

Abstract
Inflammation in the central nervous system (CNS) contributes to neurological disorders. Neuroinflammation involves the release of inflammatory molecules from glial cells such as astrocytes and microglia, and can lead to neuronal damage if unabated. In Multiple Sclerosis (MS), peripheral immune cells, including interferon-γ (IFN-γ)-producing Th1 cells, infiltrate the CNS and are important in shaping the inflammatory microenvironment, in part through cytokine-mediated interactions with glial cells. Recent evidence suggests that AMP-activated protein kinase (AMPK), a central regulator of energetic metabolism, can regulate inflammatory gene expression. In this study we have identified that IFN-γ induces biphasic AMPK signaling, suggestive of negative feedback mechanisms. Activation of AMPK suppresses several IFN-γ-induced cytokines and chemokines in primary astrocytes and microglia. IFN-γ regulates gene expression through activation of STAT1, and deletion of AMPK results in a marked increase in basal expression of STAT1. Conversely, activation of AMPK blocks IFN-γ-induced STAT1 expression. Deletion of AMPK leads to increased basal and IFN-γ-induced expression of inflammatory molecules including TNF-α, CXCL10 and CCL2. AMPK does not affect the phosphorylation of STAT1, but instead attenuates nuclear translocation of STAT1, DNA binding, and subsequent gene expression. In vivo, AMPK signaling during Experimental Autoimmune Encephalomyelitis (EAE), an animal model of MS, is down-regulated in the brain at onset and peak of disease. Diminution of AMPK signaling in vivo correlates with increased expression of IFN-γ and CCL2 in the CNS. Overall, these findings provide the first link between AMPK and STAT1, and may provide important clues about how bioenergetics and inflammation are linked.
Keywords: STAT1, astrocyte, microglia, cytokine, chemokine
INTRODUCTION
Control of inflammation in the CNS is a dynamic and essential process under physiological and pathological conditions. This complex process involves a host of cytokines, chemokines and reactive species produced by resident CNS glial cells such as astrocytes and microglia, and peripheral immune cells that invade the CNS (1). In the context of MS, a debilitating autoimmune disease involving demyelination of axons and neuronal degeneration, aberrant inflammation in the CNS contributes to disease pathology (2). IFN-γ-producing Th1 cells infiltrate the CNS leading to the activation of astrocytes and microglia which produce inflammatory molecules and chemoattractants including CCL2 and CXCL10, driving recruitment of additional cells such as macrophages, neutrophils and dendritic cells (3–6). As such, astrocytes and microglia, in response to cytokines including IFN-γ, have a central role in controlling inflammation in the CNS under neuroinflammatory conditions (7, 8).
IFN-γ is a multifaceted cytokine that is essential for proper immune function. IFN-γ is usually considered a pro-inflammatory cytokine produced by Th1 cells and natural killer (NK) cells that induces macrophage activation and upregulation of MHC molecules on antigen presenting cells (9). Beyond the well documented and critical role of IFN-γ in host defense, IFN-γ has a complex role in CNS pathology associated with inflammation. In MS, levels of IFN-γ mRNA in the cerebrospinal fluid (CSF) of MS patients correlate with disease (10), and treatment of MS patients with IFN-γ worsens disease (11), suggesting a pathogenic role. Despite this, studies in the MS animal model of EAE have demonstrated that blocking IFN-γ signaling exacerbates disease, suggesting a regulatory role (12–14). Thus, there is much interest in understanding the inflammatory and regulatory roles of IFN-γ (15). IFN-γ signals by binding to cell surface IFN-γ receptors (IFNGR1) leading to Janus Kinase 1/2 (JAK1/2)-dependent phosphorylation of STAT1 at tyrosine 701. Once phosphorylated, STAT1 homodimerizes and translocates to the nucleus where it associates with gene promoters containing GAS (gamma activated site) elements to drive expression of IFN-γ-responsive genes (16).
Adenosine monophosphate-activated protein kinase (AMPK) has been linked to the regulation of inflammatory signaling (17–24). AMPK is a heterotrimeric (α, β, and γ subunits) serine/threonine kinase critical to the maintenance of cellular energy homeostasis (25). The α subunit possesses the kinase activity and has two isoforms (AMPKα1 and AMPKα2) The β subunit functions as a scaffold molecule and the γ subunit binds AMP, ADP and ATP to detect the cellular energy state (26, 27). Increases in the AMP/ATP ratio leads to greater AMPK activity via direct allosteric activation and through a conformational change that blocks de-phosphorylation of threonine 172 (28–30). Liver kinase B1 (LKB1) is thought to be the predominant kinase responsible for the activating phosphorylation of AMPK (31); however, calmodulin-dependent protein kinase kinase-β can function as an alternative kinase to activate AMPK, independent of cellular energy content (32). Once active, AMPK phosphorylates many downstream effectors to reduce ATP consuming processes and promote ATP producing processes (33). Activation of AMPK increases the rate of glycolysis through phosphorylation of phosphofructokinase-2 (34), and decreases fatty acid synthesis through phosphorylation of acetyl-CoA carboxylase (ACC) (35). Activation of AMPK also alters the transcriptional program within the cell to provide an adaptive response in part through upregulation of PPARγ coactivator-1α (PGC-1α) (36, 37). AMPK activation has also been reported to have anti-inflammatory effects (18, 22, 24). For example, AMPK activation promotes macrophage polarization toward an anti-inflammatory M2 phenotype (22). AMPK may have a protective role in EAE as treatment of mice with the AMPK agonists AICAR or metformin attenuates EAE (38, 39) and deletion of AMPKα1 exacerbates disease (40). Further, AICAR suppresses CCL2 expression in the brain during EAE (39). AMPK modulates inflammatory gene expression through several mechanisms including suppression of NF-κB activation (41–43), phosphorylation of the transcriptional coactivator p300 (44), activation of SIRT1 (23) and in response to the combination of LPS and IFN-γ through the PI3K/Akt pathway (21). However, the role of AMPK in the regulation of the JAK/STAT1 pathway has not been examined.
In this study, we provide evidence that AMPK is a negative regulator of IFN-γ-induced gene expression. Activation of AMPK by culturing in low glucose or with the agonist AICAR suppressed IFN-γ-induced gene expression in astrocytes and microglia, while deletion of the catalytic AMPKα1 and AMPKα2 subunits promoted inflammatory gene expression. Our findings indicate that AMPK does not suppress tyrosine phosphorylation of STAT1 but rather regulates STAT1 nuclear localization in response to IFN-γ. Overall, our data suggest that AMPK is an important regulator of IFN-γ responses in the CNS.
MATERIALS AND METHODS
Reagents
Recombinant mouse IFN-γ was from R & D Systems (Minneapolis, MN). Antibodies (Ab) for P-AMPK, AMPK, P-ACC, P-STAT1, STAT1, P-STAT3, and STAT3 were from Cell Signaling (Beverly, MA); GAPDH Ab was from Abcam (Cambridge, MA); PE-conjugated anti-MHC class II Ab was from Southern Biotech (Birmingham, AL); and RNA Pol II Ab was from Covance (Princeton, NJ). Taqman primers were from Applied Biosystems (Forest City, CA). 5-Aminoimidazole-4-carboxamide-1-β-riboside, Z-Riboside (AICAR) was from EMD Millipore (Billerica, MA). AICAR was prepared fresh or as single use aliquots stored at −20°C for each experiment.
Mice and Glial Cell Preparations
C57BL/6 or AMPKα1fl/fl/AMPKα2fl/fl (45) mice were bred and housed in the animal facility at the University of Alabama in Birmingham under the care of the animal resources program. Primary murine astrocyte or microglial cultures were prepared as previously described (46, 47). Astrocytes or microglia were cultured in DMEM with 10% FBS, 16 mM HEPES, 1X non-essential amino acids, 2 mM L-Glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and 50 μg/ml gentamicin. Astrocytes were separated from microglia by shaking, and astrocyte cultures contained > 90% GFAP+ cells as determined by immunofluorescent microscopy (46). Following shaking, microglia containing supernatants were centrifuged at 1000 RPM for 5 min, and microglia were resuspended in fresh media and plated into 6-well plates.
EAE Induction and Assessment
Active EAE was induced as previously described (47). Eight- to twelve-week-old C57BL/6 mice were immunized s.c. with 200 μg of MOG35-55 emulsified in Complete Freund’s Adjuvant (supplemented with 2 mg/mL of Mycobacterium tuberculosis) and injected i.p. on days 0 and 2 with 500 ng pertussis toxin. Assessment of classical EAE was as follows: 0, no disease; 1, decreased tail tone; 2, hind limb weakness or partial paralysis; 3, complete hind limb paralysis; 4, front and hind limb paralysis; and 5, moribund state (48).
Immunoblotting
Cells were washed twice with PBS and lysed with IP lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, 1 mM phenylmethanesulfonyl fluoride, 25 μg/ml leupeptin, 25 μg/ml aprotinin and 1X phosphatase inhibitor cocktail (Pierce, Rockford, IL), as previously described (49). Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL). Equal amounts of protein from each sample were solubilized in Laemmli sample buffer (2% SDS) and heated for 5 min at 95°C. Proteins were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, the membranes were blocked in 5% milk followed by an overnight incubation at 4°C with primary Ab. Horseradish peroxidase-conjugated donkey anti-rabbit or donkey anti-mouse (1:4000 dilution) secondary Ab were incubated for 1 h at room temperature, followed by detection with enhanced chemiluminescence.
qRT-PCR
RNA was isolated using Trizol (Sigma-Aldrich) as previously described (50). RNA was quantified using a NanoDrop (NanoDrop Technologies, Wilmington DE). One μg of RNA was used for cDNA synthesis using MMLV reverse transcriptase (Promega, Madison WI). The cDNA was analyzed by quantitative PCR performed using Taqman gene expression assays according to the manufacturer’s instructions in an ABI Prismo 7500 (Applied Biosystems, Foster City, CA). Reactions were carried out in 20 μl and analyzed using the ΔΔCt method, as described previously (51).
Subcellular Fractionation
Nuclear and cytosolic fractions were prepared as previously described (52, 53) with minor modifications. Cells were washed twice with PBS and then harvested in 200 μl of lysis buffer (10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.05% Nonidet P-40, 1 mM EGTA, 1 mM sodium orthovanadate, 100 μM phenylmethanesulfonyl fluoride, 0.1 μM okadaic acid, 50 mM sodium fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin). Following lysis, the cells were centrifuged at 2700 × g for 10 min at 4°C. The supernatant was collected and centrifuged at 20,800 × g for 15 min at 4°C to obtain the cytosolic fraction. The pellet containing nuclei was washed twice in 200 μl of wash buffer (5 mM HEPES, pH 7.4, 3 mM MgCl2, 1 mM EGTA, 250 mM sucrose, 0.1% BSA, with protease and phosphatase inhibitors). The pellet was collected and the nuclei were centrifuged through a 1 M sucrose cushion (with protease and phosphatase inhibitors), at 2700 × g for 10 min at 4°C, washed in lysis buffer containing 0.05% Nonidet P-40 and the nuclei were resuspended in nuclear extraction buffer (20 mM HEPES, pH 7.9, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 100 μM phenylmethanesulfonyl fluoride, 0.1 μM okadaic acid, 50 mM sodium fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin), incubated for 30 min on ice followed by centrifugation at 20,800 × g for 15 min at 4°C, with the supernatant retained as the nuclear extract.
Chromatin immunoprecipitation
Primary astrocytes were treated as indicated and chromatin was precipitated overnight at 4°C with 5 μg of STAT1 Ab (Santa Cruz Biotechnologies, Santa Cruz, CA) or isotype-matched control IgG. The IgG/chromatin complexes were isolated with protein A coupled dynabeads (Invitrogen, Carlsbad, CA), washed extensively, and DNA eluted as previously described (54). DNA was purified using Qiagen PCR clean up columns. Purified DNA was analyzed by qPCR using sybr green (Qiagen, Germantown, MD) and the following primer pair for the CCL2 promoter: forward AGGTTCCTTGAGCCAGGGGCA, reverse AGTGCAGAGCCCTCGGGTGT.
Immunocytochemistry
Astrocytes were plated on poly-D-lysine-coated coverslips and treated as indicated followed by fixation in 4% paraformaldehyde for 20 min at 4°C, then washed with PBS. The cells were incubated in permeabilization buffer (PBS, pH 7.2, 0.5% Triton-X 100) for 10 min at room temperature. The cells were incubated for 1 h at room temperature in blocking buffer (PBS, pH 7.2, with 1% BSA, 0.2% non-fat milk). The cells were then incubated overnight with STAT1 Ab (Santa Cruz Biotechnologies, Santa Cruz, CA) diluted 1:50 in blocking buffer. The cells were washed several times with PBS and then incubated with Alexa 488-conjugated secondary Ab (Invitrogen, Carlsbad, CA) diluted 1:400 in blocking buffer for 1 h at room temperature, followed by thorough washing with PBS. Coverslips were affixed to glass slides with DAPI-containing mounting media. At least four random fields on each slide from two independent experiments were imaged using a Zeiss LSM 710 Confocal Microscope at the UAB High Resolution Imaging Facility. Nuclear and cytosolic intensities were quantified using ImageJ software.
Flow Cytometry
Cells were treated as indicated and collected by scraping in ice-cold PBS. Fc-receptors were blocked with blocking antibodies. Cells were then labeled with Alexa Fluor-conjugated anti-P-STAT1, P-STAT3 or P-STAT5 (Cell Signaling, Beverly, MA), according to the manufacturer’s protocol. For MHC staining, cells were incubated with PE-conjugated anti-MHC class II (BD Biosciences) followed by fixation in 2% paraformaldehyde. Data were collected on a BD LSR II flow cytometer, analyzed using FlowJo software and gates set based on isotype matched control antibodies.
Statistics
Data were analyzed by ordinary or repeated measures ANOVA with Student-Newman-Kuels post-test or by Student’s t test, as appropriate using Instat software. A value of p≤0.05 was considered statistically significant.
RESULTS
Energy restriction suppresses IFN-γ-induced gene expression
AMPK activation is altered by pro-inflammatory and anti-inflammatory stimuli including LPS, CNTF, IL-10 and TGF-β, among others (22, 55, 56). Here we tested if the prototypical Th1 cytokine IFN-γ could influence AMPK activity. The effects of IFN-γ were tested under two conditions, one in which AMPK activation is relatively low (high energy) and one in which AMPK activation is higher (low energy). Primary murine astrocytes cultured in normal media (high energy, 25 mM glucose/1 mM pyruvate) were stimulated with IFN-γ for 10 min to 6 h, followed by examination of AMPK and ACC phosphorylation. IFN-γ induced a biphasic response, with an initial reduction in AMPK phosphorylation at 10 and 30 min, followed by increased phosphorylation at 6 h (Figure 1A). The reduction in AMPK phosphorylation paralleled STAT1 activation (P-STAT1) and preceded IFN-γ-induced STAT1 protein expression (Figure 1A). IFN-γ stimulation significantly reduced cytosolic AMPK phosphorylation but did not affect nuclear AMPK phosphorylation under high energy conditions (Figure 1B). The inhibition of cytoplasmic AMPK phosphorylation is similar to that reported following LPS treatment of macrophages (22, 57), and the bi-phasic nature is suggestive of a negative feedback response. In cells cultured with no glucose and no pyruvate (low energy, 24 h no glucose*/no pyruvate) (*serum contributes approximately 0.6 mM glucose), conditions which stimulate AMPK activity, IFN-γ did not stimulate a biphasic response but instead increased AMPK phosphorylation throughout the time course (Figure 1A). Consistent with AMPK activation, IFN-γ stimulated rapid and prolonged phosphorylation of the AMPK substrate ACC under low energy conditions (Figure 1A). Low energy did not alter IFN-γ-induced STAT1 phosphorylation but did slightly suppress IFN-γ-induced STAT1 expression at 6 h compared to high energy conditions. Together, these data indicate that cytosolic AMPK responds to IFN-γ, and that this response is dependent on the energetic state of the cell.
Figure 1. AMPK Activation is Altered by IFN-γ and Energy Restriction Dampens IFN-γinduced Gene Expression.

A. Primary astrocytes were stimulated with IFN-γ (10 ng/ml) under high energy (25 mM glucose/1 mM pyruvate) and low energy conditions (24 h no glucose#/no pyruvate) followed by immunoblotting for P-AMPK, P-ACC, AMPKα, P-STAT1 and STAT1. B. Astrocytes were stimulated with IFN-γ (10 ng/ml) for 30 min followed by isolation of cytosolic and nuclear fractions and immunoblotting for P-AMPK, AMPK and GAPDH. C. Astroyctes were cultured as in A, were left untreated (UT) or stimulated with IFN-γ (10 ng/ml, 4 h), and gene expression was analyzed by qRT-PCR. #Media contains an additional ~0.6 mM glucose from serum. N=3, *p < 0.05.
As shown in Figure 1A, IFN-γ activates AMPK under reduced glucose/pyruvate conditions (low energy) and active AMPK has been reported to have anti-inflammatory properties (18, 20–22, 24). Therefore, we hypothesized that the cellular response to IFN-γ under these conditions would be reduced. To examine if disruption of energy metabolism could influence IFN-γ-induced gene expression, astrocytes were cultured under high energy or low energy conditions as in Figure 1C followed by stimulation with IFN-γ. Energy restriction attenuated IFN-γ-induced CCL2 and CXCL10 expression, and modestly reduced STAT1 gene expression. These data indicate that reduced energy substrate availability correlates with impaired IFN-γ-induced gene expression, and may point toward a role for AMPK in coupling bioenergetic capacity to IFN-γ-dependent inflammatory responses.
AICAR suppresses IFN-γ-induced gene expression
To test directly if AMPK could influence IFN-γ signaling, the AMPK agonist AICAR was used to induce activation of AMPK. AICAR treatment stimulated the activation of AMPK as evidenced by increased phosphorylation of AMPK and ACC (Figure 2A). AICAR-induced activation of AMPK suppressed IFN-γ-induced STAT1 expression at 8 and 16 h (Figure 2A). This regulation occurs at the mRNA level as AICAR also significantly suppressed IFN-γ-induced STAT1 mRNA in a concentration-dependent fashion (Figure 2B). To determine if forced activation of AMPK could also suppress IFN-γ-induced gene expression, astrocytes were stimulated with IFN-γ in the absence or presence of AICAR. IFN-γ increased the expression of CIITA, CXCL10, CCL2 and iNOS, and the expression of each was blocked by AICAR (Figure 2C). To test if AMPK impairs STAT1 transcriptional activity, we examined the influence of AICAR on an IFN-γ-inducible luciferase reporter (GAS-Luc) (58). Forced activation of AMPK attenuated IFN-γ-induced GAS-Luc expression by approximately 50% (Figure 2D). Next, we examined if AICAR could block STAT1 binding to the endogenous CCL2 promoter. As shown in Figure 2E, CCL2 is a STAT1 dependent gene as STAT1 siRNA effectively reduces STAT1 expression and blocks IFN-γinduced CCL2 expression. AICAR blocked basal and IFN-γ-induced STAT1 binding to the CCL2 promoter (Figure 2F). Additionally, the bi-phasic response of AMPK to IFN-γ and the findings that active AMPK antagonizes IFN-γ-induced gene expression supports the hypothesis that AMPK has a negative feedback role in IFN-γ signaling. We next examined if activation of AMPK could influence the function of other STAT family members. Stimulation of astrocytes with Oncostatin M (OSM), which activates STAT3, led to SOCS3 expression that was blocked by AICAR (Figure 2G). The pan-JAK inhibitor, P6, also blocked SOCS3 expression, confirming this gene is dependent on JAK/STAT signaling (Figure 2G). Stimulation of astrocytes with granulocyte macrophage-colony stimulating factor (GM-CSF), which activates STAT5, led to CCL2 expression that was blocked by both AICAR and P6 (Figure 2H). These data indicate that AMPK influences the function of multiple STAT family members.
Figure 2. AMPK Activation Attenuates IFN-γ-induced Inflammatory Gene Expression.

A and B. Astrocytes were stimulated with IFN-γ (10 ng/ml) in the absence or presence of the AMPK agonist AICAR (1 mM) for the indicated times, and STAT1 expression examined by immunoblotting and qRT-PCR. C. Astrocytes were stimulated with IFN-γ (10 ng/ml) for 4 h in the absence or presence of AICAR and gene expression analyzed by qRT-PCR. D. Astroyctes were transfected with an IFN-γ inducible reporter (GAS-Luciferase) followed by stimulation with IFN-γ (10 ng/ml, 16 h) with or without 1 mM AICAR. E. Astrocytes were transfected with control or STAT1 siRNA, then stimulated with IFN-γ (10 ng/ml, 4 h), and STAT1 and CCL2 levels measured by qRT-PCR. F. Astrocytes were stimulated with IFN-γ (10 ng/ml, 30 min) in the absence or presence of AICAR (1 mM), followed by chromatin immunoprecipitation with antibodies to STAT1 or control IgG. The precipitated DNA was then analyzed by PCR using primers directed to the STAT1 binding region of murine CCL2. G. and H. Astrocytes were stimulated with OSM (1 ng/ml) or GM-CSF (10 ng/ml) for 4 h in the absence or presence of AICAR (1 mM) or P6 (0.5 μM) and gene expression analyzed by qRT-PCR. N= 3, *p < 0.05.
Deletion of AMPKα1/α2 leads to increased inflammatory gene expression in primary astrocytes
Activation of AMPK impairs IFN-γ-induced gene expression, therefore we examined if genetic disruption of the AMPK pathway would have the opposite effect. Both catalytic AMPKα isoforms (AMPKα1 and AMPKα2) are expressed throughout the CNS and in astrocytes and microglia (Figure 3A). Therefore, astrocytes were isolated from mice in which both AMPKα1 and AMPKα2 are floxed (45) (referred to as AMPKαfl/fl). Primary astrocytes were isolated from AMPKαfl/fl mice and transduced with GFP or GFP-Cre recombinase adenovirus. As shown in Figure 3B, AMPKα is effectively deleted by Cre. The Ab used detects both AMPKα1 and AMPKα2. To confirm that Cre-mediated deletion of AMPKα correlates with reduced activity, astrocytes were treated with AICAR to activate AMPK; such treatment increased phosphorylation of the AMPK substrate ACC, and this was attenuated in cells with reduced AMPKα (Figure 3B).
Figure 3. Deletion of AMPKα1 and AMPKα2 in Primary Astrocytes Enhances STAT1 Expression.

A. RNA was isolated from brain (cerebrum), spinal cord and primary cultures of astrocytes or microglia. The expression of both catalytic AMPKα isoforms and the astrocyte marker GFAP was analyzed by RT-PCR. B. Isolated AMPKαfl/fl astrocytes were transduced with GFP or GFP-Cre adenovirus and stimulated with AICAR (1 mM, 1 h) 5 days post transduction followed by immunoblotting. C. Isolated AMPKαfl/fl astrocytes were transduced with GFP or GFP-Cre adenovirus, and STAT1 expression quantified by immunoblotting 5 days post transduction. D. Isolated AMPKαfl/fl astrocytes were transduced with GFP or GFP-Cre adenovirus, stimulated with IFN-γ (10 ng/ml, 4 h) 5 days post transduction, and inflammatory gene expression examined by qRT-PCR. E. The effect of AICAR on STAT1 expression is attenuated in cells lacking AMPKα. Isolated AMPKαfl/fl astrocytes were transduced with GFP or GFP-Cre adenovirus followed by treatment with IFN-γ (10 ng/ml, 16 h) in the absence or presence of AICAR (0.5 mM) and STAT1 expression measured by immunoblotting. N=3, *p < 0.05.
As shown in Figure 3C, Cre-mediated deletion of AMPKα led to a significant increase in total levels of STAT1 protein. Consistent with elevated STAT1 expression, several inflammatory genes were elevated in astrocytes with reduced AMPKα under basal conditions, including CCL2, CXCL10, IL-6, TNF-α and iNOS (Figure 3D). These findings are in line with previous reports that disruption of AMPK signaling increases inflammatory gene expression (17, 23). In addition, disruption of AMPKα enhanced a subset of IFN-γ-induced genes including CCL2, CXCL10 and iNOS. In the case of CCL2 and CXCL10, this enhancement may be due to increased basal expression. IL-6 and TNF-α were elevated in the absence of stimulus and were not further increased by IFN-γ in AMPKα deficient astrocytes. Unexpectedly, the IFN-γ-induced expression of CIITA was reduced by deletion of AMPKα, though basal expression was unaffected (Figure 3D). We next confirmed that the actions of AICAR were dependent on AMPK. Astrocytes (AMPKαfl/fl) were transduced with GFP or GFP-Cre followed by stimulation with IFN-γ in the absence or presence of AICAR. In cells transduced with GFP (left) and in cells transduced with GFP-Cre (reduced AMPKα, right), IFN-γ stimulated STAT1 expression. AICAR reduced IFN-γ-induced STAT1 by 36.5% ± 3.4% in the GFP cells, and by 18.5% ± 5.7% (n=3, p<0.05) in cells with reduced AMPKα (Figure 3E), indicating that the AICAR-mediated reduction in STAT1 occurs in an AMPK-dependent manner. Collectively, these data indicate that AMPK signaling controls basal and IFN-γ-induced STAT1 and STAT1-dependent gene expression.
AMPK controls IFN-γ-induced gene expression in primary microglia
Microglia (resident brain macrophages) have a profound influence on the inflammatory environment in the healthy and diseased brain (8). To examine if AMPK also regulates microglial responses to IFN-γ, primary microglia were stimulated with IFN-γ in the absence or presence of AICAR. AICAR activated AMPK, as demonstrated by increased phosphorylation of ACC (Figure 4A) and markedly reduced IFN-γ-induced STAT1 expression (Figure 4A, lane 3), consistent with our findings in primary astrocytes. AICAR-induced activation of AMPK also suppressed IFN-γ-induced CXCL10, TNF-α and iNOS mRNA expression, as well as reduced MHC class II expression on the surface of microglia (Figure 4B). The reduction in MHC class II expression was anticipated due to the previous observations that AICAR attenuated IFN-γ-induced CIITA expression in astrocytes (Figure 2C) and microglia (not shown). These data suggest that AMPK controls IFN-γ-induced inflammation in microglia.
Figure 4. AMPK Regulates STAT1 in Microglia.

A. Primary microglia were stimulated with IFN-γ (10 ng/ml, 16 h) in the absence or presence of AICAR (1 mM), and STAT1 expression examined by immunoblotting. B. Primary microglia were stimulated with IFN-γ (10 ng/ml, 4 h) in the absence or presence of AICAR (1 mM), and CXCL10, TNF-α and iNOS gene expression measured by qRT-PCR, and MHC class II expression (IFN-γ 10 ng/ml, 24 h) measured by flow cytometry. N=3, *p < 0.05.
AMPK attenuates STAT1 nuclear localization
IFN-γ elicits the activation of STAT1 by stimulating sequential phosphorylation, nuclear translocation and DNA binding (16). Activation of AMPK blocks STAT1 DNA binding and STAT-1 induced gene expression (Figure 2). To determine at what point AMPK interferes with the STAT1 signaling cascade, STAT1 phosphorylation and nuclear translocation were examined. Primary astrocytes were stimulated with IFN-γ in the absence or presence of AICAR followed by quantification of STAT-1 tyrosine phosphorylation by immunoblot and flow cytometry; AICAR did not significantly affect IFN-γinduced STAT1 phosphorylation (Figures 5A and B). Additionally, AICAR did not alter OSM-induced STAT3 phosphorylation or GM-CSF-induced STAT5 phosphorylation (Figure 5B). To test if AMPK influences STAT1 nuclear translocation, astrocytes were treated with IFN-γ in the absence or presence of AICAR followed by isolation of cytosolic and nuclear fractions. IFN-γ stimulated a robust increase in nuclear P-STAT1 (Figure 5C, lane 6) with a concomitant decrease in cytosolic levels of total STAT1 (lane 2). AICAR significantly attenuated total STAT1 nuclear accumulation (lane 7) and retained STAT1 in the cytosol (lane 3). Consistent with the reduction in total STAT1 in the nucleus, IFN-γ-induced nuclear P-STAT1 was also reduced by AICAR by approximately 40% (Figure 5C). Increased phosphorylation of AMPK confirms that AICAR activated AMPK. RNA pol II and GAPDH are shown as nuclear and cytosolic markers, respectively (Figure 5C). These data were confirmed by immunoflorescence microscopy. AICAR significantly attenuated the level of IFN-γ-induced nuclear STAT1 (Figures 5D and E). IFN-γ stimulated nuclear localization of STAT1 (determined by a nuclear/cytosolic staining intensity > 2) in 54% of the cells, and this was reduced to approximately 11% by co-treatment with AICAR (Figure 5E). As shown in Figure 5F, astrocytes with reduced AMPKα expression have increased levels of IFN-γ-induced nuclear STAT1 (Figure 5F, lane 8 vs. lane 6). In addition, the basal nuclear STAT1 is also elevated in astrocytes with reduced AMPK (STAT1 long exposure) (Figure 5F, lane 7 vs. lane 5). These data, together with the data in Figures 5C, D and E, suggest that AMPK impedes STAT1 nuclear localization as a means to attenuate IFN-γ-induced gene expression.
Figure 5. AMPK Signaling Does Not Influence STAT1 Phosphorylation but Attenuates Nuclear Translocation.

A. Astrocytes were stimulated with IFN-γ (10 ng/ml, 30 min) in the absence or presence of AICAR (1 mM) followed by quantification of immunoblots for P-STAT1 and STAT1. Values are expressed as the ratio of P-STAT1 to total STAT1. B. Astrocytes were stimulated with IFN-γ (10 ng/ml, 30 min), OSM (1 ng/ml, 30 min) or GM-CSF (10 ng/ml, 30 min) in the absence or presence of AICAR (1 mM) and P-STAT1, P-STAT3 and P-STAT5 measured by flow cytometry. C. Astrocytes were stimulated with IFN-γ (10 ng/ml) for 30 min in the absence or presence of AICAR (1 mM) followed by fractionation into cytosolic and nuclear fractions. STAT1, P-STAT1, AMPKα and P-AMPK subcellular distribution was examined by immunoblotting. The results for total STAT1 were quantified. The separation of GAPDH and RNA pol II demonstrates efficient separation of cytosolic and nuclear fractions, respectively. D and E. STAT1 localization was examined by immunofluorescent microscopy and quantified. The percentage of cells with nuclear/cytosolic STAT1 > 2 is reported in the table. F. AMPKαfl/fl astrocytes were transduced with GFP or GFP-Cre adenovirus followed by stimulation with IFN-γ (10 ng/ml, 30 min) and subcellular fractionation. N=3, *p < 0.05.
Reduced AMPK phosphorylation correlates with disease severity and inflammatory gene expression in EAE
To extend our findings in vivo, we examined the disease model of EAE. C57BL/6 mice were immunized with MOG peptide to induce EAE and disease severity scored as previously described (48) (Figure 6A). Brain tissue was collected at several time points during the course of disease to examine AMPK and gene expression. Consistent with previous reports (40), phosphorylated and total levels of AMPK are diminished during onset and peak of disease (days 15 and 20), but increased during remission (days 25 and 30) (Figure 6B). The changes in AMPK signaling coincided with expression of IFN-γ and the IFN-γ-induced chemokine CCL2 in the brain (Figure 6C). These data confirm that AMPK signaling is altered in the brain during EAE, and collectively suggest that AMPK and IFN-γ signaling functionally interact in the CNS.
Figure 6. Reduced AMPK Signaling Correlates With EAE Disease Severity and Inflammatory Gene Expression.
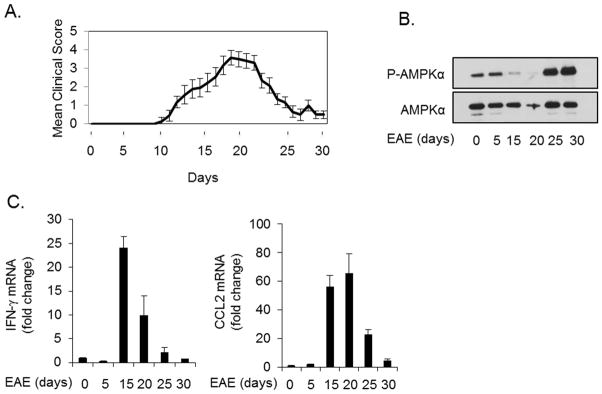
A. C57Bl/6 mice were immunized with MOG35-55 peptide to induce EAE and mice were scored daily starting at day 5. B. AMPKα phosphorylation in the brain during EAE was measured by immunoblotting. C. Brains from EAE mice were analyzed over time for IFN-γ and CCL2 expression by qRT-PCR.
Discussion
IFN-γ is a proinflammatory cytokine essential for antiviral responses, and activation/polarization of macrophages and T cells, and acts in synergy with a number of cytokines, placing it as a central cytokine controlling innate and adaptive immunity (9, 59, 60). IFN-γ elicits rapid STAT1-dependent and STAT1-independent transcriptional reprogramming leading to the expression of over 200 genes as part of an inflammatory and adaptive response (9, 16). Several studies using various inflammatory stimuli have shown that the energy sensing enzyme AMPK has anti-inflammatory actions (17–24). The anti-inflammatory actions of AMPK are thought to be largely mediated by negative regulation of the NF-κB pathway (18, 39, 41–43). The current study reveals that AMPK regulates IFN-γ-induced gene expression, including CCL2, CXCL10, iNOS and STAT1, in glial cells through modulation of STAT1.
Our findings indicate that IFN-γ stimulates a rapid and transient inhibition of cytoplasmic AMPK followed by activation at later time points. This suggests a negative feedback model in which AMPK is repressed by IFN-γ when cellular energy is abundant to allow a rapid and robust inflammatory response. As the IFN-γ response becomes prolonged (>6 h) and the energetic cost of synthesizing and secreting cytokines and chemokines becomes greater, the cell must signal for the conservation of ATP. Maintaining energetic homeostasis is essential as catastrophic energy depletion leads to necrotic cell death (61). Eukaryotes evolved the AMPK system to effectively manage energy production and utilization (33). Using AMPK to negatively regulate STAT1-induced gene expression allows the cell to mount an inflammatory response that is appropriate for the bioenergetic status of the cell. Consistent with this concept, if cells are exposed to IFN-γ when energy is limited, as in Figure 1A, AMPK is rapidly activated and the IFN-γ response is muted compared to cells cultured with high glucose and pyruvate (i.e. typical cell culture media). Collectively, our findings suggest that AMPK couples bioenergetic capacity with the STAT1-dependent IFN-γ response.
In support of this hypothesis, forced activation of AMPK with AICAR suppresses IFN-γinduced STAT1 expression. It is known that AMPK activation inhibits protein synthesis (62), and while this may partly account for suppression of STAT1, the data point toward a more selective effect than general repression of protein synthesis. Importantly, AICAR suppressed IFN-γ-induced STAT1 and other inflammatory molecules at the transcriptional level. Additionally, activation of AMPK attenuated nuclear localization of STAT1 and prevented STAT1 promoter interactions. These data suggest a selective effect of AMPK on STAT1 independent of the influence of AMPK on translation.
AMPK has been reported to regulate IL-6 signaling through suppression of STAT3 phosphorylation (20). In contrast, we observed that basal and IFN-γ-induced STAT1 phosphorylation was independent of AMPK activity. This indicates that AMPK does not regulate the upstream JAKs or recruitment of STAT1 to the IFN-γ receptor, prerequisites for IFN-γinduced phosphorylation of STAT1. The molecular mechanism by which AMPK regulates nuclear levels of STAT1 is currently unknown. AMPK has been previously shown to regulate nuclear levels of the RNA-binding protein HuR through modulation of importin alpha1; however in this context AMPK promoted nuclear localization (63, 64). STAT1 localization is facilitated by phosphotyrosine-dependent interactions with importin alpha5 (karyopherin α1) (65), and there are no reports thus far of a functional interaction between AMPK and importin alpha5. However, the possibility remains that AMPK may disrupt the interaction between STAT1 and importin alpha5. Another more generalized possibility is that AMPK may suppress an ATP-consuming step required for nuclear import as a mechanism to conserve ATP, thus blocking nuclear localization of a subset of proteins. Our findings suggest that active AMPK blocks STAT1 nuclear localization. However, there appears to be a significant fraction of activated AMPK in the nucleus (Figures 1B and 5C), and it remains possible that active AMPK is facilitating nuclear export of STAT1. Alternatively, AMPK modulates p27 localization through phosphorylation and cytoplasmic sequestration (66). Thus, a similar mechanism may exist for AMPK and STAT1. Additionally, the AMPK-dependent reduction in nuclear P-STAT1 appeared, qualitatively, less robust than AMPK-mediated suppression of gene expression. This suggests that AMPK may suppress gene expression through multiple mechanisms in addition to regulation of STAT1 nuclear localization.
AICAR is an AMP mimetic and thus has the potential to influence other enzymes sensitive to AMP levels. Recently, a small molecule AMPK agonist A769662 was described (67). This molecule selectively activates AMPK heterotrimers containing the AMPKβ1 subunit (68). We observed that A769662 was less effective than AICAR in suppressing IFN-γ-induced gene expression (not shown), suggesting that the AMPKβ1-containing pool of AMPK is either not involved in this regulation or that astrocytes have low levels of AMPKβ1. In almost every case, IFN-γ-induced genes that were inhibited by AICAR were upregulated by deletion of AMPKα, validating that these genes were regulated by AMPK. The exception was CIITA which was inhibited by AICAR-induced activation of AMPK and inhibited in cells with reduced AMPKα. Reports have indicated that AICAR can influence inflammatory signaling in an AMPK-independent fashion (69, 70). Therefore, it is possible that the AICAR-dependent influence on CIITA is independent of AMPK. This may also suggest that perturbations, either positive or negative, in AMPK activity can influence IFN-γ-induced CIITA expression. Additional studies are needed to fully determine the role of AMPK on CIITA and the CIITA-dependent MHC class II expression.
In the current study as well as others (40) it was demonstrated that activation associated phosphorylation of AMPK is markedly reduced in the brain during the onset and peak of EAE. Moreover, our results show that this reduction in AMPK activation correlates with elevated levels of IFN-γ and CCL2 in the CNS. This supports our findings that IFN-γ can modulate AMPK in a bi-phasic fashion, and that AMPK is a negative regulator of CCL2 expression. However, in this in vivo setting of a complex inflammatory milieu, IFN-γ is almost certainly not the sole determinant modulating AMPK activity. Our findings support the hypothesis that suppression of AMPK promotes a robust inflammatory response and that subsequent activation dampens this response; and is consistent with previous in vivo studies which demonstrated that the AMPK agonist and anti-diabetic drug Metfomin maintains AMPK activity in the brain, suppress inflammatory gene expression and attenuates EAE (38). The results presented here further support AMPK activation as a therapeutic anti-inflammatory target, and indicate that modulation of the IFN-γ/STAT1 axis is involved in this process.
Acknowledgments
We gratefully acknowledge Dr. Sean Morrison (University of Michigan/HHMI) for generously providing the AMPKα1fl/fl, AMPKα2fl/fl mice. Fluorescence microscopy was conducted by Shawn Williams at the UAB High Resolution Imaging Facility. Flow cytometry was conducted by Marion Spell at the UAB Center for AIDS Research Core Facility, an NIH funded program (P30 AI027767). We thank Dr. Lora L. Yanagisawa for expert technical assistance with mice.
This work was supported in part by the National Institutes of Health grants NS57563 (E.N.B.), a Collaborative MS Research Center Award from the National Multiple Sclerosis Society CA1059-A-13 (E.N.B.) and a Career Transition Award from the National Multiple Sclerosis Society TA3050-A-1 (G.P.M).
References
- 1.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman L. Multiple sclerosis: a two-stage disease. Nat Immunol. 2001;2:762–764. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- 3.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 4.Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of the interferon-gamma-inducible chemokines IP-10 and Mig and their receptor, CXCR3, in multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2000;26:133–142. doi: 10.1046/j.1365-2990.2000.026002133.x. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miljkovic D, Timotijevic G, Mostarica Stojkovic M. Astrocytes in the tempest of multiple sclerosis. FEBS Lett. 2011;585:3781–3788. doi: 10.1016/j.febslet.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 8.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 9.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi PA, Tranquill LR, McFarland HF, Cowan EP. Cytokine gene expression in cells derived from CSF of multiple sclerosis patients. J Neuroimmunol. 1998;89:198–205. doi: 10.1016/s0165-5728(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 11.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 12.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 13.Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol. 1999;163:5278–5286. [PubMed] [Google Scholar]
- 14.Sabatino JJ, Jr, Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. J Immunol. 2008;180:4451–4457. doi: 10.4049/jimmunol.180.7.4451. [DOI] [PubMed] [Google Scholar]
- 15.Muhl H, Pfeilschifter J. Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int Immunopharmacol. 2003;3:1247–1255. doi: 10.1016/S1567-5769(03)00131-0. [DOI] [PubMed] [Google Scholar]
- 16.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 17.Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, Izon D, Honeyman J, Chen ZP, van Denderen BJ, Kemp BE, Steinberg GR. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- 20.Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, Mahlapuu M. AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3) Diabetologia. 2010;53:2406–2416. doi: 10.1007/s00125-010-1856-z. [DOI] [PubMed] [Google Scholar]
- 21.Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, Reilly CM. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. 2009;156:542–551. doi: 10.1111/j.1365-2249.2009.03924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sag D, Carling D, Stout R, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285:19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardie D. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 27.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 28.Sanders M, Grondin P, Hegarty B, Snowden M, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hawley S, Boudeau J, Reid J, Mustard K, Udd L, Mäkelä T, Alessi D, Hardie D. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong S, Leiper F, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw R, Kosmatka M, Bardeesy N, Hurley R, Witters L, DePinho R, Cantley L. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hawley S, Pan D, Mustard K, Ross L, Bain J, Edelman A, Frenguelli B, Hardie D. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Hardie D. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008;32(Suppl 4):S7–12. doi: 10.1038/ijo.2008.116. [DOI] [PubMed] [Google Scholar]
- 34.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 35.Winder W, Wilson H, Hardie D, Rasmussen B, Hutber C, Call G, Clayton R, Conley L, Yoon S, Zhou B. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J Appl Physiol. 1997;82:219–225. doi: 10.1152/jappl.1997.82.1.219. [DOI] [PubMed] [Google Scholar]
- 36.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun. 2002;296:350–354. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 37.Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, Criswell DS. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
- 40.Nath N, Khan M, Rattan R, Mangalam A, Makkar RS, de Meester C, Bertrand L, Singh I, Chen Y, Viollet B, Giri S. Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem Biophys Res Commun. 2009;386:16–20. doi: 10.1016/j.bbrc.2009.05.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cacicedo JM, Yagihashi N, Keaney JF, Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–1209. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 42.Morizane Y, Thanos A, Takeuchi K, Murakami Y, Kayama M, Trichonas G, Miller J, Foretz M, Viollet B, Vavvas DG. AMP-activated protein kinase suppresses matrix metalloproteinase-9 expression in mouse embryonic fibroblasts. J Biol Chem. 2011;286:16030–16038. doi: 10.1074/jbc.M110.199398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, Chen CG, Kemp BE, Power DA. 5-aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-kappaB. Immunol Cell Biol. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Qiu J, Wang X, Xia M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler Thromb Vasc Biol. 2011;31:2897–2908. doi: 10.1161/ATVBAHA.111.237453. [DOI] [PubMed] [Google Scholar]
- 45.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- 49.Meares GP, Zmijewska AA, Jope RS. Heat shock protein-90 dampens and directs signaling stimulated by insulin-like growth factor-1 and insulin. FEBS Lett. 2004;574:181–186. doi: 10.1016/j.febslet.2004.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol. 2010;184:4898–4906. doi: 10.4049/jimmunol.1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao X, Laver T, Hong SW, Twitty GB, Jr, Devos A, Devos M, Benveniste EN, Nozell SE. An NF-kappaB p65-cIAP2 link is necessary for mediating resistance to TNF-alpha induced cell death in gliomas. J Neurooncol. 2011;102:367–81. doi: 10.1007/s11060-010-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bijur G, Jope R. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem. 2007;282:16989–17001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nozell S, Laver T, Moseley D, Nowoslawski L, De Vos M, Atkinson GP, Harrison K, Nabors LB, Benveniste EN. The ING4 tumor suppressor attenuates NF-kappaB activity at the promoters of target genes. Mol Cell Biol. 2008;28:6632–6645. doi: 10.1128/MCB.00697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med. 2006;12:541–548. doi: 10.1038/nm1383. [DOI] [PubMed] [Google Scholar]
- 56.Steinberg GR, Watt MJ, Febbraio MA. Cytokine Regulation of AMPK signalling. Front Biosci. 2009;14:1902–1916. doi: 10.2741/3350. [DOI] [PubMed] [Google Scholar]
- 57.Tadie JM, Bae HB, Deshane JS, Bell CP, Lazarowski ER, Chaplin DD, Thannickal VJ, Abraham E, Zmijewski JW. TLR4 engagement inhibits AMPK activation through a HMGB1 dependent mechanism. Mol Med. 2012;18:659–68. doi: 10.2119/molmed.2011.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wesemann DR, Qin H, Kokorina N, Benveniste EN. TRADD interacts with STAT1-alpha and influences interferon-gamma signaling. Nat Immunol. 2004;5:199–207. doi: 10.1038/ni1025. [DOI] [PubMed] [Google Scholar]
- 59.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 60.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Lieberthal W, Menza SA, Levine JS. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am J Physiol. 1998;274:F315–327. doi: 10.1152/ajprenal.1998.274.2.F315. [DOI] [PubMed] [Google Scholar]
- 62.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 63.Wang W, Fan J, Yang X, Fürer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas S, Foufelle F, Hardie D, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang W, Yang X, Kawai T, López de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Köhler M, Gorospe M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: involvement in the nuclear import of RNA-binding protein HuR. J Biol Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 65.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997;16:7067–7077. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Short JD, Houston KD, Dere R, Cai SL, Kim J, Johnson CL, Broaddus RR, Shen J, Miyamoto S, Tamanoi F, Kwiatkowski D, Mills GB, Walker CL. AMP-activated protein kinase signaling results in cytoplasmic sequestration of p27. Cancer Res. 2008;68:6496–6506. doi: 10.1158/0008-5472.CAN-07-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 68.Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, Kemp BE. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–1230. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Glund S, Treebak JT, Long YC, Barres R, Viollet B, Wojtaszewski JF, Zierath JR. Role of adenosine 5′-monophosphate-activated protein kinase in interleukin-6 release from isolated mouse skeletal muscle. Endocrinology. 2009;150:600–606. doi: 10.1210/en.2008-1204. [DOI] [PubMed] [Google Scholar]
- 70.Kuo CL, Ho FM, Chang MY, Prakash E, Lin WW. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem. 2008;103:931–940. doi: 10.1002/jcb.21466. [DOI] [PubMed] [Google Scholar]