower panels) of BODIPY stained cells are shown.
(C) RNAi-mediated depletion of several PC synthesis enzymes results in giant LDs.
(D) Choline deficiency leads to giant LDs. S2 cells were grown in normal or choline-deficient medium, oleate loaded, and analyzed as in (B).
(E and F) PC stabilizes artificial LDs better than other phospholipids. Stability and size of artificial droplets consisting of the indicated lipids were determined by fluorescence microscopy (E) and light scattering (F). Values are mean ±SD of three experiments. Bars, 5 μm.
We previously showed that giant LDs in CCT knockdown cells result from increased LD coalescence or fusion (Guo et al., 2008). SNARE proteins have been proposed to mediate LD fusion (Bostrom et al., 2007). However, codepletion of CCT1 with NSF, α-SNAP, or SNAREs had no effect on the LD phenotype (Figure S1E).
CCT2 depletion affected PC levels and LD size much less than did CCT1 depletion (Figures 1A and 1C). This suggests that CCT1 provides most of the CCT activity for LD-directed PC synthesis in S2 cells, consistent with its higher expression levels compared with CCT2 (Celniker et al., 2009). We therefore focused our studies on CCT1 (data for CCT2 are in the Supplemental Information).
Choline kinase (CK) depletion also reduced PC levels in LDs and induced giant LDs. In contrast, depletion of CDP-cholinephosphotransferase (CPT), which catalyzes the last step of PC synthesis, caused only a minor reduction of PC levels and did not affect LDs (Figures 1A and 1C). Evidently, reduction of CPT mRNA levels by ∼80% (Figure S1C) is insufficient to reduce PC synthesis enough to affect LD formation.
To confirm that the giant LDs induced by knockdown of PC synthesis enzymes was due to decreased PC synthesis, we performed add-back experiments. CCT1 depletion was rescued by adding liposomes containing PC but not those containing mostly PE (Figure S1F). Also, treatment with a CCT inhibitor, miltefosine (hexadecylphosphocholine), yielded results similar to CCT depletion during oleate loading of cells (Figure S1G). Moreover, culturing S2 cells in choline-deficient medium resulted in the formation of giant LDs, resembling those from CCT1 depletion (Figure 1D).
Depletion of SREBP or SCAP from S2 cells also leads to formation of giant LDs (Guo et al., 2008). In Drosophila, these proteins regulate phospholipid metabolism and depletion of SREBP leads to a reduction of PE (∼60%) and PC (∼45%) (Dobrosotskaya et al., 2002). Addition of PC liposomes to SCAP-depleted cells prevented the formation of giant LDs, arguing that the giant LD phenotype in these cells is also due to PC deficiency (Figure S1F).
PC Acts as a Surfactant to Prevent LD Coalescence
The knockdown experiments indicated an important role for PC in stabilizing LDs, apparently acting as a surfactant to prevent phase coalescence of the LD emulsion. To test this model, we assayed the stability of artificial droplets generated in vitro with different phospholipid compositions. Strikingly, emulsions of artificial droplets formed with PC or a PC/PE mixture reflecting the composition of LDs (PC:PE 1:2.7) remained stable for at least 3 hr. LDs formed with TG or with TG and PE alone rapidly coalesced, eventually forming a continuous lipid phase (Figures 1E and 1F). Other phospholipids that are normally present on LDs in minor amounts, such as phosphatidylinositol (PI) and phosphatidylserine (PS), protect much less against phase coalescence when added alone in excess or when added with PE in molar ratios similar to those in LDs (PE:PI:PS of 9:1.3:1, data not shown, and Jones et al., 1992) (Figure 1F). In contrast, addition of PC to this complex phospholipid mixture abolished droplet coalescence (PC:PE:PI:PS of 1:2.7:0.4:0.3; Figure 1F). With previous data on the stability of lipid-water emulsions (Saito et al., 1999), our findings indicate that PC is necessary as a surfactant that stabilizes LDs and prevents their coalescence.
Of the Kennedy Pathway Enzymes for PC Synthesis, Only CCT Localizes to LDs
We next addressed where in the cell PC synthesis for LD expansion occurs. CCT1 and CCT2 localize to LDs during oleate loading in S2 cells (Guo et al., 2008), suggesting that PC might be synthesized locally on the LD surface. However, the substrates and products of the Kennedy pathway's first two reactions (catalyzed by CK and CCT) are water soluble and could diffuse to and from their site(s) of use (Figure 2A). In contrast, diacylglycerol and PC, the substrate and product of the final CPT reaction, are lipophilic and do not diffuse through the aqueous cytoplasm. Thus, CPT localization defines the site of PC production from this pathway. Analysis of fluorescently tagged Kennedy pathway enzymes during oleate loading of S2 cells showed that CCT1 and CCT2 localized to LDs as reported (Guo et al., 2008), but that CK and CPT did not (Figure 2B). CPT was found in the ER, in agreement with its predicted bilayer-spanning topology (Figure 2B; Henneberry et al., 2000).
Figure 2. Among PC Synthesis Enzymes, CCT Uniquely Localizes to LDs.
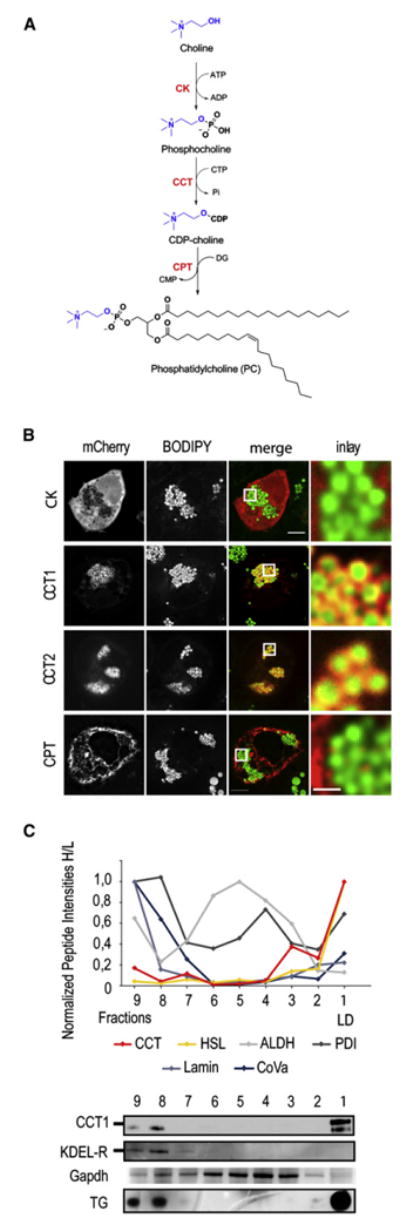
(A) Overview of the reactions of the Kennedy pathway.
(B) Transiently expressed mCherry-tagged CCT1 and CCT2, but not CK and CPT, (left panels) localize to LDs in S2 cells loaded with 1 mM oleate for 12 hr. Overlays and a magnification are shown (right two panels). Bar, 5 μm (overview) or 1μm (magnification).
(C) Endogenous CCT localizes to LDs. Normalized peptide intensities determined by LC-MS/MS are shown for fractions of a LD purification. CCT, hormone-sensitive lipase (HSL, LD marker), disulfide isomerase (PDI; ER marker), lamin (nuclear marker), cytochrome oxidase subunit Va (CoVa; mitochondrial marker), and aldehyde dehydrogenase (ALDH; cytosol marker) are shown. Bottom panels show western blots for CCT1, KDEL receptor, and Gapdh and a TLC for TG of each fraction normalized to protein.
To ensure that LD localization of CCT was not an artifact of tagged proteins, we examined the localization of endogenous CCT during LD growth. We purified LDs by differential centrifugation and floatation in a density gradient and analyzed CCT abundance in all fractions by mass spectrometry-based quantitative proteomics and immunoblotting (Figure 2C). CCT1 enrichment was found primarily in the LD fraction, mirroring the profile of TG or known LD proteins (e.g., hormone-sensitive lipase). ER proteins (e.g., protein disulfide isomerase) were present in LD fractions but in much lower amounts than in microsomes. Also marker proteins for different organelles and the cytosol (e.g., cytosolic aldehyde dehydrogenase, the nuclear protein lamin, and the mitochondrial protein cytochrome oxidase subunit Va) showed only low abundance in the LD fractions.
CCT Is Targeted to LDs when PC Levels Are Inadequate
Among PC synthesis enzymes, only CCT localized to LDs, and de novo PC synthesis required for expanding LD surfaces apparently occurs in the ER. This suggested that CCT localization to growing LDs has an alternative purpose, such as a regulatory function. Since the LD surface area increases about 10-fold with LD expansion in S2 cells (Figures 3A and 3B), and more phospholipids are needed to cover and stabilize the phase interface with the cytosol, we hypothesized that CCT detects a local PC deficiency on the expanding LD surface and is activated there to provide substrate for PC synthesis.
Figure 3. CCT1 Targets to LDs after an Initial Delay and Relocalizes to the Nucleus when Oleate Is Removed.
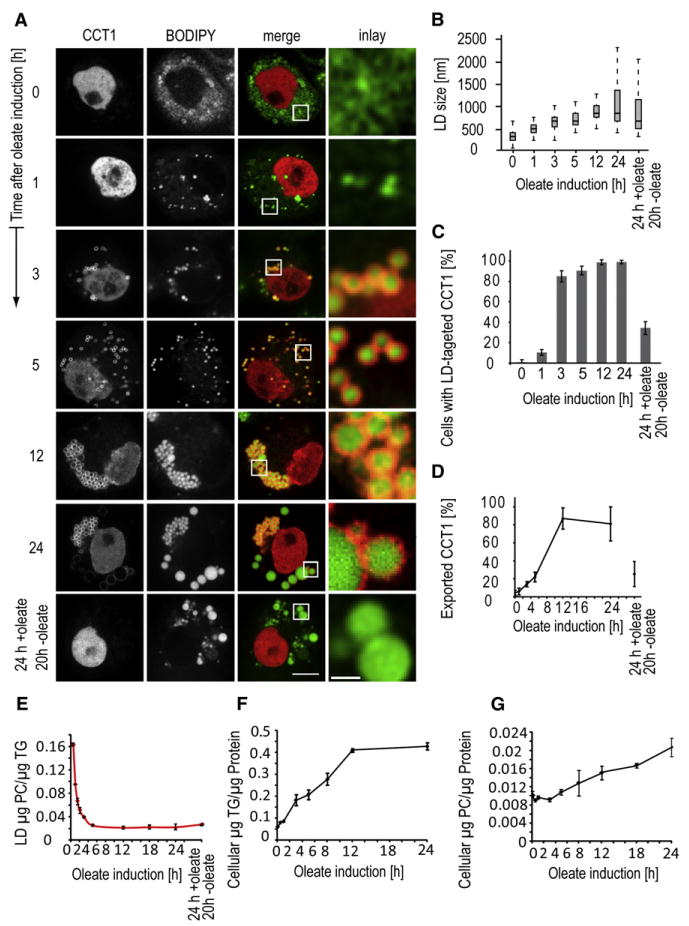
(A) mCherry-CCT1 (left panels) localizes to LDs (BODIPY, second left panels) after 3 hr of oleate loading and relocalizes to the nucleus when S2 cells are shifted to lipid-free medium. Confocal midsections of each time point are shown, as well as overlays and a magnification of LDs. Scale bar, 5 μm (overview) or 1 μm (magnification).
(B) LDs grow during oleate loading. Shown are box plots of LD diameters at different time points of oleate loading (n > 100 for each time point).
(C) Number of cells with targeting of CCT1 to LDs increases between 1 and 3 hr of oleate loading and decreases after oleate removal. Values are means ±SD of three experiments.
(D) The amount CCT1 on LDs increases in the first 12 hr of oleate loading and decreases after oleate removal. Shown is quantification of mCherry-CCT1 fluorescence intensity outside the nucleus. Values are means ±SD of three experiments.
(E) PC/TG ratio of LDs decreases for up to 5 hr of oleate loading but stabilizes afterwards. Values are mean ±SD of three experiments.
(F) Cellular TG levels increase steadily in oleate-loaded cells. Values are mean ±SD of three experiments.
(G) After an initial delay, cellular PC levels increase linearly over time in oleate-loaded cells. Values are mean ±SD of three experiments.
To test this hypothesis, we examined whether CCT1 is recruited specifically when local PC levels are insufficient, for example during LD expansion due to oleate loading. During 24 hr oleate loading, the average and maximum size of LDs steadily increased (Figures 3A and 3B). At 1 hr, small LDs were present, and CCT1 was targeted to the LD surfaces in ∼15% of cells. In contrast, CCT1 targeted to LDs in almost 90% of cells at 3 hr and essentially all cells at 5 hr (Figures 3A, 3C, and 3D). LD targeting of CCT2 followed a similar time course (Figure S2A).
During oleate loading, cellular TG synthesis increased steadily, leading eventually to a plateau of TG levels (Figure 3F). In contrast, the PC:TG ratio in LDs initially dropped rapidly but stabilized at a new, lower level after 5 hr, when essentially all CCT1 was targeted to LDs, and remained constant during the remaining 24 hr of oleate loading (Figure 3E). Total cellular PC level increased more than 2-fold after an initial lag phase, consistent with an increase in PC synthesis that balanced the TG increase, and coincided with the relocation of CCT1 to LDs (Figure 3G). The depletion of CK resulted in LD targeting of CCT at an even faster rate than that found in oleate-loaded WT cells (as judged by the rapid depletion of the nuclear CCT1; Figure S2B and data not shown), consistent with the notion that CCT binds to PC-deficient LDs.
If CCT localizes to LDs to balance the increased need for PC synthesis during their expansion, we expected it to lose this localization once LD expansion stopped. To test this prediction, we induced LD growth by treating cells for 24 hr with oleate and then stopped expansion by removing the oleate and continuing the incubation for 20 hr in lipid-free medium. After oleate removal, LDs decreased slightly in size, and CCT1 was found mostly in the nucleus, similar to its location before oleate loading (Figures 3A and 3B). Thus, the targeting of CCT1 to LDs appears to be part of a homeostatic feedback response: when LD growth reaches equilibrium and PC levels stabilize, CCT1 no longer binds to LDs and the soluble form is localized mostly in the nucleus.
CCT1 Is Mobile and Shuttles between Nucleus and Cytosol before Oleate Loading
CCT1 has a nuclear localization signal, and under basal conditions CCT1 is localized in the nucleus (DeLong et al., 2000; Guo et al., 2008). How then does CCT1 detect a need for PC at the surfaces of expanding cytoplasmic LDs and localize to this organelle? We hypothesized that CCT1 normally shuttles between the nucleus and cytosol, allowing it to survey PC levels at LD surfaces. To test this, we repeatedly bleached mCherry-CCT1 at one spot in the cytosol and monitored nuclear fluorescence (fluorescence loss in photobleaching, FLIP; Figures 4A and 4B). Almost all nuclear fluorescence was lost within ∼30 s (Movie S1). Other nuclei at a similar distance from the bleached spot, but in different cells, were not bleached, showing that cytoplasmic continuity is required for CCT1 bleaching. Thus, almost all CCT1 that appears nuclear at steady state migrated through the cytoplasmic bleaching spot within 30 s. In addition, FLIP experiments in which a nuclear spot was bleached revealed that CC1 is mobile within the nucleus (Figures 4C and 4D, Movie S2). Thus, under basal conditions, CCT1 rapidly shuttles between predominantly nuclear and less abundant cytosolic pools, affording access to LDs.
Figure 4. CCT1 Shuttles between Nucleus and Cytosol before Oleate Loading but Stably Associates with LDs during Their Expansion.
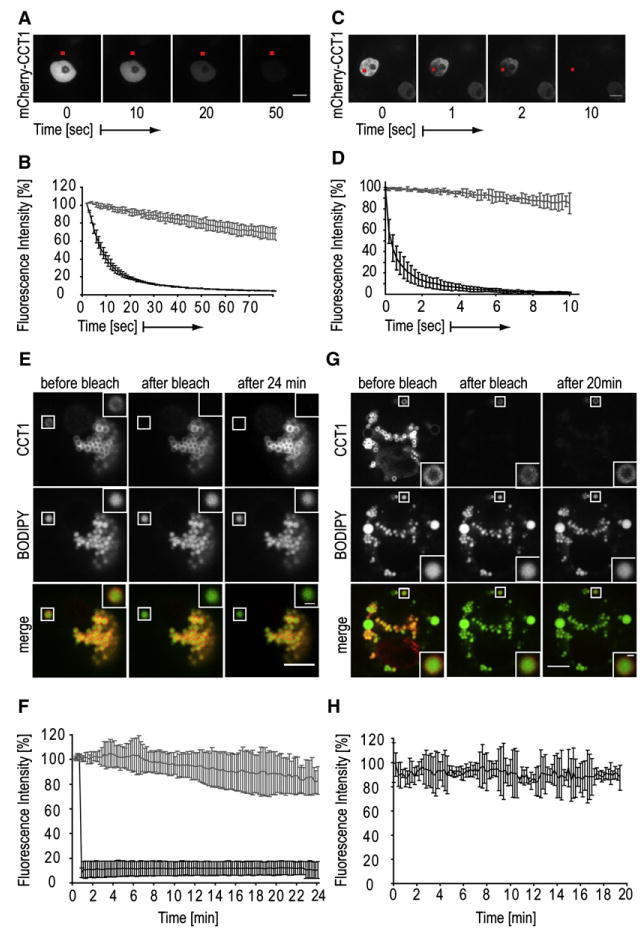
(A and B) mCherry-CCT1 shuttles between nucleus and cytosol as seen in images (A) and quantitation of three FLIP experiments (B; means ±SD are shown) in which a spot in the cytosol was repeatedly bleached.
(C and D) CCT1 is mobile in the nucleus. A FLIP experiment analogous to (A) and (B) is shown, but a spot in the nucleus was bleached repeatedly.
(E) mCherry-CCT1 fluorescence (upper panels) of a bleached LD (shown in lower panels) does not recover. Box indicates the photobleached LD, which is shown in higher magnification. Prebleach (left), immediately after bleach (middle), and postbleach (right) images of the experiment are shown.
(F) Normalized fluorescence intensity of mCherry-CCT1 on a bleached LD as in (C) over time. Values are means ±SD of three experiments.
(G) mCherry-CCT1 (upper panels) fluorescence remains stable on LD (BODIPY labeled, middle panels) in a bleached cell. Lower panels show overlays of the two channels. At time 0 min, all signal except on the indicated LD was photobleached. Prebleach (left), immediately after bleach (middle), and postbleach (right) images of the FRAP experiment are shown.
(H) Normalized fluorescence intensity of mCherry-CCT1 of experiments as in (G) over time. Values are mean ±SD of three experiments. Bars, 5 μm.
CCT Binds Stably to LDs during Their Expansion
CCT activity increases with membrane binding (Feldman et al., 1985). If CCT is recruited to LDs as part of a regulatory loop of PC synthesis, CCT1 binding to LDs should be stable at the LD surface until PC concentrations are adequate. To assess this prediction, we bleached the mCherry-CCT1 signal of one LD after oleate-mediated retargeting of CCT from the nucleus to LDs (fluorescence recovery after photobleaching, FRAP). After bleaching, the CCT1 signal was no longer detected on the selected LD (Figures 4E and 4F) and did not recover over 24 min (Movie S3). CCT2 yielded similar results (Figures S3A and S3B).
To confirm that CCT1 binds stably to growing LDs, we measured the apparent release rate of CCT1 from LDs by inverse FRAP (iFRAP). We bleached the mCherry-CCT1 signal of a whole cell except for one LD (Figures 4G and 4H). We then determined the fluorescence signal of the unbleached LD over 20 min (to correct for bleaching due to imaging, we used an unbleached cell in the same image as a control).In several cells, we found no loss of fluorescence on the unbleached LD (Movie S4). These data confirm that CCT1 is stably bound to LDs during this time period. Similar results were observed for CCT2 (Figures S3C and S3D). Thus, during LD growth, CCT enzymes stably bind to LDs and do not exchange with CCT bound to other LDs or in other pools.
CCT Binds to LDs via Its Amphipathic α Helix
We next asked which region of CCT is responsible for LD targeting. Besides an amphipathic helix in the middle of the protein, a C-terminal phosphorylation domain is implicated in CCT regulation and localization (Houweling et al., 1994). To determine which CCT1 regions mediate LD binding, we generated CCT1 mutants and assayed the ability of the expressed proteins to target to LDs (Figure 5A). Deleting the C-terminal phosphorylation domain (ΔP) had no effect on LD targeting, but CCT1 lacking the helical and phosphorylation domains(ΔHP)did not localize to LDs.LD targeting was also abolished by a point mutation, W397E,predicted to disrupt the amphipathicity of the α helix. Also, the amphipathic helical domain alone (H) fused to mCherry efficiently targeted to LDs. Thus, the CCT1 helical domain is necessary and sufficient for LD targeting.
Figure 5. CCT Activation by LD Binding Is Essential for Normal LDs.
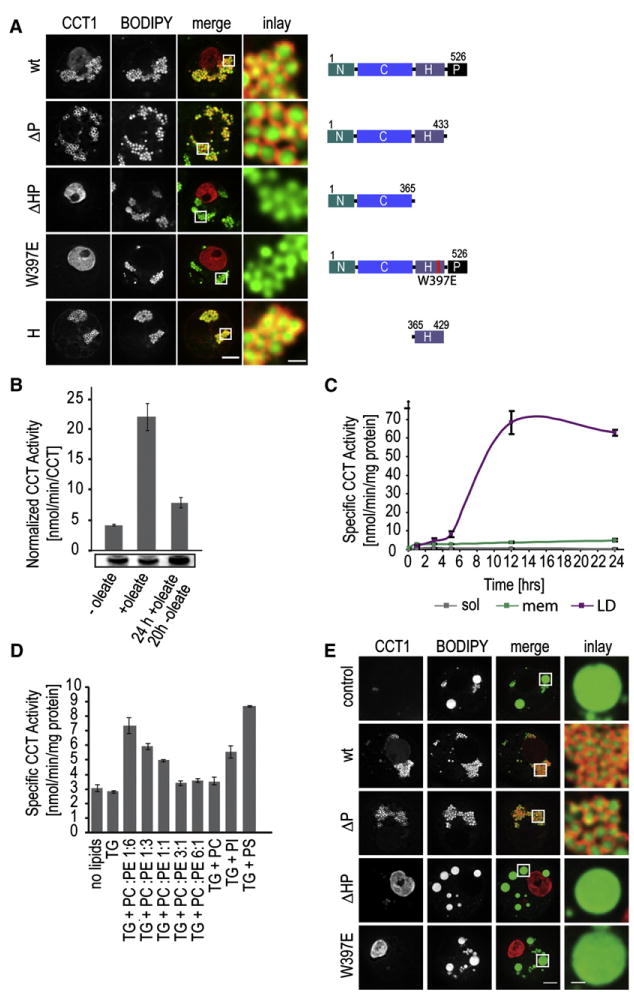
(A) Expression of different mCherry-CCT1 constructs (left panel) reveals the requirement of the helical region for LD (BODIPY stained; second left panels) binding. Overlays (merge) of the two channels and a magnification of LDs are shown. Bar, 5 μm (overview) or 1 μm (magnification).
(B) Endogenous CCT activity increases when cells are loaded with oleate and decreases in lipid-free medium. CCT activity normalized to enzyme amount before oleate loading, after 12 hr of oleate loading, and after 20 hr in lipid-free medium subsequent to 24 hr oleate loading is shown. Values are mean ±SD of three experiments.
(C) CCT is active on LDs. Specific activity of cytosol, membrane, and LD fractions of S2 cells loaded with oleate for indicated times is shown. Values are means ±SD of three experiments.
(D) CCT activity from extracts inversely correlates with PC content of added artificial droplets generated with the indicated phospholipid compositions. Values are means ±SD of three experiments.
(E) LD binding is essential for CCT1 normal LDs. Different mCherry-CCT1 constructs (left panels) were expressed in cells depleted for endogenous CCT1 by RNAi against its 3′UTR, and the ability of the mutants to rescue the LD phenotype (LDs stained with BODIPY; second left panels)was tested. Merged images and magnifications of LDs are shown. Bar, 5 μm (overview) or 1 μm (magnification).
In vitro, the binding of CCT to liposomes activates the enzyme through direct interaction with membrane bilayers (Taneva et al., 2003). We determined if CCT interacts similarly with the phospholipid monolayer of LDs in vitro. We used affinity-purified CCT2 (Figure S4A) (which was easier to produce than CCT1 and shares a highly conserved membrane-binding domain) and incubated it with artificial LDs. We then assayed for protein binding to LDs by floatation. A significant fraction of CCT2, but not a GFP control protein, floated with artificial droplets, indicating that CCT2 interacts directly with monolayer-delimited droplets (Figure S4B).
LD Binding Activates CCT
Our model predicts that LD binding activates CCT. Consistent with this notion, oleate loading activated endogenous CCT 4-fold (and transfected mCherry-CCT1 3-fold; Figure 5B and Figure S5A). CCT induction was due to the LD-targeted enzyme pool, because CCT activity increased only in the LD fractions (Figure 5C). CCT activation during oleate loading occurred with a delay of several hours and increased concurrently with CCT localization to LDs (Figures 3 and 5C). CCT1 relocalization to LDs also coincided with the decrease of the PC:TG ratio (Figure 3E). In addition, after removal of oleate from the medium (when CCT is no longer localized to LDs [Figure 3A]), CCT activity decreased almost to basal levels (Figure 5B).
To directly determine if PC-deficient LDs activate CCT, we mixed S2 cell extracts with artificial droplets of different compositions and measured CCT activity. Droplets made with PC alone did not activate CCT (Figure 5D). Strikingly, however, decreasing the molar PC:PE ratio markedly activated endogenous CCT. Droplets made with other phospholipids (e.g., PS or PI) also activated CCT. Adding other metabolites, such as oleate or DG, to artificial LDs shifted the baseline of CCT activity slightly or acted inhibitory, respectively, but did not change the response to decreasing levels of PC (data not shown). Moreover, mutant CCT1 (W397E), which does not bind LDs, was not activated by PC-deficient artificial droplets (Figure S5B), consistent with the requirement for droplet binding to mediate enzyme activation. Our data on CCT activation by PC-deficient LDs are consistent with CCT activation by liposomes (Tilley et al., 2008) and show that CCT is activated by PC deficiency on LDs in vivo and in vitro.
LD Binding Is Essential for CCT1 Function during LD Expansion
If CCT is regulated by subcellular relocalization during LD expansion, LD binding of CCT should be crucial for its function in this process. To test this prediction, we determined whether LD binding of CCT1 is required for normal LD formation with RNAi rescue experiments. We depleted endogenous CCT1 by RNAi against the 3′ UTR of its mRNA, which led to the formation of giant LDs during oleate loading as expected. This phenotype was rescued by expressing RNAi-resistant mCherry-CCT1 or mCherry-CCT1ΔP (Figure 5E). In contrast, constructs of CCT1 that cannot bind to LDs (ΔHP and W397E) did not rescue the giant LD phenotype (Figure 5E). Importantly, both the ΔHP and W397E mutants shuttled normally between the nucleus and cytoplasm, as determined by FLIP analysis, and had access to LDs (data not shown). These results indicate that LD binding via the a helix is necessary for CCT function in LD expansion. Also, the overexpression of CCT2, which does not localize to the nucleus and is present in low amounts in S2 cells, fully rescued CCT1 depletion (Figure S5C). Thus, the nuclear-cytoplasmic shuttling of CCT1 is apparently required only to make the enzyme available in the cytoplasm during LD expansion.
CCT1 Regulates LD Size and TG Storage In Vivo
We next tested whether the function of CCT in LD expansion is relevant under physiological conditions. We determined LD morphology in Drosophila expressing CCT1-directed shRNA in larval fat body. Larvae expressing the CCT1-directed shRNA exhibited many large LDs, similar to findings in PC-deficient cells or PC-deficient artificial LDs (Figures 1C, 1E, and 6A), showing conservation of CCT function in flies. In addition, larvae stored more TG if CCT1 was depleted from the fat body (Figure 6B), consistent with our previous finding that the large LDs of CCT1-depleted S2 cells are resistant to lipolysis (Guo et al., 2008). As a physiological consequence, flies with fat bodies depleted of CCT1 not only stored more TG (which is likely released more slowly) but also lived longer during starvation (Figure 6C).
Figure 6. CCT Is Important for LD Homeostasis In Vivo and in Mammalian Cells.
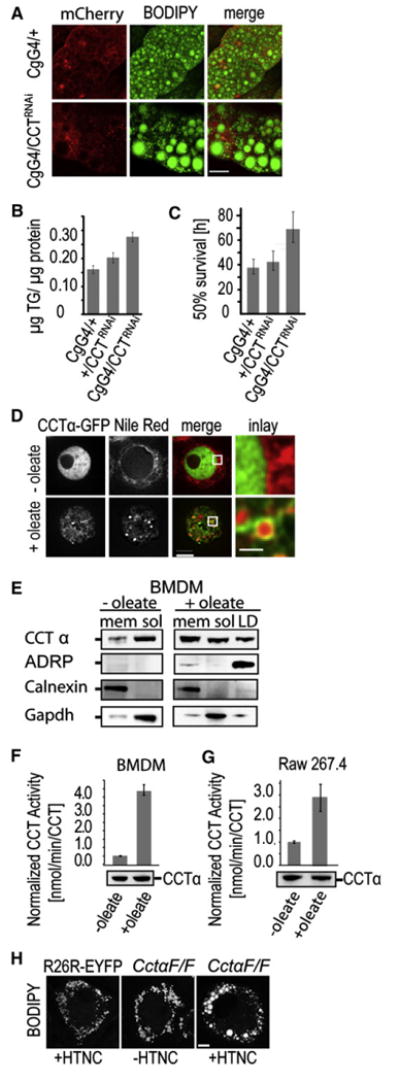
(A) Flies with CCT1 knockdown in larval fat body have giant LDs there. LDs in fat body of CgG4/+ and CgG4/CCTRNAi flies were stained with BODIPY (middle). Expression of the UAS-Gal driver was monitored by mCherry levels (left). Merged images are shown on the right. Bar, 10 μm.
(B) Flies with CCT1 knockdown in larval fat body have increased TG content. Lipids from third-instar larval from Cg /+, +/CCTRNAi, and CgG4/CCTRNAi flies were quantified by TLC.
(C) Flies with CCT1 knockdown in larval fat body are more resistant to starvation. Fifty percent survival rates of CgG4/+, +/CCTRNAi, and CgG4/CCTRNAi flies are indicated. Values are mean ±SD of three experiments.
(D) CCTα-GFP (left) targets to LDs (Nile red; middle) and membranes in oleate-loaded Raw267.4 macrophages. Merged images (second right panels) and magnifications (right panels) are shown. Bar, 5 μm (overview) or 1 μm (magnification).
(E) CCTα moves from the soluble fraction to membranes and LDs during oleate loading of BMDM. The indicated fractions were probed for CCT α and the marker proteins ADRP, calnexin, and Gapdh.
(F and G) CCTα is activated by oleate loading in BMDMs and Raw267.4 macrophages. Values are mean ±SD of three experiments analogous to Figure 5B.(H) CCTα knockdown in HTCN-treated BMDM from CctαF/F mice results in giant LDs. Confocal images of BODIPY-stained untreated BMDM of CctαF/F mice or of HTNC-treated R26R-EYFP are shown as controls. Bar, 5 μm.
CCT Targeting and Function in LD Stabilization Are Conserved in Mammalian Cells
To determine if CCT localization to LDs in Drosophila cells is conserved in mammals, we expressed murine CCTα in S2 cells. Like the Drosophila protein, CCTα localized to expanding LDs (Figure S6A). CCTα also localized to LDs in response to oleate loading of murine macrophage cells (Raw267.4; Figure 6D). In macrophages, CCTα also localized partly to a reticular structure, likely the ER (Figures 6D). In bone marrow-derived macrophages (BMDMs), we assessed CCTα localization by subcellular fractionation (Figure 6E). Before oleate loading, the enzyme was found mostly in the soluble fraction. However, during oleate loading, CCTα redistributed to the LD and microsomal fractions. To test whether this relocalization corresponded to CCT activation, we measured CCT activity normalized to protein levels and found 8- and 3-fold activation in BMDMs or Raw267.4 cells, respectively (Figures 6F and 6G, respectively). The localization and activation of CCTα to LDs were not specific to macro-phages. Neuronal N2a cells exhibited similar and even more pronounced recruitment and activation (Figures S6B–S6D). Furthermore, in vitro assays with N2a cell lysate showed CCT activation by artificial droplets with low amounts of PC (Figure S6E) similar to Drosophila CCT (Figure 5D).
To determine if the functional requirements of CCT in LD expansion are conserved in mammals, we analyzed LD morphology in BMDMs that lacked CCTα. Macrophages from mice homozygous for a conditional Cctα allele (CctαF/F) (Jacobs et al., 2004) or control mice (R26R-eYFP) were treated with a cell-penetrating version of Cre recombinase (HTNC) to inactivate the floxed allele (Peitz et al., 2007). Recombination efficiency was routinely >90%, leading to more than 85% reduction of CCTα mRNA levels in BMDMs compared with controls, and immunoblotting showed nearly complete depletion of the protein (Figures S7A and S7B). Of the BMDMs with deleted CCTα, 66% had highly enlarged LDs in response to oleate loading (Figure 6H), similar to findings in CCT1-depleted Drosophila S2 cells. Thus, the functional requirement of CCT in LD expansion is apparently conserved in murine macrophages.
Discussion
LDs are highly dynamic cellular organelles that change their volume dramatically according to the metabolic state. Our studies reveal important insights into the coordination of phospholipid synthesis with LD expansion. We show that PC is crucial to stabilize growing LDs and prevent their coalescence. We also show that CCT enzymes localize to and are activated at the surfaces of growing LDs with relatively low PC. CCT activation on surfaces of expanding LDs levels increases the flux through the Kennedy pathway, which generates PC to coat growing LDs and prevent their coalescence. In this manner, the activation of the pathway occurs in response to a sensed localized need and proceeds until homeostasis is restored. This model agrees with previous observations for CCT activation by membrane binding, which suggested CCT1 regulation functions similarly to homeostatically control membrane expansion (e.g., during cell proliferation; Jamil et al., 1990; Northwood et al., 1999).
Despite the clustering of LDs with oleate loading in Drosophila cells, expanding LDs rarely fuse (Guo et al., 2008). This is surprising, since it is thermodynamically favorable for a liquid-liquid biphasic system, such as a TG emulsion in water, to coalesce into single oil and water phases to minimize the interface between the phases and the resulting surface tension. Indeed, we found that TG droplets generated in vitro coalesced rapidly, similar to LDs that form in cells with diminished PC synthesis (Figures 1C, 1E, and 1F; Guo et al., 2008), and PC but not other phospholipids provided in physiological amounts prevented this coalescence. This difference in properties of phospholipids is likely explained by the biophysical properties of PC, which has a larger head group and is more cylindrical than the smaller head groups and more conical shapes of other phospholipids (Vance and Vance, 2008). During the transition state of LD coalescence, strong bending of the monolayer occurs at the interface, and such bending is easier to achieve with conical lipids, such as PE (Kabalnov and Wennerström, 1996; Saito et al., 1999). In contrast, the higher energy of the coalescence transition state of hydrophobic phases covered with cylindrical PC increases the activation energy and thus may prevent coalescence (Kabalnov and Wennerström, 1996). Another stabilizing factor may be the ability of PC to shield underlying TG, which would promote coalescence. For example, phase-boundary surfaces composed of PE and triolein have 28% exposed TG, whereas in PC and triolein mixtures only 3% of the surface is exposed TGs (Saito et al., 1999). We note that although our results highlight the role of PC in stabilizing LDs and preventing coalescence, they do not exclude a role for PE in cells, which may have been insufficiently depleted to observe a LD phenotype.
Rapid LD expansion during lipid loading confronts cells with a huge demand for additional PC to cover the LD surface and thus prevent coalescence. In our study, average LD diameters increased from 300 nm to >900 nm during oleate loading, correlating to a more than 9-fold surface expansion. Our data show that the activation of CCT, the rate-limiting enzyme of PC synthesis, by LD binding leads to increased PC synthesis (Figure 7). At steady state before oleate loading, CCT1 shuttles between the nucleus and cytoplasm without associating with membranes and thus being only moderately active (Feldman et al., 1985; Vance and Pelech, 1984). As PC deficiency develops on expanding LDs and phospholipid monolayer properties change, binding sites for CCT are generated. In agreement with this model, liposome membranes with low amounts of PC bind CCT more strongly than other membranes (Helmink and Friesen, 2004; Jamil et al., 1990), and we found similar results for PC-deficient artificial LDs. CCT binding to LDs then activates the enzyme, and since CCT catalyzes the rate-limiting step of PC synthesis, its activation increases flux through the Kennedy pathway, thereby increasing PC synthesis (Vance et al., 1980). PC is then trafficked, through unknown mechanisms to LD surfaces where it prevents coalescence.
Figure 7. Model of CCT Regulation during LD Expansion.
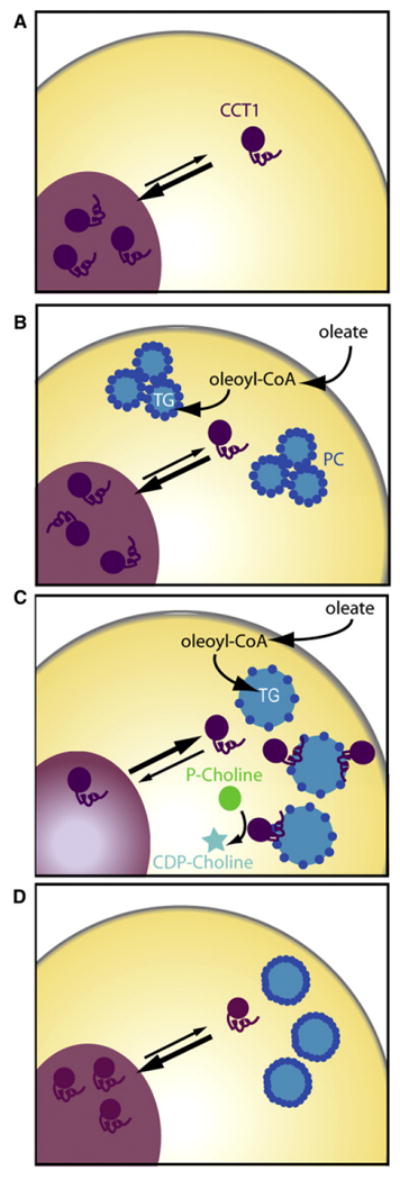
(A–D) See the Discussion for details.
Several finding suggest that CCT regulation by LD binding is likely part of a dynamic, homeostatic mechanism. First, CCT is targeted to LDs only after a delay and PC levels are sufficiently depleted. Second, the PC:TG ratio in LDs is maintained at a constant level during LD growth once CCT targets to LDs. Third, CCT1 targeting to LDs is reversible, either by release from LDs or through degradation of LD-associated CCT1. The mechanism for the latter finding is unclear, but it will be interesting to determine how the loss of CCT1 is achieved and which CCT1 domains are sufficient for mediating this loss.
How does CCT recognize expanding LD surfaces? With liposomes, CCT binds to and is activated by membranes of low PC content or those enriched in DG or oleate (Weinhold et al., 1991, 1994; Yao et al., 1990). Our data for artificial LDs are most consistent with PC deficiency per se triggering LD binding of CCT. The notion that CCT is regulated specifically by PC deficiency at expanding LDs is also supported by findings that DG accumulates at early time points of LD expansion (Kuerschner et al., 2008), before CCT localizes to LDs. Importantly, oleate and DG did not alter the regulation of CCT in vitro but only shifted the baseline of activity (data not shown). Our results also show that CCT1 accomplishes binding to LDs through its amphipathic helix, similar to how it binds to membrane bilayers (Cornell and Vance, 1987; Feldman et al., 1985). PC-poor membranes display a number of biophysical properties that may serve to recruit CCT binding (Jamil et al., 1990; Weinhold et al., 1994). Intriguingly, low lateral surface pressure of a PC-poor LD monolayer could expose TG on the LD surface, which the amphipathic CCT a helix may bind, analogous to the mechanism of binding of amphipathic α-helical peptides and apolipoproteins to oil/water inter-phases (Small et al., 2009).
Many of our findings in S2 cells were conserved in vivo and in mammalian cells. For example, CCT1 depletion from Drosophila larval fat body resulted in grossly enlarged LDs with increased TG and resistance to starvation. Resistance to starvation may indicate that energy stores are released more slowly from the CCT-depleted fat body, since a similar phenotype results from inactivating lipolysis by mutating Brummer, the major TG lipase in Drosophila fat body (Gronke et al., 2005). We also found in macrophages that CCTα was recruited to the surfaces of expanding LDs and that the depletion of CCTα resulted in large LDs. In mammals, a link between PC and LD biology was established decades ago. Studies in 1932 showed that adding choline to the diets of rats reduced hepatic steatosis induced by a high-fat diet (Best et al., 1932). Additionally, a choline-deficient diet is commonly used to induce fatty liver, full of large LDs. Similarly, a murine knockout of CCTα specifically in liver, where it is responsible for roughly 70% of PC synthesis (Sundler and Akesson, 1975), leads to hepatic TG accumulation, similar to the phenotype of CCT1 depletion in Drosophila cells (Guo et al., 2008; Jacobs et al., 2004). Our observations suggest that the steatosis of CCTα deficiency in the liver is mediated in part by the formation of large LDs.
PC content might also be involved in regulating LD size in adipocytes. Mammalian adipocytes are characterized by the presence of usually only one, very large, LD, suggesting a relationship to surface PC levels. However, the nature of this relationship is unclear. In contrast to Drosophila cells, mammalian adipocytes express phosphatidylethanolamine methyltransferase (PEMT), which methylates PE to form PC (Vance et al., 1997) and may contribute additional PC to regulate LD size (Hörl et al., 2011). Moreover, proteins, such as CIDEC, that promote the formation of unilocular LDs in adipocytes (Nishino et al., 2008) may locally modulate PC content at the LD surface.
Our findings concerning CCTα in macrophages may have clinical relevance. During the formation of macrophage foam cells, a hallmark of atherosclerotic lesions, CCT activation and PC synthesis help protect macrophages from apoptosis caused by elevated levels of unesterified cholesterol (Zhang et al., 2000). Although part of this activation is mediated by dephosphoylation of CCT (Wang and Kent, 1995), the PC homeostatic mechanism we describe may be important in enabling macrophages to optimally function in dealing with lipid overload in atherosclerotic lesions.
In summary, our studies uncover a crucial role for PC in maintaining the stability of LDs in emulsions and in cells, and we identified a regulatory mechanism that depends on the conditional localization of the rate-limiting enzyme of PC synthesis to a subcellular compartment to control local lipid contents and hence the size of this organelle. This mechanism thus directly couples the regulation of a biochemical pathway to organelle expansion.
Experimental procedures
Microscopy
Cells were prepared for fluorescence microscopy, including staining with BODIPY, as described (Guo et al., 2008), and imaged with a spinning-disk confocal microscope (100 × 1.4 NA oil immersion objective [Olympus], iMIC [Till], CSU22 [Yokugawa], iXonEM 897 [Andor]). 16-bit images were collected with Image iQ (version 1.9; Andor) and deconvoluted (Huygens, SVI).
TLC and Lipid Measurements
Lipids were extracted (Folch et al., 1957), separated on silica TLC plates (Merck) with chloroform/methanol/ammonia solution (25%) (50:25:6; per volume) for phospholipids or petroleum ether/diethyl ether/acetic acid (70:30:2; per volume) for neutral lipids, and detected by cerium molybdate staining. Bands were identified by comparing with standards. PC and TG levels were quantified from extracted lipids as described (Nanjee and Miller, 1996).
Emulsion and Liposome Preparation
Lipid emulsions and liposomes were prepared by mixing lipids in chloroform/ methanol (2:1); drying them under a stream of N2; resuspending them in 150 mM NaCl, 50 mM Tris/HCl (pH 7.5), 1 mM EDTA, 1 mM DTT, 50 mM PMSF; and sonication. For emulsions, the molar ratio of TG to total phospholipids was 2:5. Contaminating vesicles were removed and LDs were concentrated by ultracentrifugation at 100,000 × g for 15 min.
Cellular Fractionation and LD Purification
Drosophila S2 cells were harvested; washed with ice-cold PBS; resuspended in 2 ml 200 mM Tris/HCL (pH 7.5), 2 mM MgAc, and Complete Protease Inhibitor (Roche); lysed with a tissue homogenizer; and fractionated by three centrifugation steps at 3,000, 20,000, and 100,000 × g. The supernatant was adjusted to 1 M sucrose, layered under a sucrose step gradient, and centrifuged at 200,000 × g. Six fractions of the gradient and the three pellet fractions of the differential centrifugation steps were analyzed by MS-based proteomics and western blot with antibodies against CCTα (McCoy et al., 2006), Gapdh (LSBio), KDEL-receptor (Abcam), and Calnexin (Abcam). Similarly, mammalian cells were harvested; washed with ice-cold PBS; resuspended in 2 ml of 150 mM NaCl, 50 mM Tris/HCl (pH 7.5), 1 mM EDTA, 1 mM DTT, 50 mM PMSF; lysed by sonication; and fractionated into LDs and soluble and membrane fractions by centrifugation at 100,000 × g. The top LD fraction was washed in buffer to remove cytosolic contamination.
CCT Activity Assay
CCT activity in total cell lysate or fractions was determined as described (Pelech et al., 1981) and normalized when indicated to the amount of CCT determined by western blot.
Transgenic Flies
Fly stocks were raised on standard corn meal-agar medium at room temperature. Male UAS-RNAi CCT1 transgenic flies (ID 18628) from the Vienna Drosophila RNAi center were crossed with CgGal4 (Asha et al., 2003) UAS-mCherry reporter strain virgins. Third-instar larvae of the progeny were dissected in PBS, stained with BODIPY, and imaged with a confocal microscope (LSM 510, Zeiss). UAS-RNAi and CgGal4 flies were both extensive backcrossed to wild-type strain white1118 Berlin. For starvation assays, virgin flies starved in vials with 1% agar surviving flies were scored every 8 hr.
Supplementary Material
Acknowledgments
We thank Dr. R. Mallampalli for CCTα antibodies, Dr. J. Friesen for CCT2 expression baculovirus, Dr. N. Ridgway for the CCTα-GFP clone, Dr. T. Kornberg for the UAS-mCherry transgenic flies, Dr. U. Heberlein for white1118 Berlin flies, Dr. H. Wang for experimental help, and Drs. G. Howard and S. Ordway for editorial assistance. The National Institutes of Health (NIH) (grant DK-056084 to R.V.F.), German Research Foundation (DFG; T.C.W.), J. David Gladstone Institutes (R.V.F.), and Canadian Institutes of Health Research (D.E.V.) supported this work. D.E.V. is a Scientist of the Alberta Heritage Foundation for Medical Research.
Footnotes
Supplemental Information: Supplemental Information includes seven figures, four movies, three tables, Supplemental Experimental Procedures, and Supplemental References and can be found with this article online at doi:10.1016/j.cmet.2011.07.013.
References
- Asha H, Nagy I, Kovacs G, Stetson D, Ando I, Dearolf CR. Analysis of Ras-induced overproliferation in Drosophila hemocytes. Genetics. 2003;163:203–215. doi: 10.1093/genetics/163.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best CH, Hershey JM, Huntsman ME. The effect of lecithine on fat deposition in the liver of the normal rat. J Physiol. 1932;75:56–66. doi: 10.1113/jphysiol.1932.sp002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom P, Andersson L, Rutberg M, Perman J, Lidberg U, Johansson BR, Fernandez-Rodriguez J, Ericson J, Nilsson T, Boren J, et al. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat Cell Biol. 2007;9:1286–1293. doi: 10.1038/ncb1648. [DOI] [PubMed] [Google Scholar]
- Buhman KK, Chen HC, Farese RV., Jr The enzymes of neutral lipid synthesis. J Biol Chem. 2001;276:40369–40372. doi: 10.1074/jbc.R100050200. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell R, Vance DE. Binding of CTP: phosphocholine cytidylyltransferase to large unilamellar vesicles. Biochim Biophys Acta. 1987;919:37–48. doi: 10.1016/0005-2760(87)90215-3. [DOI] [PubMed] [Google Scholar]
- DeLong CJ, Qin LY, Cui Z. Nuclear localization of enzymatically active green fluorescent protein-CTP: phosphocholine cytidylyltransferase alpha fusion protein is independent of cell cycle conditions and cell types. J Biol Chem. 2000;275:32325–32330. doi: 10.1074/jbc.M004644200. [DOI] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, Rawson RB. Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science. 2002;296:879–883. doi: 10.1126/science.1071124. [DOI] [PubMed] [Google Scholar]
- Esko JD, Raetz CR. Mutants of Chinese hamster ovary cells with altered membrane phospholipid composition. Replacement of phosphatidylinositol by phosphatidylglycerol in a myo-inositol auxotroph. J Biol Chem. 1980;255:4474–4480. [PubMed] [Google Scholar]
- Feldman DA, Rounsifer ME, Weinhold PA. The stimulation and binding of CTP: phosphorylcholine cytidylyltransferase by phosphatidylcholine-oleic acid vesicles. Biochim Biophys Acta. 1985;833:429–437. doi: 10.1016/0005-2760(85)90100-6. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, Muller G, Jackle H, Kuhnlein RP. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 2005;1:323–330. doi: 10.1016/j.cmet.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Friesen JA. Characterization of a lipid activated CTP:phosphocholine cytidylyltransferase from Drosophila melanogaster. Biochim Biophys Acta. 2004;1683:78–88. doi: 10.1016/j.bbalip.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Henneberry AL, Wistow G, McMaster CR. Cloning, genomic organization, and characterization of a human cholinephosphotransferase. J Biol Chem. 2000;275:29808–29815. doi: 10.1074/jbc.M005786200. [DOI] [PubMed] [Google Scholar]
- Hörl G, Wagner A, Cole LK, Malli R, Reicher H, Kotzbeck P, Köfeler H, Höfler G, Frank S, Bogner-Strauss JG, et al. Sequential synthesis and methylation of phosphatidylethanolamine promote lipid droplet biosynthesis and stability in tissue culture and in vivo. J Biol Chem. 2011;286:17338–17350. doi: 10.1074/jbc.M111.234534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houweling M, Jamil H, Hatch GM, Vance DE. Dephosphorylation of CTP-phosphocholine cytidylyltransferase is not required for binding to membranes. J Biol Chem. 1994;269:7544–7551. [PubMed] [Google Scholar]
- Jacobs RL, Devlin C, Tabas I, Vance DE. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- Jamil H, Yao ZM, Vance DE. Feedback-regulation of Ctpphosphocholine cytidylyltransferase translocation between cytosol and endo-plasmic-reticulum by phosphatidylcholine. J Biol Chem. 1990;265:4332–4339. [PubMed] [Google Scholar]
- Jones HE, Harwood JL, Bowen ID, Griffiths G. Lipid composition of subcellular membranes from larvae and prepupae of Drosophila melanogaster. Lipids. 1992;27:984–987. doi: 10.1007/BF02535576. [DOI] [PubMed] [Google Scholar]
- Kabalnov A, Wennerström H. Macroemulsion stability. The oriented wedge theory revisited. Langmuir. 1996;12:276–292. [Google Scholar]
- Kuerschner L, Moessinger C, Thiele C. Imaging of lipid biosynthesis: how a neutral lipid enters lipid droplets. Traffic. 2008;9:338–352. doi: 10.1111/j.1600-0854.2007.00689.x. [DOI] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy DM, Fisher K, Robichaud J, Ryan AJ, Mallampalli RK. Transcriptional regulation of lung cytidylyltransferase in developing transgenic mice. Am J Respir Cell Mol Biol. 2006;35:394–402. doi: 10.1165/rcmb.2005-0401OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessinger C, Kuerschner L, Spandl J, Shevchenko A, Thiele C. Human lysophosphatidylcholine acyltransferases 1 and 2 are located in lipid droplets where they catalyze the formation of phosphatidylcholine. J Biol Chem. 2011;286:21330–21339. doi: 10.1074/jbc.M110.202424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanjee MN, Miller NE. Sequential microenzymatic assay of cholesterol, triglycerides, and phospholipids in a single aliquot. Clin Chem. 1996;42:915–926. [PubMed] [Google Scholar]
- Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S, Matsuki Y, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northwood IC, Tong AHY, Crawford B, Drobnies AE, Cornell BB. Shuttling of CTP: phosphocholine cytidylyltransferase between the nucleus and endoplasmic reticulum accompanies the wave of phosphatidylcholine synthesis during the G(0) → G(1) transition. J Biol Chem. 1999;274:26240–26248. doi: 10.1074/jbc.274.37.26240. [DOI] [PubMed] [Google Scholar]
- Peitz M, Jager R, Patsch C, Jager A, Egert A, Schorle H, Edenhofer F. Enhanced purification of cell-permeant Cre and germline transmission after transduction into mouse embryonic stem cells. Genesis. 2007;45:508–517. doi: 10.1002/dvg.20321. [DOI] [PubMed] [Google Scholar]
- Pelech SL, Pritchard PH, Vance DE. cAMP analogues inhibit phosphatidylcholine biosynthesis in cultured rat hepatocytes. J Biol Chem. 1981;256:8283–8286. [PubMed] [Google Scholar]
- Saito H, Kawagishi A, Tanaka M, Tanimoto T, Okada S, Komatsu H, Handa T. Coalescence of lipid emulsions in floating and freezethawing processes: examination of the coalescence transition state theory. J Colloid Interface Sci. 1999;219:129–134. doi: 10.1006/jcis.1999.6452. [DOI] [PubMed] [Google Scholar]
- Small DM, Wang L, Mitsche MA. The adsorption of biological peptides and proteins at the oil/water interface. A potentially important but largely unexplored field. J Lipid Res. 2009;(Suppl. 50):S329–S334. doi: 10.1194/jlr.R800083-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundler R, Akesson B. Biosynthesis of phosphatidylethanolamines and phosphatidylcholines from ethanolamine and choline in rat liver. Biochem J. 1975;146:309–315. doi: 10.1042/bj1460309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneva S, Johnson JE, Cornell RB. Lipid-induced conformational switch in the membrane binding domain of CTP:phosphocholine cytidylyltransferase: a circular dichroism study. Biochemistry. 2003;42:11768–11776. doi: 10.1021/bi035234k. [DOI] [PubMed] [Google Scholar]
- Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- Tilley DM, Evans CR, Larson TM, Edwards KA, Friesen JA. Identification and characterization of the nuclear isoform of Drosophila melanogaster CTP:phosphocholine cytidylyltransferase. Biochemistry. 2008;47:11838–11846. doi: 10.1021/bi801161s. [DOI] [PubMed] [Google Scholar]
- Vance DE, Pelech SL. Enzyme translocation in the regulation of phosphatidylcholine Biosynthesis. Trends Biochem Sci. 1984;9:17–20. [Google Scholar]
- Vance JE, Vance DE. Functional roles of lipids in membranes. In: Vance DE, Vance JE, editors. In Biochemistry of Lipids, Lipids and Membranes. London: Elsevier; 2008. pp. 1–39. [Google Scholar]
- Vance DE, Trip EM, Paddon HB. Poliovirus increases phosphatidylcholine biosynthesis in HeLa cells by stimulation of the rate-limiting reaction catalyzed by CTP: phosphocholine cytidylyltransferase. J Biol Chem. 1980;255:1064–1069. [PubMed] [Google Scholar]
- Vance DE, Walkey CJ, Cui Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim Biophys Acta. 1997;1348:142–150. doi: 10.1016/s0005-2760(97)00108-2. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr The life of lipid droplets. Biochim Biophys Acta. 2009;1791:459–466. doi: 10.1016/j.bbalip.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kent C. Effects of altered phosphorylation sites on the properties of CTP:phosphocholine cytidylyltransferase. J Biol Chem. 1995;270:17843–17849. doi: 10.1074/jbc.270.30.17843. [DOI] [PubMed] [Google Scholar]
- Weinhold PA, Charles LG, Feldman DA. Microsomal CTP:choline phosphate cytidylyltransferase: kinetic mechanism of fatty acid stimulation. Biochim Biophys Acta. 1991;1086:57–62. doi: 10.1016/0005-2760(91)90154-a. [DOI] [PubMed] [Google Scholar]
- Weinhold PA, Charles L, Feldman DA. Regulation of CTP: phosphocholine cytidylyltransferase in HepG2 cells: effect of choline depletion on phosphorylation, translocation and phosphatidylcholine levels. Biochim Biophys Acta. 1994;1210:335–347. doi: 10.1016/0005-2760(94)90238-0. [DOI] [PubMed] [Google Scholar]
- Yao ZM, Jamil H, Vance DE. Choline deficiency causes translocation of CTP:phosphocholine cytidylyltransferase from cytosol to endoplasmic reticulum in rat liver. J Biol Chem. 1990;265:4326–4331. [PubMed] [Google Scholar]
- Zhang D, Tang W, Yao PM, Yang C, Xie B, Jackowski S, Tabas I. Macrophages deficient in CTP:phosphocholine cytidylyltransferasealpha are viable under normal culture conditions but are highly susceptible to free cholesterol-induced death. Molecular genetic evidence that the induction of phosphatidylcholine biosynthesis in free cholesterol-loaded macrophages is an adaptive response. J Biol Chem. 2000;275:35368–35376. doi: 10.1074/jbc.M007099200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.