

Abstract
mTOR is activated in epilepsy, but the mechanisms of mTOR activation in post-traumatic epileptogenesis are unknown. It is also not clear whether mTOR inhibition has an anti-epileptogenic, or merely anticonvulsive effect. The rat hippocampal organotypic culture model of post-traumatic epilepsy was used to study the effects of long-term (four weeks) inhibition of signaling pathways that interact with mTOR. Ictal activity was quantified by measurement of lactate production and electrical recordings, and cell death was quantified with lactate dehydrogenase (LDH) release measurements and Nissl-stained neuron counts. Lactate and LDH measurements were well correlated with electrographic activity and neuron counts, respectively. Inhibition of PI3K and Akt prevented activation of mTOR, and was as effective as inhibition of mTOR in reducing ictal activity and cell death. A dual inhibitor of PI3K and mTOR, NVP-BEZ235, was also effective. Inhibition of mTOR with rapamycin reduced axon sprouting. Late start of rapamycin treatment was effective in reducing epileptic activity and cell death, while early termination of rapamycin treatment did not result in increased epileptic activity or cell death. The conclusions of the study are as follows: (1) the organotypic hippocampal culture model of post-traumatic epilepsy comprises a rapid assay of anti-epileptogenic and neuroprotective activities and, in this model (2) mTOR activation depends on PI3K-Akt signaling, and (3) transient inhibition of mTOR has sustained effects on epilepsy.
Introduction
Epileptogenesis, or the development of epilepsy, after brain injury is characterized by a gradual and continual increase in seizure probability (Salazar et al., 1985; Williams et al., 2009; Raymont et al., 2010). One of the characteristics of the epileptic brain is axon sprouting that has been best described at the mossy fiber pathway in the hippocampus (Cronin and Dudek, 1988; Sutula et al., 1989; Okazaki et al., 1995).
The mTOR pathway is transiently activated after experimental epileptogenic brain injuries (Buckmaster et al., 2009; Zeng et al., 2009; Huang et al., 2010) and in the genetic syndrome of tuberous sclerosis, in which intractable epilepsy is a prominent feature (Crino, 2011; McDaniel and Wong, 2011; Staley et al., 2011). mTOR inhibition suppresses mossy fiber sprouting in models of acquired epilepsy (Buckmaster et al., 2009) and reduces spontaneous seizures in the kainate model of chronic epilepsy (Zeng et al., 2009; Huang et al., 2010). Anti-epileptogenic effects were more variable in the pilocarpine model (Buckmaster et al., 2009; Tang et al., 2012) and the acute kindling model (Sliwa et al., 2012; van Vliet et al., 2012).
Unlike traditional anticonvulsants, rapamycin has no immediate effects on seizures but rather alters hippocampal circuitry (McDaniel and Wong, 2011). mTOR inhibition could prevent or reverse post-traumatic changes in brain circuitry that lead to an epileptic state (Crino, 2011; Lew and Buckmaster, 2011; McDaniel and Wong, 2011). However, rapamycin, the drug that is widely used to inhibit mTOR, is a potent immunosuppressant (Chueh and Kahan, 2005), and the mTOR pathway has a broad role in regulation of protein synthesis and other growth-related cellular processes (Sarbassov et al., 2005; Caron et al., 2010). To develop more specific treatments, a better understanding of mTOR activation in epilepsy is needed.
The mTOR pathway has been extensively studied in cancer (Chiang and Abraham, 2007; Guertin and Sabatini, 2007; Engelman, 2009; Borders et al., 2010). Among regulators of mTOR containing, rapamycin-sensitive protein complex (mTORC1) are Raf kinase (RAF) mitogen-activated protein kinase kinase (MEK)—ERK (RAF-MEK-ERK) and phosphatidylinositol 3-kinase (PI3K)—Akt pathways, and metabolite availability sensed through pathways involving AMPK, REDD1, and others.
PI3K signaling has recently been found to be necessary for mTOR activation by pentylenetetrazole-induced seizures (Zhang and Wong, 2012), but upstream activators of mTOR in post-traumatic epileptogenesis remain largely unknown (McDaniel and Wong, 2011), and effects of mTOR inhibitors on epilepsy have varied depending on the timing of intervention and the model. To study mTOR signaling in epilepsy, we used an accelerated in vitro model of post-traumatic epilepsy that captures critical features of clinical epileptogenesis, including gradual onset, axon sprouting, spontaneous electrographic seizures, seizure clustering, status epilepticus, and subsequent activity-dependent cell death, response to anticonvulsants, and emergence of anticonvulsant resistance (Dyhrfjeld-Johnsen et al., 2010; Berdichevsky et al., 2012). We developed a novel assay of epileptic activity based on seizure-dependent increases in extracellular lactate concentration (During et al., 1994) to quantify epilepsy in this preparation, and combined it with estimates of cell death based on release of the enzyme lactate dehydrogenase (LDH).
Materials and Methods
Organotypic cultures.
We generated organotypic hippocampal slice cultures from postnatal day 7 or 8 male Sprague Dawley rats, and maintained them in poly-d-lysine-coated, 6-well tissue culture plates on a rocking platform at <1 cycle/min, in 37°C and 5% CO2 in Neurobasal-A/B27 (Invitrogen, prepared according to manufacturer's specifications), with 0.5 mm GlutaMAX and 30 μg/ml gentamicin (Invitrogen). All animal use protocols were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital and were conducted in accordance with the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Drugs and lactate and LDH assays.
Rapamycin, SP 600125, U0126, and LY 294002 were purchased from Tocris Bioscience, and NVP-BEZ235 was purchased from BioVision. All drugs were dissolved in dimethylsulfoxide (DMSO) and applied to cultures starting on 3 d in vitro (DIV), unless otherwise specified. Control cultures were treated with 0.1% DMSO as vehicle. Since organotypic cultures from different animals develop epilepsy at slightly different rates, all experiments used control slices from the same animal. Lactate and LDH concentrations were measured in used culture medium with kits from BioVision. Lactate concentrations were calculated relative to known lactate standards (BioVision), while LDH concentrations were calculated in terms of arbitrary units (a.u.), normalized to the 6–28 DIV average of LDH concentration in control (epileptic) culture supernatant.
Electrophysiology and propidium iodide staining.
Activity was recorded with tungsten microelectrodes in CA1 pyramidal layer while cultures were maintained in an interface chamber perfused with Neurobasal-A at 37°C. Chronic multiple electrode array (MEA) recordings were performed with custom-designed chips (Berdichevsky et al., 2012), on which organotypic cultures were maintained as described above. Lactate concentrations in the culture supernatant after 3 or 4 d in the incubator were compared with MEA electrical recordings from the same cultures. Dead cell stain was performed using propidium iodide (PI) (Invitrogen; 1 mg/ml stock) added to live cultures for a final 1:250 dilution. Cultures were incubated for 1 h, imaged on an inverted confocal microscope (Zeiss), and analyzed as described previously (Berdichevsky et al., 2012). Numbers of dead cells were counted during medium changes between 6 and 25 DIV, and compared with relative LDH concentrations in used culture medium from the same cultures during the same time period.
Nissl staining.
Cultures were fixed in 4% paraformaldehyde, permeabilized for 1 h in 0.3% Triton-X on a rocking platform, and stained with PI (Invitrogen; 1 mg/ml stock, diluted 1:250 in PBS) for 5 h. Cultures were mounted, and images were acquired with a 40× objective on a confocal microscope (Leica) with appropriate laser wavelength and filters. Optical stacks spanning the entire thickness of the slice were recorded, with z-direction step size of 3 μm.
Immunohistochemistry.
Cultures were fixed and permeabilized. Blocking buffer (10% goat serum in 0.05% Triton-X in PBS) was applied for 2 h after permeabilization. Cultures were incubated with primary antibody (1:100 anti-phospho-S6 (Ser235/236); Cell Signaling Technology, or 1:200 anti-MAP2 and 1:500 anti-SMI-312; Covance) overnight at 4°C, washed, and incubated with appropriate secondary antibodies conjugated with Alexa Fluor 488, 568, or 647 (Life Technologies) overnight. Cultures were then washed, counterstained with Nissl stain (only for p-S6), mounted, and imaged with a confocal scanning microscope (Leica or Zeiss).
Western Blots and analysis.
Cultures were homogenized, and protein extracts were loaded into 12% Tris-Glycine polyacrylamide gels (Novex gels; Invitrogen) and separated by electrophoresis. Proteins were then transferred to PVDF membranes (Millipore) and stained with a primary antibody mixture (1:500 rabbit polyclonal anti-phospho-S6 (Ser 235/236) or anti-phospho-S6 (Ser 240/244), and 1:500 mouse monoclonal anti-S6, Cell Signaling Technology, in blocking buffer) followed by appropriate secondary antibodies from LI-COR. Membranes were imaged on a two-channel infrared scanner (Odyssey; LI-COR) and bands were quantified with LI-COR software.
Electrographic analysis.
Ictal events (electrographic seizures) were defined as either paroxysmal events of much larger amplitude than background multiple unit activity and lasting longer than 10 s, or shorter paroxysmal events that occurred with frequency of at least 2 Hz for at least 10 s. Interictal events (events between seizures) were defined as paroxysmal events of <200 ms duration occurring at a frequency of <2 Hz (Dyhrfjeld-Johnsen et al., 2010; Berdichevsky et al., 2012). In those cases where ictal events were preceded or followed by interictal activity, start and end of an ictal event were defined as time points where paroxysmal event frequency exceeded or decreased <2 Hz, respectively.
Axon sprouting quantification.
We took phase contrast micrographs of the slice and surrounding area with sprouted axons at 20× magnification. Approximately 30–40 micrographs were needed to cover the sprouting area. We stitched the images together in Adobe Photoshop CS2, using an automated batch stitching tool. We then traced the slice outline, and generated equidistant expanding contours from the slice outline at 200 pixel steps, using Fiji (extension of ImageJ software; National Institutes of Health, Bethesda, MD). Neurites crossing the contours were marked and counted using Cell Counter plug-in in Fiji. We selected crossing counts at the contour at 900 μm distance from the edge of the slice to quantify the number of sprouted axons, since sprouting dendrites are not long enough to extend so far from the neuronal soma (Taylor et al., 2005). Axon crossings were subdivided by the proximal hippocampal area: CA1 and subiculum, CA3, or dentate gyrus (DG).
Statistical analysis.
We used Student's t test for two-variable comparisons, and one-way ANOVA with Holm–Sidak post hoc analysis for multiple variable comparisons. For correlation analysis, we used Pearson Product Moment to calculate correlation coefficients and p values.
Results
Lactate and LDH concentrations reflect ictal activity and ictal cell death
In organotypic hippocampal cultures, interictal and ictal activities follow a latent period of ∼7 d after dissection injury (Fig. 1A). We measured lactate and LDH concentrations in media from organotypic cultures that were chronically treated by 100 μm phenytoin, 3 mm kynurenic acid (KYNA), or vehicle. Data from 7, 10, 14, 17, 21, 24, 28, and 32 DIV were combined into four time points by arithmetic mean, DIV <{7,10}>, <{14,17}>, <{21,24}>, and <{28,32}> (Fig. 1 B,C). Lactate and LDH concentrations in control (vehicle) cultures were significantly higher than in cultures treated by either phenytoin or KYNA (n = 12 cultures from three animals, each condition).
Figure 1.
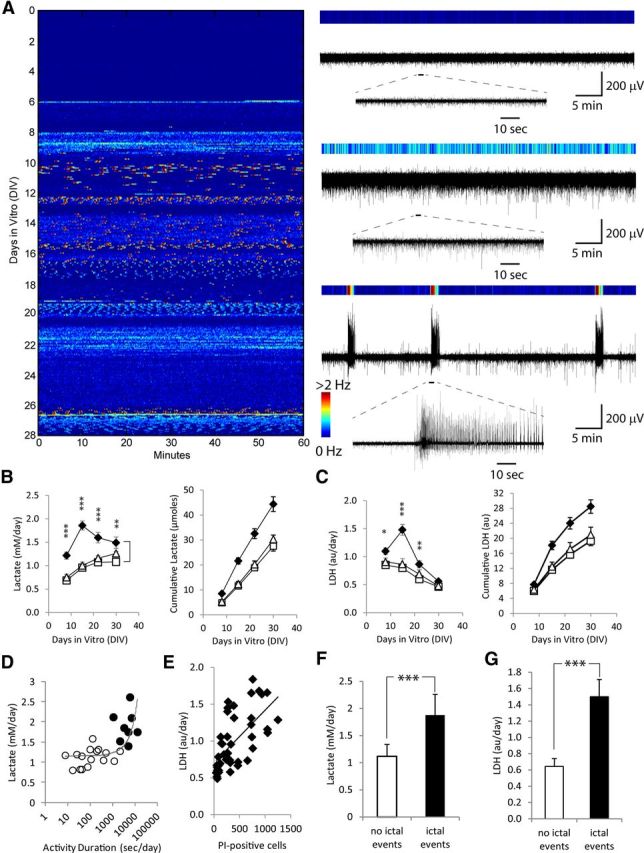
Validation of lactate and LDH measurements as biomarkers for epileptic activity and cell death. A, Representative time course of epileptogenesis in an organotypic hippocampal culture. Colors correspond to the frequency of paroxysmal event occurrence in 10 s bins, with examples shown on the right. Deep blue, multiunit activity (top trace); light blue or yellow, interictal activity (middle trace); red, electrographic seizures (ictal events, bottom trace). B, C, time course of lactate (B) and LDH (C) concentration and cumulative production in epileptic controls (♦) and cultures treated with phenytoin (▵), and KYNA (□); n = 12 each condition. Error bars indicate SEM. Statistical significance is shown for epileptic controls versus KYNA-treated cultures, with *p < 0.05, **p < 0.01, and ***p < 0.001. Statistically significant differences between epileptic controls and phenytoin-treated cultures were the same as control versus KYNA, except for lactate at DIV <{28,32}> and LDH at DIV <{21,24}> and <{28,32}>, where no statistically significant differences were found. D, Relationship of lactate concentration and activity, during time periods when only physiological or interictal activity was present (○) or when ictal activity was present (●). Lactate concentrations were plotted versus the duration of electrical activity, with each data point corresponding to the time between medium changes. Exponential fit line is shown. E, Relationship between LDH concentration in culture supernatant and the number of dead (PI-positive) cells. LDH concentrations and numbers of PI-stained dead cells were measured in the same cultures, with each time point again corresponding to the time between medium changes (LDH accumulated in the culture medium since last medium change vs number of PI cells at the time of medium change). Linear fit line is shown. F, Lactate production (average ± SD, ***p < 0.001) during time periods with and without ictal vents, from data shown in D. G, LDH release (average ± SD, ***p < 0.001) from cultures during time periods with and without ictal events, from data in E.
In epileptic control cultures, when ictal activity was present, lactate production was 1.88 ± 0.38 mm/d (Fig. 1F; average ± SD, n = 8 periods between medium changes, seven cultures), while in the absence of ictal events, lactate was produced at 1.12 ± 0.22 mm/d (p < 0.001, t test; average ± SD, n = 16 periods between medium changes in seven slice cultures). Lactate production and duration of electrical activity were strongly correlated (Fig. 1D; n = 7 cultures, Pearson r = 0.622, p < 0.01), as were LDH concentrations and PI counts (Fig. 1E; n = 6 cultures, data from seven medium changes, r = 0.635, p < 0.001). LDH release during time periods when ictal events occurred was significantly higher than LDH release during periods with no ictal events (Fig. 1G; p < 0.001, n = 6 cultures).
Manipulation of mTOR signaling: effects on ictal activity and ictal cell death
We tested the effects of inhibition of kinases (Table 1) on the development of epilepsy (Fig. 2A–F; n = 3 each condition). Inhibition of JNK had no significant effect, while inhibition of MEK had no significant effect on lactate production before 20 DIV. However, inhibition of PI3K, Akt, mTOR, and dual inhibition of PI3K-mTOR significantly decreased lactate production starting from 6 DIV (p < 0.05), and LDH release starting from 9–10 DIV (p < 0.05). We repeated the experiments with LY 294002 and rapamycin (n = 3) and again found significant inhibition of lactate production (from 6 DIV, p < 0.05) and LDH release (from 10 DIV, p < 0.05) relative to controls. We also tested the dose–response of API-2 and NVP-BEZ235 (n = 3 per condition), and found dose-dependent inhibition of both lactate and LDH (Fig. 3). We calculated lactate and LDH concentrations in drug-treated cultures as percentage of paired controls at the day of peak lactate concentration (maximum ictal activity; Fig. 2G). Significant differences were found between controls and rapamycin (n = 6, p < 0.001), 0.5 μm API-2 (n = 3, lactate p < 0.01, LDH p < 0.001), LY 294002 (n = 6, p < 0.001), and 40 nm NVP-BEZ235 (n = 3, p < 0.001).
Table 1.
Inhibitor names, concentrations, and targets
| Target name | Target abbreviation | Drug name | Concentration |
|---|---|---|---|
| c-Jun N-terminal kinase | JNK | SP 600125 | 10 μm |
| MAP kinase kinase | MEK | U0126 | 20 μm |
| Mammalian target of rapamycin | mTOR | Rapamycin | 20 nm |
| Protein kinase B | Akt | API-2 | 0.5 μm |
| Phosphoinositide 3-kinase | PI3K | LY 294002 | 10 μm |
| Dual inhibitor | PI3K-mTOR | NVP-BEZ235 | 40 nm |
Figure 2.

Screen of inhibitors. A, SP 600125, JNK inhibitor. B, U0126, MEK inhibitor. C, Rapamycin, mTOR inhibitor. D, API-2, Akt inhibitor. E, LY 294002, PI3K inhibitor. F, NVP-BEZ235, dual PI3K/mTOR inhibitor. n = 3 per data point, for cultures with vehicle (●) and inhibitor treatment (○) lactate, and for cultures with vehicle (♢) and inhibitor treatment (♦) LDH. Cumulative averages of lactate and LDH production are plotted in each figure as solid gray line (vehicle-treated cultures) or dashed gray line (inhibitor-treated cultures). G, Summary of the data at the day of peak lactate production. N = 3 for both controls and treated slices for all groups except rapamycin (n = 6) and LY 294002 (n = 6). Error bars indicate SD.
Figure 3.

Lactate and LDH concentrations show dose–response to varying concentrations of inhibitors. A, Dose–response to chronic API-2 treatment, n = 3 per point/condition. B, Dose–response to chronic NVP-BEZ235 treatment, n = 3 per point/condition. Concentrations are shown in the figure. Error bars indicate SD.
We recorded electrographic activity in rapamycin, LY 294002, and control cultures between 10 and 15 DIV (Fig. 4A–E). Ictal activity was present in >85% of treated and control cultures. However, there were significant 62 and 45% reductions in the number of ictal events per hour in rapamycin (n = 16) and LY 294002-treated cultures (n = 14), respectively, versus controls (n = 13; p < 0.001). Ictal durations were unchanged (p = 0.21) between controls (36.7 ± 3.3 s), rapamycin (32.9 ± 3.8 s), and LY 294002-treated cultures (28 ± 2.4 s, average ± SEM). The total time cultures spend in ictus per hour was significantly increased in controls versus rapamycin and LY 294002-treated cultures (p < 0.001).
Figure 4.

Electrical recordings and neuronal counts confirm that LY 294002 (LY) and rapamycin (RAPA) treatments reduce number of ictal events and promote neuronal survival. A, Typical ictal event in an epileptic control culture. B, Representative electrical recordings from control, rapamycin, and LY 294002-treated cultures. Rapamycin and LY 294002 treatments reduce seizure frequency (C), but not seizure duration (D), with the overall effect of reducing the time spent in ictal state (E). **p < 0.01, ***p < 0.001. F, Representative images of Nissl-stained organotypic hippocampal cultures at 28 DIV. Scale bars, 50 μm. G, Neuronal counts revealed that more neurons survive in CA3c and CA1 of rapamycin and LY 294002-treated cultures compared with epileptic controls, n = 3 per condition, **p < 0.01, ***p < 0.001.
Pyramidal neuronal layers were well organized in control epileptic cultures at 28 DIV (Fig. 4F); however, the number of surviving CA3c neurons at 28 DIV was >50% higher in cultures treated with either rapamycin or LY 294002 versus controls (Fig. 4 G; p < 0.01, n = 3). The number of surviving CA1 neurons in rapamycin and LY 294002-treated cultures was >100% higher than in controls (p < 0.01, n = 3). In a separate experiment, we found similar numbers of surviving neurons in control cultures, but 70–230% more surviving neurons in NVP-BEZ235-treated cultures compared with controls in CA3c (p < 0.01, n = 3) and in CA3b (p < 0.05, n = 3).
Effect of rapamycin wash-in and wash-out on epileptic activity
Rapamycin treatment from 3 to 28 DIV had the same effect as described above (n = 6 cultures from two additional experiments; Fig. 5A). Late application of rapamycin from 10 to 28 DIV resulted in significantly lower lactate concentrations compared with controls from 17 to 28 DIV, and in significantly lower LDH concentrations from 17 to 24 DIV (n = 6; Fig. 5B). Early termination of rapamycin treatment (rapamycin applied 3–17 DIV; Fig. 5C) did not increase the rate of subsequent lactate accumulation (1.19 ± 0.314 mm/d on 17 DIV, 1.16 ± 0.448 mm/d on 28 DIV, average ± SD, n = 6, p = 0.9) or LDH accumulation (0.29 ± 0.2 a.u./d on 17 DIV, 0.29 ± 0.16 a.u./d on 28 DIV, average ± SD, n = 6, p = 0.97).
Figure 5.

Effects of different time courses of rapamycin (RAPA) treatment on progression of epilepsy. A, Rapamycin is applied 3 to 28 DIV, 10–28 DIV (B), 3–17 DIV (C). Control lactate concentrations are plotted as ●, while lactate concentrations in the rapamycin experimental group are plotted as ○. LDH concentrations are plotted as ♦ for control group, and ♢ for rapamycin group. N = 6 per condition, per data point, *p < 0.05, **p < 0.01, ***p < 0.001. D, Representative electrical recording of a culture that was chronically treated with rapamycin, showing no change in activity upon rapamycin wash-out. E, Results of rapamycin washout in three cultures, ○ represents power of electrical activity in rapamycin, and ● power of activity after wash-out. F, Comparison of seizure activity in rapamycin-treated cultures at 17 DIV (empty circle), and cultures in which rapamycin treatment was discontinued starting at 17 DIV for 11 d (filled circle).
We did not see a significant change in the power of electrographic activity after 1 h of rapamycin wash-out following chronic rapamycin treatment (14—24 DIV) compared with electrographic activity in rapamycin (Fig. 5D,E; n = 3, p = 0.122, paired t test). We also did not see significant changes in time seizing (duration of ictal activity per hour of recording) in cultures treated with rapamycin from 3 to 17 DIV and recorded on 17 DIV versus cultures treated with rapamycin from 3 to 17 DIV and recorded after 11 d of wash-out (i.e., control conditions) on 28 DIV (Fig. 5F; same experimental design as Fig. 5C; n = 18 rapamycin-treated 17 DIV cultures and n = 19 28 DIV cultures in which rapamycin treatment was discontinued on 17 DIV (11 d wash), p = 0.447, Mann–Whitney rank sum test).
Chronic rapamycin treatment decreases axon sprouting
We quantified the number of sprouted axons at 8 DIV (Fig. 6). We confirmed that neurites crossing contours 900 μm away from the edge of organotypic culture were axons by staining for SMI-312 (axon marker; Ulfig et al., 1998; Chung et al., 2003) and MAP-2 (dendritic marker). Both of these markers stain only neuronal, and not glial, processes. Representative images are shown for an area close to the edge of the slice (Fig. 6D), and an area 900 μm away from the edge of the slice (Fig. 6E). Large numbers of SMI-312-positive axonal processes, but only a few MAP-2-positive dendritic processes, leave stratum oriens and extend outside of the slice (Fig. 6D), confirming that most of the sprouted processes outside of the slice are axons. Further away from the slice, all of the processes are positive for SMI-312, and none are positive for MAP-2 (Fig. 6E), confirming that only sprouting axons cross the 900 μm contour. Axon sprouting was significantly reduced in rapamycin-treated cultures compared with controls (Fig. 6F; average ± SEM, n = 8 cultures, p < 0.05). Rapamycin treatment did not significantly reduce axon sprouting from CA1 and subiculum (p = 0.381), but did significantly reduce sprouting from CA3 and DG areas (p < 0.01 in CA3, p < 0.05 in DG; Fig. 6G). The lack of effect in area CA1 may reflect the much higher neuronal density in this area in the rapamycin-treated cultures (Fig. 4F,G). Termination of rapamycin treatment for 11 d did not result in significant new axon sprouting (n = 8 cultures, comparison of rapamycin-treated cultures at 17 DIV, and same cultures at 28 DIV after 11 d of rapamycin wash-out, p = 0.686, paired t test; Fig. 6H).
Figure 6.
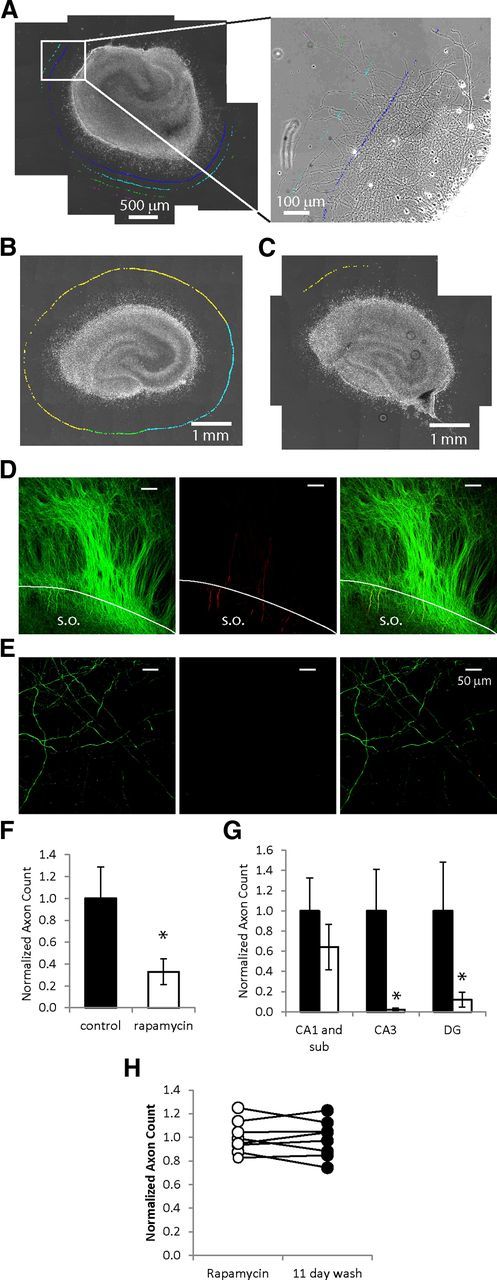
mTOR inhibition reduces axon sprouting. A, Left, Stitched micrographs show organotypic hippocampal culture and surrounding area with sprouted axons. The inset shows sprouting axons with quantification markers at different distances from the culture. B, C, Representative images of a control culture (B) and rapamycin-treated culture (C) are shown at 8 DIV, with marks corresponding to axons crossing the contour 900 μm away from CA3 (blue), DG (green), or CA1 and subiculum (yellow) border. D, Representative SMI-312 (green) and MAP-2 (red) staining of processes at the edge (white line) of an 8 DIV organotypic culture; right image is merged SMI-312 and MAP-2 staining (stratum oriens, SO). E, Representative SMI-312 (green, left image) and MAP-2 (red, middle image), and merged SMI-312/MAP-2 (right image) staining of processes 900 μm away from the edge of an 8 DIV organotypic culture. F, Comparison of the numbers of axons crossing 900 μm contour in rapamycin or vehicle-treated cultures (n = 8, p < 0.05), G, Numbers of axons originating from different areas of the hippocampal culture, *p < 0.05. H, Comparison of the numbers of axons crossing 900 μm contour in rapamycin-treated cultures at 17 DIV (empty circles) versus axons in the same cultures 11 d after discontinuation of rapamycin treatment (filled circles).
LY 294002 and rapamycin reduce phosphorylation of S6 ribosomal protein
Phosphorylation of S6 (Ser 235/236) significantly increased on day 1 after dissection versus immediately after dissection (Fig. 7A; n = 4, p < 0.01). The ratio of pS6 to S6 then decreased by 7 DIV, and significantly increased again by 21 DIV (1.00 ± 0.30 vs 1.88 ± 0.43, 7 and 21 DIV, respectively, average ± STD, n = 4, p < 0.05). Phosphorylation of S6 at Ser 235/236 was significantly reduced in pyramidal cells in rapamycin and LY 294002-treated cultures at 7 DIV compared with controls (Fig. 7 B; n = 3). The ratio of phospho-S6 (Ser 235/236) versus total S6 was reduced in rapamycin and LY 294002-treated cultures (94 and 81%, respectively, p < 0.001, n = 9, Fig. 7C). The ratio of phospho-S6 (Ser 240/244) versus total S6 was also significantly reduced in rapamycin and LY 294002-treated cultures (99 and 91%, respectively, p < 0.001, n = 6; Fig. 7D).
Figure 7.
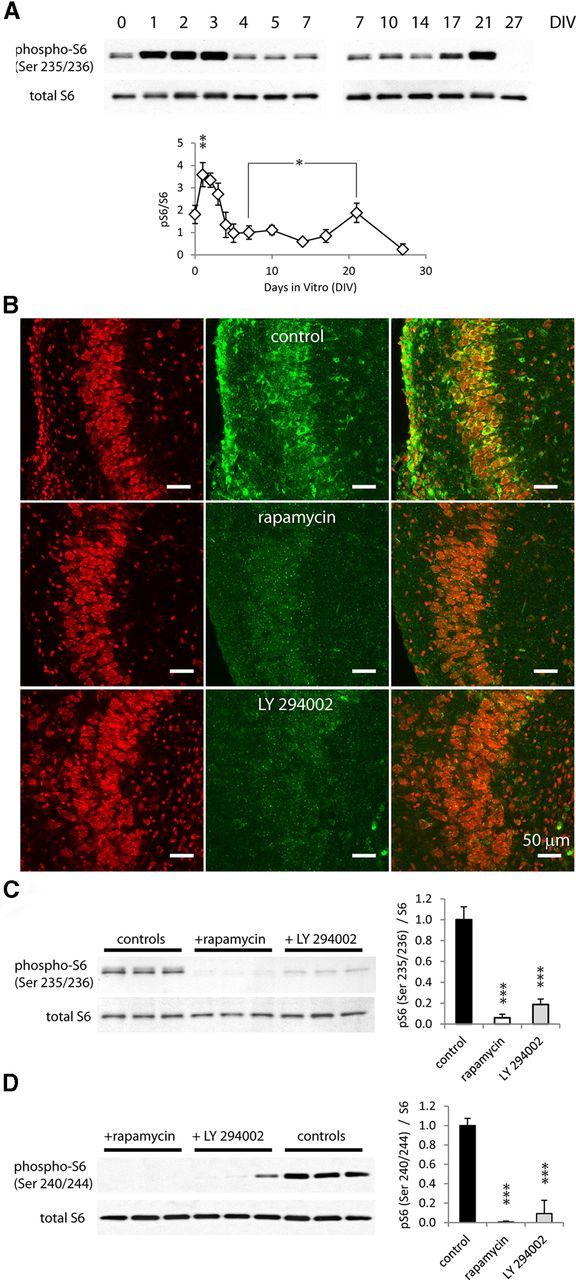
Phosphorylation of S6 ribosomal protein is reduced with chronic rapamycin or LY 294002 treatment. A, Changes in S6 phosphorylation with time in vitro. Top: Representative Western blots of phospho-S6 (Ser 235/236) and total S6, from 0 to 7 DIV, and from 7 to 27 DIV. Bottom, Ratios of phospho-S6 (Ser 235/236) to total S6 are plotted (n = 4, average +/− STD, *p < 0.05, **p < 0.01). B, Immunohistochemistry micrographs of cultures with different treatments, Nissl stain is shown in red and phosphor-S6 is in green. C, Representative Western blot of pS6 (Ser235/236) and total S6 and comparison of pS6 (Ser 235/236)/S6 ratio in cultures treated with vehicle, rapamycin, or LY 293002. D, Representative Western blot of pS6 (Ser 240/244) and total S6, and comparison of pS6 (Ser 240/244)/S6 ratio in cultures treated with vehicle, rapamycin, or LY 294002 (***p < 0.001).
Discussion
Key findings
We found that lactate and LDH assays of culture media sampled at twice-weekly media changes provided useful estimates of ictal activity and ictal cell death in organotypic hippocampal cultures as they became epileptic in the first 10 DIV. PI3K and mTOR inhibition reduced S6 phosphorylation, and PI3K, Akt, mTOR, and combined mTOR/PI3K inhibition reduced cell death, axon sprouting, and the severity of epilepsy. Wash-out experiments indicated that mTOR inhibition had no direct anticonvulsant effect, and that the anti-epileptogenic and neuroprotective effects of mTOR inhibition persisted for 11 d after wash-out.
Lactate and LDH assays
In the organotypic hippocampal culture model of pediatric post-traumatic epilepsy, interictal and ictal activities appear during the second week after initial injury (Dyhrfjeld-Johnsen et al., 2010). Ictal activity is accompanied by a significant increase in activity-dependent cell death, and both ictal activity and cell death are prevented by KYNA and phenytoin (Pozzo Miller et al., 1994; Berdichevsky et al., 2012). If untreated, ictal activity persists for longer than 4 weeks in vitro (Fig. 1A). The time course of epileptogenesis in cultures from different animals varied slightly, similar to variances previously found in epileptogenesis in individual animals subject to the same insult (Williams et al., 2009; Kadam et al., 2010). For this reason, all experiments were controlled by comparing vehicle- and inhibitor-treated slices from the same animal.
We found increased lactate production due to ictal activity, and increased LDH release due to activity-dependent cell death. Ictal lactate accumulation occurs in humans (During et al., 1994; Castillo et al., 2001; Mercimek-Mahmutoglu et al., 2012) and in animal models of epilepsy (Chapman et al., 1977; Fujikawa et al., 1988; Wasterlain et al., 2010). LDH release is a widely used marker of cell death (Decker and Lohmann-Matthes, 1988; Bonfoco et al., 1995). Measurements of lactate and LDH concentrations in organotypic hippocampal cultures are faster and less labor intensive than electrographic recordings and dead cell counts, enabling rapid analysis of chronic drug effects including dose–response (Fig. 3). Lactate and LDH levels were significantly reduced by phenytoin and KYNA acid, known inhibitors of ictal activity and cell death in organotypic cultures (Berdichevsky et al., 2012). Lactate and LDH assays were also congruent with electrographic and Nissl measurements of rapamycin and LY 294002 effects on epileptiform activity and neuronal survival. These findings support the utility of lactate and LDH measurements as biomarkers of the progression of epilepsy in this model.
Total lactate production by an organotypic hippocampal culture depends on the number of surviving neurons in the culture. It has been previously shown that spontaneous seizures in organotypic cultures result in ongoing neural death (Pozzo Miller et al., 1994; Berdichevsky et al., 2012), leaving fewer neurons to make lactate. This may result in decreased lactate production such as that seen in vehicle-treated cultures in Figures 1B and 2A–F between 3 and 4 weeks in vitro. To interpret whether this decrease corresponds to a decrease in epileptic activity or a decrease in the number of neurons, LDH assays of cell death are necessary. The time course of LDH production in vehicle-treated epileptic cultures in Figures 1C and 2A–F shows significant increases in cell death after the end of latent period. Cell death due to seizures is ongoing and cumulative (Fig. 1C), and by 28 DIV results in a dramatic reduction in the number of surviving neurons (Fig. 4F,G; Berdichevsky et al., 2012). In these cultures, lactate production is reduced due to lower number of neurons, but ictal activity has not ceased. In contrast, cultures treated with KYNA, phenytoin, or inhibitors of PI3K-Akt-mTOR pathway have reduced or no ictal activity, and correspondingly, no seizure-dependent cell death (as shown by LDH assays). Enhanced neuron survival in these cultures (Figs. 4F,G) results in higher rates of physiological lactate production, because there are more neurons to make lactate. At later time points (fourth week in vitro), lactate production from a chronically epileptic slice with fewer surviving neurons may be the same as the rate of physiological lactate production in a treated slice. Thus, differences in lactate production diminish over time, and analysis of cell death via LDH assay is necessary for interpretation of lactate assay as a marker of ictal activity.
mTOR and epilepsy
Lactate and LDH levels were dramatically reduced once ictal events began to occur (9–17 DIV) in cultures treated with inhibitors of PI3K, Akt, and mTOR. The reductions in lactate and LDH due to rapamycin (mTOR), API-2 (Akt), and LY 294002 (PI3K) treatments were similar and confirmed by direct measurements of electrographic activity and cell survival, which suggested that PI3K-Akt signaling was responsible for most of mTOR activation as measured by physiological effects on epilepsy and cell death. The phosphorylation level of S6 ribosomal protein (a downstream effector of mTOR pathway phosphorylated through S6K) is a widely used marker of mTOR activation (Guertin and Sabatini, 2007; Buckmaster et al., 2009; Zeng et al., 2009). Phosphorylation of S6 has been reported to increase after epileptogenic brain injury (Buckmaster et al., 2009; Zeng et al., 2009; Huang et al., 2010; Zhang and Wong, 2012) and hypoxic seizures (Talos et al., 2012). Interestingly, phosphorylation of S6 was biphasic in our experiments: a transient increase immediately after initial injury (dissection) followed by a second increase during the period of ictal activity and ictal cell death. These data suggest that injury, whether caused by physical trauma or by ictal activity, can increase mTOR activation. On the other hand, S6 remained moderately phosphorylated throughout the latent period, suggesting that mTOR activation may play a role in circuit reorganization before appearance of spontaneous seizures. Chronic LY 294002 treatment reduced phosphorylation of S6 at 7 DIV (near the end of the pre-ictal period) as effectively as chronic rapamycin treatment, again suggesting that most of mTOR activation during the pre-ictal period occurs through PI3K. Finally, a dual PI3K-mTOR inhibitor NVP-BEZ235 (Maira et al., 2008) reduced lactate and LDH concentrations as much as PI3K or mTOR inhibitors alone. The absence of additive or synergistic effects confirms that anti-epileptic effects of PI3K inhibition are due to inactivation of mTOR. Our findings demonstrate that mTOR activation in post-traumatic epileptogenesis is strongly PI3K dependent, suggesting that activation of PI3K by growth factors may be an important contributing factor to the development of epilepsy after trauma. Considering the complexity and feedbacks present in mTOR signaling (Guertin and Sabatini, 2007) and involvement of PI3K and Akt in mTOR activation, dual inhibitors such as NVP-BEZ235 or other drugs with similar mechanisms (Engelman, 2009; Borders et al., 2010) may prove preferable to rapamycin in treatment of epilepsy.
Anti-epileptogenic effects of mTOR inhibition
mTOR inhibition could be either anti-epileptogenic (preventing the development of epilepsy), or merely anticonvulsant. Data from acute applications of rapamycin in vitro do not suggest a direct anticonvulsant effect (Daoud et al., 2007; Rüegg et al., 2007). Structural changes such as the reduction of mossy fiber sprouting after chronic mTOR inhibition suggest an anti-epileptogenic effect (Buckmaster et al., 2009; Zeng et al., 2009; Huang et al., 2010). In our model, mTOR inhibition with rapamycin also led to reduced sprouting at 8 DIV. In the organotypic hippocampal culture model of epileptogenesis, 8 DIV is the latest time point at which this comparison can be made without confounding by the onset of seizures and ictal cell death, which may produce additional sprouting. Our results suggest that inhibition of mTOR signaling after injury, but before onset of seizures, reduces injury-induced axon sprouting. Suppression of mossy fiber sprouting required ongoing treatment with rapamycin (Buckmaster et al., 2009), suggesting that temporary mTOR inhibition does not have sustained anti-epileptogenic effects. However, we found that after early termination of rapamycin treatment, lactate production, and LDH release remained suppressed for at least 11 d. It is important to note that at later time points, rapamycin-treated cultures produce low levels of lactate despite retaining many more neurons than control (epileptic) cultures that have experienced significant ictal cell death. Lactate production is similar in rapamycin-treated and vehicle-treated cultures at later time points (fourth week in vitro) due to significant neural death in vehicle-treated cultures from seizures, and enhanced neuronal survival in rapamycin-treated cultures, as discussed above. Low lactate production from large numbers of neurons is consistent with strong attenuation of ictal activity in rapamycin-treated cultures, even after rapamycin wash-out. Reductions in axon sprouting and electrographic seizures persisted after rapamycin wash-out and confirmed results of lactate and LDH assays. These findings imply that in our model, transient inhibition of mTOR has sustained effects on epilepsy. Finally, it was reported that rapamycin reduces seizure activity even when given after the development of spontaneous recurrent seizures (Huang et al., 2010). Our findings were similar: epileptiform activity and resulting cell death were both significantly reduced by late rapamycin treatment (starting on 10 DIV) compared with epileptic controls (Fig. 5B), suggesting that rapamycin was effective even in this relatively late stage of epileptogenesis. While these findings may have clinical implications for treatment of acquired epilepsy, they suggest that epileptogenesis may be a more dynamic process than previously imagined. Ictal cell death (Berdichevsky et al., 2012) may alter epileptic networks in a fashion that requires ongoing circuit plasticity to maintain epilepsy, and this ongoing epileptogenesis might be blocked by late mTOR inhibition.
Utility and limitations of the organotypic slice preparation for mTOR signaling
Hippocampal organotypic slice cultures provide an exceptionally rapid model of epileptogenesis that retains key features of this process (Dyhrfjeld-Johnsen et al., 2010; Berdichevsky et al., 2012; Sabolek et al., 2012). Because the slices are created at P7, this is a model of epileptogenesis after pediatric brain injury, and some of our results may be specific to the pediatric population. The slice cultures have microglia but few other elements of the immune system and no blood–brain barrier, so anti-inflammatory and vascular effects of mTOR inhibition can be largely excluded. The accessibility of the preparation lends itself to rapid assays of both seizure activity and neuroprotection. The high throughput of this approach is particularly important for the exploration of the interaction of brain injury, epilepsy, and the effect of manipulation of complex signaling systems such as the mTOR pathway, in which not only the point of pathway intervention but also the timing relative to injury are important variables.
Footnotes
This study was supported by National Institutes of Health Grants R21 NS072258 and R01 NS077908 (National Institute of Neurological Disorders and Stroke).
References
- Berdichevsky Y, Dzhala V, Mail M, Staley KJ. Interictal spikes, seizures and ictal cell death are not necessary for post-traumatic epileptogenesis in vitro. Neurobiol Dis. 2012;45:774–785. doi: 10.1016/j.nbd.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borders EB, Bivona C, Medina PJ. Mammalian target of rapamycin: biological function and target for novel anticancer agents. Am J Health Syst Pharm. 2010;67:2095–2106. doi: 10.2146/ajhp100020. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Ghosh S, Matsuoka Y, Ashton-Beaucage D, Therrien M, Lemieux S, Perreault C, Roux PP, Kitano H. A comprehensive map of the mTOR signaling network. Mol Syst Biol. 2010;6:453. doi: 10.1038/msb.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo M, Smith JK, Kwock L. Proton MR spectroscopy in patients with acute temporal lobe seizures. AJNR Am J Neuroradiol. 2001;22:152–157. [PMC free article] [PubMed] [Google Scholar]
- Chapman AG, Meldrum BS, Siesjö BK. Cerebral metabolic changes during prolonged epileptic seizures in rats. J Neurochem. 1977;28:1025–1035. doi: 10.1111/j.1471-4159.1977.tb10665.x. [DOI] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Chueh SC, Kahan BD. Clinical application of sirolimus in renal transplantation: an update. Transpl Int. 2005;18:261–277. doi: 10.1111/j.1432-2277.2004.00039.x. [DOI] [PubMed] [Google Scholar]
- Chung RS, Vickers JC, Chuah MI, West AK. Metallothionein-IIA promotes initial neurite elongation and postinjury reactive neurite growth and facilitates healing after focal cortical brain injury. J Neurosci. 2003;23:3336–3342. doi: 10.1523/JNEUROSCI.23-08-03336.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–742. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats. Brain Res. 1988;474:181–184. doi: 10.1016/0006-8993(88)90681-6. [DOI] [PubMed] [Google Scholar]
- Daoud D, Scheld HH, Speckmann EJ, Gorji A. Rapamycin: brain excitability studied in vitro. Epilepsia. 2007;48:834–836. doi: 10.1111/j.1528-1167.2006.00976.x. [DOI] [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- During MJ, Fried I, Leone P, Katz A, Spencer DD. Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J Neurochem. 1994;62:2356–2361. doi: 10.1046/j.1471-4159.1994.62062356.x. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol. 2010;27:418–424. doi: 10.1097/WNP.0b013e3181fe0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Vannucci RC, Dwyer BE, Wasterlain CG. Generalized seizures deplete brain energy reserves in normoxemic newborn monkeys. Brain Res. 1988;454:51–59. doi: 10.1016/0006-8993(88)90802-5. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam SD, White AM, Staley KJ, Dudek FE. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 2010;30:404–415. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew FH, Buckmaster PS. Is there a critical period for mossy fiber sprouting in a mouse model of temporal lobe epilepsy? Epilepsia. 2011;52:2326–2332. doi: 10.1111/j.1528-1167.2011.03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, García-Echeverría C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett. 2011;497:231–239. doi: 10.1016/j.neulet.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Horvath GA, Coulter-Mackie M, Nelson T, Waters PJ, Sargent M, Struys E, Jakobs C, Stockler-Ipsiroglu S, Connolly MB. Profound neonatal hypoglycemia and lactic acidosis caused by pyridoxine-dependent epilepsy. Pediatrics. 2012;129:e1368–1372. doi: 10.1542/peds.2011-0123. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- Pozzo Miller LD, Mahanty NK, Connor JA, Landis DM. Spontaneous pyramidal cell death in organotypic slice cultures from rat hippocampus is prevented by glutamate receptor antagonists. Neuroscience. 1994;63:471–487. doi: 10.1016/0306-4522(94)90544-4. [DOI] [PubMed] [Google Scholar]
- Raymont V, Salazar AM, Lipsky R, Goldman D, Tasick G, Grafman J. Correlates of posttraumatic epilepsy 35 years following combat brain injury. Neurology. 2010;75:224–229. doi: 10.1212/WNL.0b013e3181e8e6d0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabolek HR, Swiercz WB, Lillis KP, Cash SS, Huberfeld G, Zhao G, Ste Marie L, Clemenceau S, Barsh G, Miles R, Staley KJ. A candidate mechanism underlying the variance of interictal spike propagation. J Neurosci. 2012;32:3009–3021. doi: 10.1523/JNEUROSCI.5853-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/WNL.35.10.1406. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post-treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012;509:105–109. doi: 10.1016/j.neulet.2011.12.051. [DOI] [PubMed] [Google Scholar]
- Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–125. doi: 10.1542/peds.2010-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, Klein PM, Lan VJ, Joseph A, Jensen FE. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One. 2012;7:e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Long H, Zeng C, Li Y, Bi F, Wang J, Qian H, Xiao B. Rapamycin suppresses the recurrent excitatory circuits of dentate gyrus in a mouse model of temporal lobe epilepsy. Biochem Biophys Res Commun. 2012;420:199–204. doi: 10.1016/j.bbrc.2012.02.143. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Nickel J, Bohl J. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res. 1998;291:433–443. doi: 10.1007/s004410051013. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, Forte G, Holtman L, den Burger JC, Sinjewel A, de Vries HE, Aronica E, Gorter JA. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–1263. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Thompson KW, Suchomelova L, Niquet J. Brain energy metabolism during experimental neonatal seizures. Neurochem Res. 2010;35:2193–2198. doi: 10.1007/s11064-010-0339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wong M. Pentylenetetrazole-induced seizures cause acute, but not chronic, mTOR pathway activation in rat. Epilepsia. 2012;53:506–511. doi: 10.1111/j.1528-1167.2011.03384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]