

Abstract
The nonreceptor tyrosine kinase BMX (bone marrow tyrosine kinase gene on chromosome X) is abundant in various cell types and activated downstream of phosphatidylinositol-3 kinase (PI3K) and the kinase Src, but its substrates are unknown. Positional scanning peptide library screening revealed a marked preference for a priming phosphorylated tyrosine (pY) in the −1 position, indicating that BMX substrates may include multiple tyrosine kinases that are fully activated by pYpY sites in the kinase domain. BMX phosphorylated focal adhesion kinase (FAK) at Tyr577 subsequent to its Src-mediated phosphorylation at Tyr576. Loss of BMX by RNA interference or by genetic deletion in mouse embryonic fibroblasts (MEFs) markedly impaired FAK activity. Phosphorylation of the insulin receptor in the kinase domain at Tyr1189 and Tyr1190, as well as Tyr1185, and downstream phosphorylation of the kinase AKT at Thr308 were similarly impaired by BMX deficiency. However, insulin-induced phosphorylation of AKT at Ser473 was not impaired in Bmx knockout MEFs or liver tissue from Bmx knockout mice, which also showed increased insulin-stimulated glucose uptake, possibly because of decreased abundance of the phosphatase PHLPP (PH domain leucine-rich repeat protein phosphatase). Thus, by identifying the pYpY motif as a substrate for BMX, our findings suggest that BMX functions as a central regulator among multiple signaling pathways mediated by tyrosine kinases.
INTRODUCTION
Tec kinases, including TEC (tec protein tyrosine kinase), BTK (Bruton's tyrosine kinase), ITK [interleukin 2 (IL2)–inducible T cell kinase], TXK (TXK tyrosine kinase), and BMX (bone marrow tyrosine kinase gene on chromosome X), are a family of nonreceptor tyrosine kinases expressed primarily in hematopoietic cells that function in signaling pathways initiated by various extracellular stimuli. They are related in structure to the nonreceptor tyrosine kinases Src, CSK (C-terminal Src kinase), and ABL (c-Abl oncogene 1) in having Src homology 3 (SH3) domain followed by Src homology 2 (SH2) and tyrosine kinase domains (Fig. 1A). However, the Tec kinases lack the C-terminal tyrosine that negatively regulates Src kinases through an intramolecular interaction with the SH2 domain, and the extent to which they are autoinhibited in the basal state is not yet clear (1–4). The Tec kinases are also unique in having a pleckstrin homology (PH) domain, which is located at the N terminus and mediates membrane targeting in response to phosphatidylinositol-3 kinase (PI3K) activation by binding to phosphatidylinositol 3,4,5-triphosphate (PIP3) (5). This membrane targeting, which may also be facilitated by SH2 domain– and SH3 domain–mediated interactions, results in Src-mediated phosphorylation of a tyrosine in the kinase domain that activates the enzyme. Mutations in BTK, which is restricted to B cells and signals downstream of the B cell antigen receptor, are the cause of X-linked agammaglobulinemia (6). ITK, abundant in T cells, is similarly linked to the T cell antigen receptor, and its loss in mice results in a variety of T cell defects (7). Whereas these Tec kinases in lymphoid cells presumably phosphorylate multiple substrates, the only identified direct substrate is PLCg1 (phospholipase C γ1) (8).
Fig. 1. Identification of the BMX substrate motif.

(A) Schematic of the BMX structure. (B) Biotinylated peptide libraries that were phosphorylated using wild-type BMX (BmxWT) or kinase-deficient BMX (BmxKD) and then captured on membranes and analyzed. Representative images from one experiment are shown, and two additional independent experiments are shown in fig. S2. (C) Optimal BMX substrates based on either phosphorylated Tyr (pY) or isoleucine (I) at the −1 position. (D) In vitro kinase assay using the optimal pYY or IY peptide (Bmxtide 1 or 2, respectively) and BmxWT or BmxKD. (E) BMX substrate motif preference at the −2 position. BMX substrate motif peptide pool with Ala (A), Asp (D), Glu (E), Ser (S), or Thr (T) at the −2 position (and other positions fixed as in Bmxtide 1) was phosphorylated using BmxWT and then subjected to MS. The preference toward different amino acids at the −2 position was quantified as the percentage of dual tyrosine phosphorylation among all peptides with the corresponding amino acid at the −2 position and the priming pY.
BMX, also termed ETK (epithelial tyrosine kinase), was originally identified as a tyrosine kinase expressed in the myeloid cell lineage. However, in contrast to other Tec kinase family members, it is broadly expressed by cell types outside the lymphoid and myeloid lineage including arterial endothelium and epithelial cells (9–12). BMX also lacks a proline-rich motif between the SH3 and SH2 domains that is present in other Tec kinases, which may mediate an inhibitory interaction with the SH3 domain (13–15). Similarly to other Tec kinases, BMX can be activated downstream of PI3K by PH domain–mediated membrane targeting and Src-mediated phosphorylation of a kinase domain tyrosine (Tyr566) (5, 16). Alternatively, the BMX PH domain can bind to the FERM domain of focal adhesion kinase (FAK) and be activated by Src, which associates with FAK in response to integrin signaling (17). FAK can then phosphorylate a site on the BMX PH domain (Tyr40), which may relieve an inhibitory interaction between the PH domain and the kinase domain and facilitate phosphorylation by Src (17).
BMX-deficient mice have only modest defects that include decreased ischemia-induced angiogenesis (10, 18–20), impaired inflammatory responses (21), and decreased pressure overload-induced cardiac hypertrophy (22). However, increasing evidence indicates that BMX has roles in modulating multiple cellular processes including proliferation, differentiation, motility, and apoptosis (23–28), and increased BMX abundance has been implicated in neoplasia in skin (29) and prostate (30, 31). BMX has been reported to regulate multiple proteins including TNFR2 (tumor necrosis factor receptor 2), PAK1 (p21-activated protein kinase 1), p53, PIM1 (serine/threonine-protein kinase pim-1), and STAT3 (signal transducer and activator of transcription 3) (20, 24, 26, 32–34). More recently, BMX-dependent phosphorylation of STAT3 was found to be critical for the maintenance of glioblastoma stem cells (35). However, despite the apparent role of BMX in cell proliferation, transformation, and other processes, the spectrum of direct downstream targets of BMX remains elusive. We identified a novel substrate motif for BMX, pTyr-Tyr (herein referred to as pYY), and show that BMX can modulate the activation of multiple tyrosine kinases by phosphorylating the second tyrosine in kinase domain pYpY motifs that are required for full activity.
RESULTS
Determination of BMX substrate specificity
Determining which proteins are in vivo substrates of kinases has been a major bottleneck for elucidating signal transduction pathways. To gain insight into the substrates of BMX, we used a modified positional scanning peptide library approach to determine whether it has an optimal substrate phosphorylation motif (36). As a source of enzymes, we used LNCaP prostate cancer cells stably transfected with either wild-type purified 3xFlag-BMX (BmxWT) or kinase-deficient 3xFlag-BMX that harbors a K445M mutation (BmxKD) (37) (fig. S1). The assay used 198 biotinylated peptide libraries, each containing a tyrosine fixed at the central position and one additional position fixed to one of the 20 naturally occurring amino acids (Fig. 1B). All other positions contained a degenerate mixture of amino acids (excluding serine, threonine, and cysteine). Phosphothreonine and phosphotyrosine (herein referred to as pY) residues were included at the fixed positions to identify motifs for kinases that may require priming phosphorylation events. To avoid effects due to enzyme recruitment through SH2 or SH3 domains on a solid surface, we performed kinase assays in solution using [γ-32P]adenosine triphosphate ([γ-32P]ATP) with the active and kinase-deficient BMX. Biotinylated peptides then were captured with an avidin-coated membrane, and the preference for each amino acid at each position was determined by the relative level of radiolabel incorporation.
Wild-type BMX exhibited strong sequence selectivity at positions surrounding the phosphorylation site, whereas kinase-deficient BMX exhibited minimal activity (Fig. 1B), confirming that peptides were not being phosphorylated by a contaminating kinase. BMX showed a dramatic preference for a priming pYat position −1 and less of a marked preference for isoleucine at the −1 position. The kinase also preferred substrates with acidic residues at position −2 and showed less of a marked preference for additional tyrosine residues at −4, +2, or +3 and for tryptophan at the +1 position. Data from three independent library screens (Fig. 1B and fig. S2) were quantified to yield consensus optimal substrates with either pY (peptide 1; matrix shown in table S1) or isoleucine at the −1 position (“IY,” peptide 2) (Fig. 1C).
Using in vitro kinase assays, we confirmed that purified BMX (but not the kinase-deficient K445M mutant BMX) could phosphorylate the pY peptide (peptide 1) but not the IY peptide (peptide 2) (Fig. 1D). Although this did not rule out an IY-containing motif for BMX, we subsequently focused on peptide 1 and the pYY motif. Because peptide 1 contained four tyrosines that were potential phosphorylation sites, we next analyzed phosphorylated peptide 1 in vitro using mass spectrometry (MS). In addition to the nonphosphorylated peptide and the pYY peptide (phosphorylated at the priming −1 tyrosine), the only peptide detected was tyrosine-phosphorylated at both the central and −1 positions (pYpY). We also used MS to assess the effects of varying the −2 position in peptide 1. We modified the −2 position in peptide 1 to Ala, Asp, Glu, Ser, or Thr. A mixture of these peptides was then phosphorylated in vitro by BMX, and using MS, we determined the ratio of pYpY versus pYY for each peptide and found that BMX had a preference for Asp or Glu at the −2 position, and less of a preference for Thr or Ser (Fig. 1E). Together, these results indicated that pYY was a core substrate motif for BMX.
BMX phosphorylation of kinase domain pYY sites in multiple tyrosine kinases
Using MS data compiled in the PhosphoSitePlus database (http://www.phosphosite.org), we screened for proteins containing adjacent phosphotyrosines (pYpY) as potential BMX substrates. Strikingly, many receptor and nonreceptor tyrosine kinases met this BMX substrate motif requirement (table S2). The pYpY sites are highly conserved and, in the receptor tyrosine kinases, are located in the kinase domains. Previous studies have shown that these tyrosines can undergo autophosphorylation (or Src-mediated phosphorylation in the case of FAK) through dimerization in response to hormone or growth factor binding and that phosphorylation of both tyrosines is required for full kinase activity (38–42). However, additional kinases have not been implicated in this phosphorylation.
To validate the predicted substrate motif and determine whether BMX could phosphorylate these sites in tyrosine kinases after a priming phosphorylation, we used several available commercial antibodies recognizing the dual phosphotyrosine (pYpY) in specific tyrosine kinases. To assess BMX phosphorylation of MET (receptor tyrosine kinase c-MET), we cotransfected cells with expression vectors encoding MET and either wild-type or kinase-deficient BMX (or a control vector), and the cells were cultured in medium supplemented with 10% fetal bovine serum (FBS). Whole-cell lysates immunoblotted with an antibody recognizing the pYpY (pTyr1234 and pTyr1235, herein called pTyr1234/1235) in the MET kinase domain showed increased abundance of this dual-phosphorylated tyrosine motif in cells transfected with wild-type BMX compared with cells transfected with either control vector or kinase-deficient BMX (Fig. 2A, left panels). Similar results were obtained when we first immunoprecipitated MET and then blotted for pTyr1234/1235, supporting the specificity of the pTyr1234/1235 antibody for MET (Fig. 2A, right panels). Abundance of the dual-phosphorylated MET motif was decreased in cells transfected with the kinase-deficient BMX compared with control-transfected cells, possibly reflecting a dominant-negative effect on the endogenous BMX.
Fig. 2. BMX phosphorylation of MET, FAK, FGFR1, and ACK1.

(A) 293 cells were cotransfected with MET and wild-type BMX (BmxWT), kinase-deficient BMX (BmxKD), or empty vector control plasmids (Ctrl). Whole-cell lysates or immunoprecipitated (IP) MET was then immunoblotted as indicated. Detection of phosphorylated MET (pMET) was generally lower after MET immunoprecipitation, which may reflect either phosphatase activity or decreased reactivity of the MET antibody toward pMET. (B) HEK 293 cells were transfected with BmxWT, BmxKD, or empty vector control plasmids and either serum-starved for 48 hours (SFM) or serum-starved then stimulated with HGF for 10 min, and whole-cell lysates were then immunoblotted for endogenous pTyr1234 or pTyr1234/1235 MET. (C) 293 cells were cotransfected with 3xFlag-FAK and BmxWT, BmxKD, or empty vector control plasmids. Whole-cell lysates or Flag-immunoprecipitated FAK was immunoblotted. (D) 293 cells were transfected and treated with FBS as in (B), and whole-cell lysates were then immunoblotted for endogenous pTyr576/577 or pTyr576 FAK. (E and F) 293 cells were cotransfected with FGFR1 (E) or Myc-tagged ACK1 (F) and BmxWT, BmxKD, or empty vector control plasmids. Whole-cell lysates or immunoprecipitated FGFR1 (E) or Myc-tagged ACK1 (F) was immunoblotted. Band intensity ratios for the phosphorylated versus the total protein in this experiment are shown in lower panels. From three independent experiments, the mean band intensity ratio for pFGFR1 in cells transfected with BmxWT versus BmxKD was 2.0 ± 0.28 SEM (P < 0.05), and for pACK1 in cells transfected with BmxWT versus BmxKD, it was 11.5 ± 3.09 SEM (P < 0.05).
Because this dual tyrosine phosphorylation stimulates MET kinase activity, including its ability to autophosphorylate (or more precisely to transphosphorylate unphosphorylated MET), we anticipated that BMX would also increase the abundance of MET phosphorylated at Tyr1234. In deed, cells transfected with wild-type BMX also had increased abundance of this single Tyr1234-phosphorylated MET, assessed with an antibody selective for the pTyr1234 site (Fig. 2A). To further assess the requirement for a priming phosphorylation and whether BMX enhanced MET transphosphorylation, we next examined cells cultured in serum-free medium (SFM) and stimulated with hepatocyte growth factor (HGF). As expected, the basal activity of MET transphosphorylation, assessed by the abundance of pTyr1234 MET, was low in SFM-cultured cells and increased in response to HGF (Fig. 2B, lane 1 versus lane 4), which stimulates the autophosphorylation of MET at Tyr1234. BMX only slightly increased pTyr1234 and pTyr1234/1235 abundance in SFM-cultured cells, indicating that BMX did not directly phosphorylate Tyr1234. In contrast, BMX markedly increased the abundance of pTyr1234/1235 when cells were treated with HGF. This result is consistent with the requirement for a priming phosphorylation of the −1 tyrosine (Tyr1234) by MET through autophosphorylation or transphosphorylation, with BMX then fully activating MET by phosphorylating Tyr1235.
Cotransfection of wild-type BMX with FAK (3xFlag-tagged) similarly increased the abundance of dual-phosphorylated Tyr576/577 in the FAK kinase domain (Fig. 2C). This could be detected in the whole-cell lysates and in the anti-Flag immunoprecipitated fraction. In contrast, phosphorylation of Tyr576 (which is mediated by Src rather than by FAK autophosphorylation) was not markedly increased by BMX, and there was no detectable Tyr577 monophosphorylation, consistent with the notion that BMX phosphorylates Tyr577 only after a priming phosphorylation of Tyr576. Transfected BMX also increased the dual phosphorylation (pTyr576/577) of endogenous FAK in cells cultured in SFM, without increasing Src-mediated basal Tyr576 phosphorylation (Fig. 2D). As expected, stimulation with serum increased Src-mediated phosphorylation of FAK at Tyr576. Consistent with the pYY priming motif, this increase in pTyr576 was associated with a marked increase in pTyr576/577 in BMX-transfected cells (Fig. 2D). Using other available pYpY-specific antibodies in cells cultured under basal conditions (10% serum without further stimulation), we found that BMX modestly increased the abundance of dual-phosphorylated FGFR1 (fibroblast growth factor receptor 1) (Fig. 2E) and more markedly increased pYpY in the nonreceptor tyrosine kinase ACK1 (activated Cdc42-associated kinase 1) (Fig. 2F). Although some of these effects of BMX overexpression could be nonphysiological or indirect, these findings in conjunction with the BMX substrate motif data indicate that BMX can mediate the phosphorylation of pYY sites in kinase domains.
Confirmation that BMX phosphorylates the pYY site in the FAK kinase domain
We next focused on FAK because previous data showed that the PH domain of BMX can interact with the FERM domain of FAK, with subsequent membrane recruitment and activation of BMX by FAK-associated Src (17). Immunoblotting with a general anti-pY antibody that is not site-specific showed that tyrosine phosphorylation of endogenous, immuno-precipitated FAK was increased in cells overexpressing wild-type, but not kinase-deficient, BMX (Fig. 3A). It should be noted that we also observe substantial tyrosine phosphorylation of BMX, which may reflect both autophosphorylation and phosphorylation by other tyrosine kinases that are activated in cells overexpressing wild-type BMX.
Fig. 3. FAK is phosphorylated by BMX at Tyr577 after priming by pTyr576.

(A) Endogenous FAK was immunoprecipitated from LNCaP cells stably overexpressing wild-type (WT) or kinase-deficient (KD) BMX and blotted for pTyr. Arrows indicate the positions of FAK, BMX, and a BMX degradation product in the blot. (B) Recombinant Src-phosphorylated FAK was phosphorylated in vitro with recombinant BmxWT or BmxKD and analyzed by MS. Phosphopeptides detected in the BmxWT reaction are shown (pY and pYpY sites are underlined). (C) MS spectrum for the dual-phosphorylated (pTyr576/577 or pYpY) peptide. (D) Relative phosphorylated peptide signals in the BmxWT versus BmxKD samples quantified using the isotope-free liquid chromatography-tandem MS (LC/MS/MS) method. (E) 293 cells were cotransfected with the BmxKD or BmxWT plasmid and either a Flag wild-type or mutant FAK vector. Whole-cell lysates (top panels) or anti-Flag immunoprecipitated Flag-FAK (bottom panels) was then immunoblotted for total FAK and pTyr576/577 FAK.
To confirm that BMX was phosphorylating FAK at Tyr577 strictly after a priming phosphorylation at Tyr576 and to control for contaminating kinases, we used MS and a commercial recombinant FAK that was initially phosphorylated in vitro by Src and subjected to in vitro kinase reactions with purified wild-type or kinase-deficient BMX. The initial MS analyses were not comprehensive because of limited peptide coverage, but multiple tyrosine-phosphorylated peptides were identified. Those of FAK identified in the wild-type BMX reaction are shown in Fig. 3B, and complete phosphorylation maps of FAK with wild-type or kinase-deficient BMX are in fig. S3. As expected, we found tyrosine phosphorylation at Tyr576 alone, reflecting the in vitro phosphorylation mediated by Src. Dual phosphorylation at Tyr576/577 was observed with wild-type BMX, and we did not detect phosphorylation at Tyr577 alone. This latter result is again consistent with BMX requiring a priming phosphorylation and is also consistent with previous data showing that Tyr576 in FAK is phosphorylated before Tyr577 (39).
To generate more quantitative data, we used an isotope-free targeted mass spectrometric approach to quantify relative ratios of peptides phosphorylated at Tyr576, Tyr577, or both sites (spectra for the pTyr576/577 peptide are shown in Fig. 3C). Among all detected peptides covering this site in the wild-type BMX reaction, 81% were phosphorylated only at Tyr576, and 8.4% were dually phosphorylated at Tyr576 and Tyr577. In contrast, although 86% of peptides covering this site in the kinase-deficient BMX reaction were similarly phosphorylated at Tyr576 (mediated by the Src pretreatment), there were no peaks above background (0.66%) detected for the pTyr576/577 peptide, indicating an increase of at least 12-fold by wild-type BMX (Fig. 3D). Smaller increases in phosphorylation also were found at two other sites targeted for analysis: Tyr194 and Tyr441 (Fig. 3D). Tyr441 is an IY site, which may be an alternative BMX substrate as suggested from our peptide array (Fig. 1B).
Finally, we used a series of Flag-tagged FAK mutants to validate both the selectivity of the antibody for dual-phosphorylated FAK (pTyr576/577) and the in vitro FAK phosphorylation results. Cells were cotransfected with wild-type or mutant FAK vectors and wild-type or kinase-deficient BMX, and lysates were immunoblotted for pTyr576/577 FAK. The abundance of pTyr576/577 increased in cells expressing wild-type FAK and BMX, but this increase was impaired or abrogated by Y576A and Y577A mutations in FAK (Fig. 3E, upper panels). The low background of pTyr576/577 FAK detected in these mutant-overexpressing cells was likely from background reactions between BMX and endogenous FAK because pTyr576/577 FAK was not observed in these cells when we examined anti-Flag immunoprecipitates (Fig. 3E, lower panels). In contrast, Y194A or Y441A mutations did not prevent BMX-induced pTyr576/577. These results support the specificity of the antibody and further establish that BMX can phosphorylate FAK at Tyr577 in cells subsequent to the priming phosphorylation at Tyr576.
Enhancement of endogenous FAK activity by BMX-mediated phosphorylation
Previous studies have shown that BMX expression is increased in prostate cancer and that transgenic BMX overexpression in mouse prostate tissue induces intraepithelial neoplasia (30). Therefore, we next addressed the role of endogenous BMX in inducing the phosphorylation and activity of endogenous FAK. Using targeted small interfering RNA (siRNA), BMX was depleted in two human prostate cancer cell lines (LNCaP and VCS2) that normally have a relatively high abundance of endogenous BMX. LNCaP cells, which are PTEN (phosphatase and tensin homolog)–deficient and subsequently have increased activation of the PI3K pathway, were transfected with BMX-targeted or control siRNA and serum-starved for 72 hours, followed by serum stimulation and immunoblotting for pTyr576/577 FAK. BMX knockdown did not prevent the initial increase of this dual phosphorylation at 5 to 10 min after treatment with serum, but it decreased the duration of the response (Fig. 4A). In VCS2 cells (derived from a VCaP xenograft that relapsed after castration) (43), BMX depletion decreased dual phosphorylation of FAK at Tyr576/577 under serum-starved conditions and also impaired the response to serum stimulation (Fig. 4B). Because FAK links both growth factor and matrix or integrin stimulation to intracellular signals that promote cell migration, we also analyzed matrix-stimulated phosphorylation of FAK in VCS2 cells. BMX knockdown reduced the abundance of pTyr576/577 FAK in basal and fibronectin (FN)–stimulated conditions (Fig. 4C), indicating that BMX mediates FAK phosphorylation in response to physiological stimuli.
Fig. 4. Endogenous BMX regulates FAK through phosphorylation of the Tyr576/577 site.

(A and B) BMX-targeted or control siRNA–transfected LNCaP cells (A) or VCS2 cells (B) were serum-starved for 72 hours and then serum-stimulated for the indicated times, and whole-cell lysates were immunoblotted as indicated. Band intensity ratios in this experiment for the phosphorylated versus total FAK are shown in lower panels. At 20 to 30 min of serum stimulation in three independent experiments, the mean pFAK band intensity ratio in LNCaP cells transfected with control versus BMX siRNA was 2.03 ± 0.27 SEM (P < 0.05). From three independent VCS2 cell experiments at 10 to 20 min, this ratio was 2.77 ± 0.59 SEM (P < 0.05) for the control versus BMX siRNA–transfected cells. (C) BMX-targeted or control siRNA–transfected VCS2 cells were serum-starved for 72 hours, and then trypsinized and kept in suspension for 1 hour followed by plating onto FN-coated dishes for the indicated times. From three independent experiments at 30 to 45 min after plating, the mean of pFAK band intensity ratios for the control versus BMX siRNA–transfected cells was 1.51 ± 0.11 SEM (P < 0.05). (D) Bmx+ or Bmx− male MEFs were suspended for 1 hour (Ctrl) and then plated onto FN-coated dishes for 30 min (FN). A blot representative of three independent experiments is shown.
Because we could not completely ablate the abundance of BMX in LNCaP or VCS2 cells, we next turned to mouse embryonic fibroblasts (MEFs) from Bmx knockout mice (note that Bmx is on the X chromosome). Previous studies found that Bmx− mice had only a defect in ischemia-induced angiogenesis, which could reflect decreased FAK activity and migration of endothelial cells (18). Male littermates that were wild type for Bmx (Bmx+) or Bmx negative (Bmx−) and females that were heterozygous for Bmx (Bmx+/−) were identified by genotyping and used to generate short-term MEF lines (fig. S4). Notably, we detected lower basal abundance of pTyr576/577 FAK in Bmx− MEFs compared to Bmx+ MEFs (Fig. 4D). Moreover, the FN-induced increase in pTyr576/577 FAK was markedly blunted in Bmx− MEFs. Finally, the FAK-mediated phosphorylation of the scaffold protein p130Cas also was markedly impaired in Bmx− MEFs, consistent with the requirement for pTyr576/577 dual phosphorylation to fully activate FAK (Fig. 4D). Together, these findings indicate that BMX mediates the dual tyrosine phosphorylation and activation of FAK.
Because the Src-mediated phosphorylation of FAK at Tyr576 occurs at the plasma membrane, we predicted that the increased pTyr576/577 FAK in BMX-transfected cells would also be localized at the plasma membrane. To test this hypothesis, we used immunofluorescence to assess the cellular localization of transfected BMX and endogenous pTyr576/577 FAK. Human 293 cells transfected with wild-type BMX showed increased abundance of pTyr576/577 FAK compared with cells transfected with kinase-deficient BMX, and this dual-phosphorylated FAK was localized primarily at the plasma membrane (Fig. 5A). To further assess the localization of BMX and pTyr576/577 FAK in response to a physiological FAK stimulus, we introduced scratch wounds into COS7 cells transfected with wild-type or kinase-deficient BMX. Immunostaining performed at about 3 hours after wounding again showed that dual phosphorylation of FAK at Tyr576/577 was increased in the wild-type versus kinase-deficient BMX-transfected cells (Fig. 5B). The cellular distribution of BMX in these cells was variable, but there was substantial plasma membrane localization in most cells (Fig. 5B and fig. S5). In contrast, pTyr576/577 FAK in the wild-type BMX-transfected cells was consistently localized at the leading edge of cells migrating toward the wound (Fig. 5B and fig. S5). Quantitative fluorescence intensity profiles confirmed that pTyr576/577 was increased at the leading edge in wild-type BMX transfected cells (Fig. 5C) and showed that colocalization of BMX and pTyr576/577 was clearly increased in lamellipodia at the leading edge versus random areas of the cell (Fig. 5D). These results further indicate that BMX phosphorylated Tyr577 after a priming phosphorylation at Tyr576, which is mediated by Src at the leading edge in migrating cells.
Fig. 5. BMX regulates FAK-mediated migration.

(A) 293 cells transfected with BmxWT or BmxKD were immunostained for BMX and pTyr576/Y577 FAK. Scale bars, 20 μm. Images are representative fields from one of three independent experiments. (B) Scratch wounds were introduced into COS7 cells transfected and immunostained as in (A), and pictures of the leading edge (arrows) were taken after 3 hours. Scale bars, 20 μm. Boxed region is magnified in the middle row. Images are representative fields from one of three independent experiments. (C) BMX (green) and pTyr576/Y577 FAK (red) fluorescence intensities along the axis defined by the white arrows in (B) were assessed using Zeiss LSM (WT, above; KD, below). Circles denote the leading edge. Data are results from scanning 10 cells each transfected with BmxWT or BmxKD. (D) Colocalization of BMX and pTyr576/Y577 FAK in lamellipodia at leading edges (n = 22) versus random areas (n = 20) from three independent experiments was assessed using Volocity. (E) Scratch wounds were introduced into confluent Bmx+/−, Bmx+, or Bmx− MEFs, and leading edges were photographed at 0 and 9 hours. Migrated distance was normalized to the #1 Bmx− group. Data are means ± SEM from three experiments. ***P < 0.001. (F) Scratch wounds were introduced into confluent Bmx− MEFs stably overexpressing BmxWT, BmxKD, or empty vector (EV) and photographed as in (E). Migrated distance was normalized to the EV group. Data are means ± SEM from three independent experiments. *P < 0.05. (G) Bmx− MEF cells stably overexpressing BmxWT were preincubated with FAK inhibitor (PF562271) for 4 hours. Scratch wounds were introduced and photographed as in (E). Migrated distance was normalized to the control group. Data are means ± SEM from three experiments. ***P < 0.001.
We next performed quantitative wound healing assays to determine the influence of endogenous BMX on cell migration. Scratch wounds were introduced on MEFs grown to confluency, and pictures of the leading edge were taken at time points 0 and 9 hours. Bmx− male MEFs showed impaired wound healing compared to Bmx+ male MEFs and Bmx+/− female MEFs (Fig. 5E). BMX depletion by siRNA similarly impaired wound healing in LNCaP cells (fig. S6). This migration defect in Bmx− MEFs was corrected by transfection with wild-type, but not kinase-deficient, BMX (Fig. 5F). Moreover, this stimulatory effect of BMX could be blocked by an antagonist of FAK (PF562271), further indicating that it was mediated through FAK (Fig. 5G). Together, these findings show that BMX activated FAK by phosphorylating Tyr577 subsequent to a priming phosphorylation by Src at Tyr576.
BMX modulation of insulin signaling
An additional candidate substrate for BMX was the insulin receptor (InsR), which, in addition to pTyr1189/1190 in its kinase domain, undergoes phosphorylation at Tyr1185 to achieve full activation (table S2) (41, 44–46). As expected, immunoblotting for pTyr1189/1190 and pTyr1185 InsR in control LNCaP cells (transfected with nontargeting siRNA) showed that insulin rapidly increased the abundance of pTyr1189/1190 and pTyr1185 (Fig. 6A). BMX depletion by siRNA impaired the insulin-stimulated increase in pTyr1189/1190 and markedly impaired the increase in pTyr1185. Tyr1185 is preceded by an isoleucine, suggesting that it may be an IY motif substrate for BMX. However, its decreased phosphorylation may instead be secondary to the decreased pTyr1189/1190 and subsequently decreased autophosphorylation or transphosphorylation of Tyr1185.
Fig. 6. BMX regulates the phosphorylation and signaling of the InsR.

(A) LNCaP cells transfected with BMX-targeted or nontargeted control siRNA were serum-starved for 48 hours, then stimulated with insulin (Ins; 100 nM) for 0 to 30 min, and immunoblotted for pTyr1189/1190 or pTyr1185 InsR. Intensities of the pInsR Tyr1189/1190 versus total InsR bands in this experiment are in the bottom panel. From three independent experiments at 5 to 10 min of stimulation, the mean pInsR Tyr1189/1190 intensity in the control versus BMX siRNA–transfected cells was 2.13 ± 0.42 SEM (P < 0.05). (B) Bmx+ and Bmx− MEFs were serum-starved for 48 hours and then stimulated with insulin for 10 min, and whole-cell lysates were immunoblotted as indicated. From three independent experiments, the mean pInsR Tyr1189/1190 intensity in control versus BMX siRNA–transfected cells treated with 100 nM insulin was 2.49 ± 0.13 SEM (P < 0.001). (C) Whole-cell lysates from independent Bmx+ and Bmx− MEF cell lines or livers were blotted as indicated. (D) After an overnight fast, 8- to 10-month-old Bmx+ and Bmx− mice were injected intraperitoneally with glucose (2 g/kg). Liver was harvested after 15 min, and lysates were immunoblotted. The normalized mean pTyr1189/1190 intensity in the three Bmx+ versus three Bmx− mice was 1.12 versus 0.41 (P < 0.01). (E) Equal amounts of liver protein extracts from Bmx+ and Bmx− mice at 15 min after glucose challenge were blotted for GSK3β pSer9 and S6 pSer235/236. Signals were quantified and further normalized to actin, and quartile plots are shown (*P < 0.05, **P < 0.01).
Insulin-stimulated phosphorylation of InsR was similarly impaired in Bmx− MEFs despite higher abundance of total InsR in Bmx− compared with Bmx+ MEFs (Fig. 6B). Insulin-stimulated phosphorylation of the kinase AKT at Thr308, which is mediated by PDK1 (phosphoinositide-dependent kinase 1) immediately downstream of PI3K, was also decreased in Bmx− MEFs, as was the phosphorylation of the AKT substrate GSK3β (glycogen synthase kinase 3β) and the downstream mTORC1 (mammalian target of rapamycin complex 1) substrate S6 kinase (Fig. 6B). In contrast, basal and insulin-stimulated AKT phosphorylation at Ser473, which is mediated by mTORC2 and generally correlates with Thr308 phosphorylation, was not decreased in the Bmx− MEFs (Fig. 6B). AKT is dephosphorylated at Ser473 by the PH domain leucine-rich repeat protein phosphatase (PHLPP) (47). By immunoblotting with an antibody that recognizes both the α and β isoforms of PHLPP1 and PHLPP2, we detected a major band at the predicted mass of PHLPP1β (about 170 kD). PHLPP abundance was markedly decreased in three independent Bmx− MEF lines, which also had increased abundance of InsR compared to Bmx+ MEFs (Fig. 6C). Moreover, we also observed increased abundance of total InsR and decreased PHLPP in vivo in livers from Bmx− mice (Fig. 6C), indicating that the selective increase of pSer473 AKT may be a result of decreased abundance of PHLPP in Bmx− mice.
Compensatory mechanisms in Bmx− mice mediating increased insulin sensitivity
The increased abundance of InsR and decreased abundance of PHLPP appear to be adaptations in Bmx− MEFs and mice to restore InsR signaling and downstream PI3K-AKT pathway activity, and these observations support a physiological role for BMX in regulating these pathways. To assess the effects of the loss of BMX on insulin signaling in vivo, we examined liver tissue from mice that were sacrificed 15 min after injection with glucose to stimulate insulin secretion. As observed in the Bmx− MEFs, phosphorylation of InsR at Tyr1189/1190 and that of AKT at Thr308 were reduced, whereas the abundance of pSer473AKT was increased (Fig. 6D). Moreover, analysis of phosphorylation of GSK3β at Ser9 (a direct AKT substrate) and of S6 kinase (a TORC1 substrate downstream of AKT) in livers from a series of Bmx+ and Bmx− mice at 15 min after a glucose challenge showed that AKT activity was increased in the Bmx− mice (Fig. 6E), confirming results obtained in MEFs.
To determine whether these changes in Bmx− mice had effects on glucose regulation, we did glucose tolerance testing. Fasting glucose levels were comparable in Bmx− and Bmx+ mice, but peak serum glucose concentrations in response to a glucose challenge were significantly lower in the Bmx− mice (Fig. 7A). Moreover, we found that serum insulin concentration in response to a glucose challenge was lower in the Bmx− mice, indicating that these mice had increased sensitivity to insulin (Fig. 7B). Finally, to directly assess systemic responses to insulin, we measured serum glucose concentrations after insulin injection. In Fig. 7C, the decline in serum glucose was greater in the Bmx− mice, confirming their increased insulin sensitivity. These findings demonstrate that loss of BMX leads to adaptations that include increased abundance, but decreased insulin-stimulated activity, of InsR and decreased abundance of PHLPP, which paradoxically enhance responses to insulin.
Fig. 7. Bmx− mice are hypersensitive to insulin.
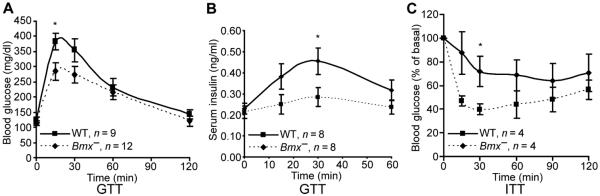
(A and B) Bmx+ and Bmx− mice were injected with glucose, and concentrations of serum glucose (A) and insulin (B) were measured at the indicated times. GTT, glucose tolerance test. (C) After a 4-hour fast, 8- to 10-month-old Bmx+ and Bmx− mice were injected with insulin (0.75 U/kg), and blood glucose was measured at the indicated times. ITT, insulin tolerance test. Data are means ± SEM from the indicated number of mice for each group. *P < 0.05.
DISCUSSION
BMX has been implicated in multiple cellular pathways, but its direct substrates have remained obscure. The unique pYY substrate preference of BMX demonstrated in this study suggested that BMX may mediate the phosphorylation of pYpY sites in the kinase domains of multiple receptor tyrosine kinases. Previous studies of InsR and other receptor tyrosine kinases have established that the first Y undergoes autophosphorylation or transphosphorylation in response to ligand binding and that phosphorylation of the second Y is required for full kinase activity. In the case of FAK, the first Y is phosphorylated by Src, but phosphorylation of the second Y is required for full kinase activity (38, 40, 41, 44–46, 48). Although this second Y may also undergo autophosphorylation, we propose that BMX functions to amplify receptor tyrosine kinase signaling by phosphorylation of the second Y subsequent to the priming phosphorylation of the first Y. Because BMX activity is activated by PI3K and Src, this function of BMX provides the cell with a mechanism to broadly modulate the activity of multiple tyro-sine kinases in response to stimulation by PI3K or Src.
Using available pYpY-specific antibodies, we confirmed that phosphorylation of these sites in the tyrosine kinases MET, FAK, FGFR1, ACK1, and InsR was increased by BMX. We further used MS to confirm that BMX phosphorylates Tyr577 in the FAK kinase domain subsequent to a priming phosphorylation at Tyr576 by Src. BMX may associate constitutively with some tyrosine kinases through its SH3 domain, or it may be recruited through its SH2 domain in response to an initial priming phosphorylation so that it can mediate transphosphorylation to achieve full activation. The interaction with FAK may be unique because it appears to be mediated by the FERM domain of FAK and the PH domain of BMX, and Src recruitment to this complex may serve to both activate BMX and phosphorylate the priming Tyr576 in the FAK kinase domain (17).
Although depletion of BMX suppresses multiple signal transduction pathways, we found that loss of BMX activity, as occurs in Bmx− mice, led to adaptations that include decreased abundance of PHLPP, which may actually enhance downstream signaling in response to some stimuli. The translation of PHLPP is positively regulated by mTORC1, consistent with a feedback loop wherein loss of BMX and subsequent decreases in receptor tyrosine kinase signaling to mTORC1 through PI3K-AKT and MAPKs (mitogen-activated protein kinases) result in decreased mTORC1 activity, decreased PHLPP, and increased pSer473AKT. Remarkably, whereas BMX deletion decreased insulin-stimulated activation of InsR, these adaptations (which also include increased abundance of InsR) actually increased sensitivity to insulin in vitro and in vivo in Bmx− mice. Other factors regulating insulin signaling, such as IRS-1 phosphorylation, also may be altered in Bmx− mice and are currently being examined.
BMX has been reported to modulate the activity of multiple pathways and contribute to tumorgenesis in cell line models (26, 32, 34, 49). Increased expression of endogenous BMX occurs in keratinocytes during wound healing, and transgenic over-expression of BMX in keratinocytes results in increased proliferation, chronic inflammation, and angiogenesis (29). BMX has also been implicated in prostate cancer progression (30) and may directly phosphorylate androgen receptor (31). Most recently, increased BMX in glioblastoma multiforme has been linked to increased STAT3 activation and maintenance of tumor stem cells (35). Although sites phosphorylated on STAT3 do not fit the pYpY or pIpY motifs, we cannot rule out STAT3 as a direct BMX substrate under some conditions. As well, BMX could possibly activate STAT3 through JAKs (Janus kinases) that do contain pYpY motifs in their kinase domain. Alternatively, BMX may indirectly increase STAT3 phosphorylation by contributing to the broad increases in the activities of multiple receptor tyrosine kinases that have been reported in glioblastoma multiforme and prostate cancer.
Further studies are clearly needed to establish the spectrum of BMX substrates, in particular cells and tissues, and to establish mechanistic links between phosphorylation of these substrates and the diverse functions attributed to BMX. Nonetheless, this study has revealed a new mechanism of action for BMX that has broad implications for the regulation of multiple signal transduction pathways. The central role of BMX in modulating tyrosine kinase signal transduction pathways indicates that BMX inhibitors may be efficacious in many cancers that are characterized by increased tyrosine kinase signaling. Conversely, chronic exposure to BMX inhibitors may enhance signaling downstream of some receptor tyrosine kinases and could be efficacious in some diseases including type 2 diabetes characterized by insulin resistance.
MATERIALS AND METHODS
Materials
Antibodies against pFAK (pTyr576/577), pIGF1R (pTyr1165/1166) or InsR (pTyr1189/1190), pMET (pTyr1234/1235), pGSK3β (pSer9), and pp130Cas (pTyr410) were from Cell Signaling Technology. Antibodies against Flag M2, FAK, pFAK (pTyr576), pFAK (pTyr577), BMX, and β-actin were from Santa Cruz Biotechnology. Antibodies against pTyr (4G10) and tubulin were from Millipore, and antibody against pMET (Tyr1234) was from GeneTex. Bmxtides were from GenScript Corporation. BMX-targeted and control siRNAs were from Dharmacon. 3xFlag-FAK was produced by inserting FAK (pCMV-SPORT6-FAK, Open Biosystems) into p3XFlag-CMV (Sigma) between the Hind III and Bam H1 sites, and mutants were then generated with QuikChange Site-Directed Mutagenesis Kit (Stratagene). Purified FAK that was activated by Src was from Invitrogen. Bmx− mice were provided by K. Alitaro (University of Helsinki, Finland).
Cell culture and transfection
Human embryonic kidney (HEK) 293 and COS7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS (HyClone). LNCaP and VCS2 cells were cultured in RPMI 1640 supplemented with 10% FBS. Primary MEFs were isolated from E13.5 embryos derived from a Bmx+/− female and a Bmx− male breeding pair. The cells were trypsinized and cultured in DMEM supplemented with 10% FBS. The first generation was genotyped, and cells were used through no more than five generations. Transfections were performed with Lipofectamine 2000 (Invitrogen). For wound healing assays, cells were grown to confluence, and pipette tips were used to injure the cell surface. Fresh medium was then added, and the injured areas were photographed at the time points indicated.
Immunoblotting and immunofluorescence
Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 1 mM β-glycerophosphate, 1 mM pyrophosphate, 100 mM sodium fluoride, 1 mM Na3VO4, and protease inhibitors]. Lysates were sonicated for 10 s and centrifuged at 13,000 rpm at 4°C for 15 min. For anti-3xFlag immunoprecipitation, equal amounts of protein (1 to 5 mg) were mixed with 20 to 50 μl of anti-3xFlag–conjugated (M2) agarose beads and incubated at 4°C overnight with continuous agitation. The beads were washed extensively with RIPA buffer followed by tris-buffered saline (TBS) buffer, and beads were eluted with 2x Laemmli sample buffer. Samples were boiled for 5 min and then resolved on 4 to 12% NuPAGE gels (Invitrogen) followed by membrane transfer. Membranes were blocked with 5% milk (or 5% bovine serum albumin for phosphospecific antibodies) in TBS/0.1% Tween 20 at room temperature for 1 hour and incubated with primary antibodies overnight at 4°C. Membranes were then incubated with secondary antibodies at room temperature for 1 hour and developed by enhanced chemiluminescence. All blots are representative of at least three experiments.
Mice were fasted overnight, injected intraperitoneally with glucose (2 g/kg), and sacrificed after 15 min to assess the effects of BMX on InsR in vivo. Tissues were rapidly removed, frozen in liquid nitrogen, and homogenates were immunoblotted as above.
For immunofluorescence, cells grown on glass coverslips were fixed with formaldehyde and then incubated with primary antibodies followed by secondary antibodies conjugated with Alexa Fluor (Invitrogen); the nuclei were stained with Hoechst. All micrographs were taken at the same confocal microscope setting.
In vitro kinase assays
Purified BMX was mixed with substrate (FAK or Bmxtides), kinase buffer [final 20 mM Hepes (pH 7.5), 10 mM MgCl2, 20 mM β-glycerophosphate, 1 mM dithiothreitol, 20 μM ATP, 5 mM Na3VO4], and 1 μCi of [γ-32P] ATP (omitted for cold in vitro kinase assays) for 30 min at 30°C. Reactions were stopped with 10 mM EDTA and Laemmli sample buffer. Samples were resolved by 4 to 12% NuPAGE gel and visualized by autoradiography. The positional scanning peptide library assay was performed as described previously (36, 50). Labeled peptide libraries were spotted onto avidin-coated filter sheets (Promega SAM2 Biotin Capture Membrane), which were washed, dried, and exposed to a phosphorimager screen.
Tandem mass spectrometry (LC/MS/MS)
For MS experiments, FAK protein was separated with SDS–polyacrylamide gel electrophoresis, stained with Coomassie blue, and the FAK band was excised from the gel. Samples were subjected to reduction with dithiothreitol, alkylation with iodoacetamide, and in-gel digestion with trypsin or chymotrypsin overnight at pH 8.3, followed by reversed-phase microcapillary LC/MS/MS. LC/MS/MS was performed with an Easy-nLC nanoflow high-performance liquid chromatograph (Proxeon Biosciences) with a self-packed 75-mm internal diameter × 15-cm C18 column coupled to an LTQ Orbitrap XL mass spectrometer (Thermo Scientific) in the data-dependent acquisition and positive ion mode at 300 nl/min. Peptide ions from BMX-predicted phosphorylation sites were also targeted in MS/MS mode for quantitative analyses. MS/MS spectra collected via collision-induced dissociation in the ion trap were searched against the concatenated target and decoy (reversed) single-entry FAK and full Swiss-Prot protein databases with Sequest (Proteomics Browser Software, Thermo Scientific) with differential modifications for Ser, Thr, or Tyr phosphorylation (+79.97) and the sample processing artifacts Met oxidation (+15.99), deamidation of Asn and Gln (+0.984), and Cys alkylation (+57.02).
Phosphorylated and unphosphorylated peptide sequences were identified if they initially passed the following Sequest scoring thresholds against the target database: 1+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, P ≥ 5; 2+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, P ≥ 5; 3+ ions, Xcorr ≥ 2.60, Sf ≥ 0.4, P ≥ 5 against the target protein database. Passing MS/MS spectra were manually inspected to be sure that all b and y fragment ions aligned with the assigned sequence and modification sites. Determination of the exact sites of phosphorylation was aided with FuzzyIons and GraphMod, and phosphorylation site maps were created with the ProteinReport software (Proteomics Browser Software Suite, Thermo Scientific). False discovery rates of peptide hits (phosphorylated and unphosphorylated) were estimated below 1.5% based on reversed database hits (51–54).
Relative quantification of phosphorylation sites
For relative quantification of phosphorylated peptide signals, an isotope-free (label-free) method was used by first integrating the total ion counts (TIC) for each MS/MS sequencing event during a targeted ion MS/MS (TIMM) experiment or a data-dependent acquisition. For each targeted phosphorylation site, ratios of the phosphorylated peptide signal (TIC of phosphorylated form) to the total peptide signal (TIC of phosphorylated form + TIC of nonphosphorylated form) for both the untreated and treated samples were calculated according to the following equation: TICPO4/(TICPO4 + TICnonPO4) = ratio of phosphopeptide signal (RPO4).
These ratios of phosphopeptide signal were then compared to the same phosphopeptide ratios from the untreated samples according to the following equation: [(RPO4Unstimulated/RPO4Stimulated) − 1] × 100 = % change in phosphorylation upon treatment. Whereas a direct comparison of phosphopeptide signals between different experimental conditions is not accurate because of differences in sample content, a comparison of the relative ratios of the phosphorylated to nonphosphorylated peptide forms between samples is an accurate measure of signal change because the total peptide signal (modified and unmodified) is measured. The above calculations were performed manually with Microsoft Excel and an automated in-house-developed software called Protein Modification Quantifier v1.0 (Beth Israel Deaconess Medical Center) (55–58).
Metabolic assays
Mice aged 8 to 10 months were used for metabolic assays. For glucose tolerance test, mice were fasted for 16 hours, blood was drawn, and mice were injected intraperitoneally with dextrose (2 g/kg). Blood glucose and insulin measurement were obtained from the tail vein with OneTouch Ultra (LifeScan) and Ultra Sensitive Mouse Insulin ELISA Kits (Crystal Chem). Tissues were collected 15 min after injection and frozen in liquid nitrogen until analysis. For insulin tolerance tests, mice were fasted for 4 hours and were given human recombinant insulin (0.75 U/kg body weight) (Invitrogen) intraperitoneally. Blood glucose level was monitored at the indicated times.
Statistical analysis
Unless otherwise indicated, results are expressed as means ± SE. Statistical significance was determined by a two-sided Student's t test, with P < 0.05 considered statistically significant.
Supplementary Material
Acknowledgments
We acknowledge X. Yang for help with MS experiments. We thank K. Alitaro (University of Helsinki, Finland) for Bmx− mice. Myc-ACK1 plasmid was a gift from W. Yang (Geisinger System Services).
Funding: This work was supported by NIH grants P01CA089021 (S.P.B. and L.C.C.), P01CA120964-04 (J.M.A.), and R01GM56203 (L.C.C.); NIH Dana-Farber/Harvard Cancer Center Support Grant P30CA006516-46 (J.M.A.); Department of Defense grants W81XWH-08-1-0675 (S.P.B.) and W81XWH-09-1-0435 (S.P.B.); NIH K99 grant K99CA166507 (C.C.); and the Prostate Cancer Foundation.
Footnotes
SUPPLEMENTARY MATERIALS www.sciencesignaling.org/cgi/content/full/6/277/ra40/DC1
Author contributions: S.C. devised and conducted experiments, interpreted results, and co-authored the manuscript. X.J. initiated the project, devised and conducted experiments, and interpreted results. C.A.G. devised and conducted peptide library experiments and interpreted results. J.M.A. conducted and interpreted MS studies. N.I.S. assisted with experiments and interpreted results. C.C. devised and conducted experiments and interpreted results. L.C.C. devised peptide library experiments and interpreted results. S.P.B. devised experiments, interpreted results, and co-authored the manuscript.
Competing interests: The Beth Israel Deaconess Medical Center has submitted a patent for therapeutic use of BMX inhibitors.
Data and materials availability: MS data have been deposited in the PRIDE (PRoteomics IDEntifications) Data Repository with accession number 4172.
REFERENCES AND NOTES
- 1.Afar DE, Park H, Howell BW, Rawlings DJ, Cooper J, Witte ON. Regulation of Btk by Src family tyrosine kinases. Mol. Cell. Biol. 1996;16:3465–3471. doi: 10.1128/mcb.16.7.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- 3.Nore BF, Mattsson PT, Antonsson P, Backesjö CM, Westlund A, Lennartsson J, Hansson H, Löw P, Rönnstrand L, Smith CI. Identification of phosphorylation sites within the SH3 domains of Tec family tyrosine kinases. Biochim. Biophys. Acta. 2003;1645:123–132. doi: 10.1016/s1570-9639(02)00524-1. [DOI] [PubMed] [Google Scholar]
- 4.Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, Scharenberg AM, Kinet JP, Witte ON. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4:515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- 5.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19:5651–5661. doi: 10.1038/sj.onc.1203958. [DOI] [PubMed] [Google Scholar]
- 6.de Weers M, Mensink RG, Kraakman ME, Schuurman RK, Hendriks RW. Mutation analysis of the Bruton's tyrosine kinase gene in X-linked agammaglobulinemia: Identification of a mutation which affects the same codon as is altered in immunodeficient xid mice. Hum. Mol. Genet. 1994;3:161–166. doi: 10.1093/hmg/3.1.161. [DOI] [PubMed] [Google Scholar]
- 7.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, Sher A, Varmus HE, Lenardo MJ, Schwartzberg PL. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 9.Chott A, Sun Z, Morganstern D, Pan J, Li T, Susani M, Mosberger I, Upton MP, Bubley GJ, Balk SP. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am. J. Pathol. 1999;155:1271–1279. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajantie I, Ekman N, Iljin K, Arighi E, Gunji Y, Kaukonen J, Palotie A, Dewerchin M, Carmeliet P, Alitalo K. Bmx tyrosine kinase has a redundant function downstream of angiopoietin and vascular endothelial growth factor receptors in arterial endothelium. Mol. Cell. Biol. 2001;21:4647–4655. doi: 10.1128/MCB.21.14.4647-4655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson D, He F, Pretlow T, Kung HJ. A tyrosine kinase profile of prostate carcinoma. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5958–5962. doi: 10.1073/pnas.93.12.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamagnone L, Lahtinen I, Mustonen T, Virtaneva K, Francis F, Muscatelli F, Alitalo R, Smith CI, Larsson C, Alitalo K. BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene. 1994;9:3683–3688. [PubMed] [Google Scholar]
- 13.Pursglove SE, Mulhern TD, Mackay JP, Hinds MG, Booker GW. The solution structure and intramolecular associations of the Tec kinase SRC homology 3 domain. J. Biol. Chem. 2002;277:755–762. doi: 10.1074/jbc.M108318200. [DOI] [PubMed] [Google Scholar]
- 14.Joseph RE, Andreotti AH. Conformational snapshots of Tec kinases during signaling. Immunol. Rev. 2009;228:74–92. doi: 10.1111/j.1600-065X.2008.00740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takesono A, Finkelstein LD, Schwartzberg PL. Beyond calcium: New signaling pathways for Tec family kinases. J. Cell Sci. 2002;115:3039–3048. doi: 10.1242/jcs.115.15.3039. [DOI] [PubMed] [Google Scholar]
- 16.Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3644–3649. doi: 10.1073/pnas.95.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen R, Kim O, Li M, Xiong X, Guan JL, Kung HJ, Chen H, Shimizu Y, Qiu Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 2001;3:439–444. doi: 10.1038/35074500. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Luo Y, Tang S, Rajantie I, Salven P, Heil M, Zhang R, Luo D, Li X, Chi H, Yu J, Carmeliet P, Schaper W, Sinusas AJ, Sessa WC, Alitalo K, Min W. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J. Clin. Invest. 2006;116:2344–2355. doi: 10.1172/JCI28123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, Min W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J. Biol. Chem. 2003;278:51267–51276. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 20.Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: Role in endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2002;22:7512–7523. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottar-Guillier M, Dodeller F, Huesken D, Iourgenko V, Mickanin C, Labow M, Gaveriaux S, Kinzel B, Mueller M, Alitalo K, Littlewood-Evans A, Cenni B. The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J. Immunol. 2011;186:6014–6023. doi: 10.4049/jimmunol.1002813. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell-Jordan SA, Holopainen T, Ren S, Wang S, Warburton S, Zhang MJ, Alitalo K, Wang Y, Vondriska TM. Loss of Bmx nonreceptor tyrosine kinase prevents pressure overload-induced cardiac hypertrophy. Circ. Res. 2008;103:1359–1362. doi: 10.1161/CIRCRESAHA.108.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu T, Thotala D, Geng L, Hallahan DE, Willey CD. Bone marrow X kinase–mediated signal transduction in irradiated vascular endothelium. Cancer Res. 2008;68:2861–2869. doi: 10.1158/0008-5472.CAN-07-5743. [DOI] [PubMed] [Google Scholar]
- 24.Jiang T, Guo Z, Dai B, Kang M, Ann DK, Kung HJ, Qiu Y. Bi-directional regulation between tyrosine kinase Etk/BMX and tumor suppressor p53 in response to DNA damage. J. Biol. Chem. 2004;279:50181–50189. doi: 10.1074/jbc.M409108200. [DOI] [PubMed] [Google Scholar]
- 25.Kim O, Yang J, Qiu Y. Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J. Biol. Chem. 2002;277:30066–30071. doi: 10.1074/jbc.M201713200. [DOI] [PubMed] [Google Scholar]
- 26.Bagheri-Yarmand R, Mandal M, Taludker AH, Wang RA, Vadlamudi RK, Kung HJ, Kumar R. Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity of breast cancer cells. J. Biol. Chem. 2001;276:29403–29409. doi: 10.1074/jbc.M103129200. [DOI] [PubMed] [Google Scholar]
- 27.Chau CH, Chen KY, Deng HT, Kim KJ, Hosoya K, Terasaki T, Shih HM, Ann DK. Coordinating Etk/Bmx activation and VEGF upregulation to promote cell survival and proliferation. Oncogene. 2002;21:8817–8829. doi: 10.1038/sj.onc.1206032. [DOI] [PubMed] [Google Scholar]
- 28.Semaan N, Alsaleh G, Gottenberg JE, Wachsmann D, Sibilia J. Etk/BMX, a Btk family tyrosine kinase, and Mal contribute to the cross-talk between MyD88 and FAK pathways. J. Immunol. 2008;180:3485–3491. doi: 10.4049/jimmunol.180.5.3485. [DOI] [PubMed] [Google Scholar]
- 29.Paavonen K, Ekman N, Wirzenius M, Rajantie I, Poutanen M, Alitalo K. Bmx tyrosine kinase transgene induces skin hyperplasia, inflammatory angiogenesis, and accelerated wound healing. Mol. Biol. Cell. 2004;15:4226–4233. doi: 10.1091/mbc.E04-03-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai B, Kim O, Xie Y, Guo Z, Xu K, Wang B, Kong X, Melamed J, Chen H, Bieberich CJ, Borowsky AD, Kung HJ, Wei G, Ostrowski MC, Brodie A, Qiu Y. Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 2006;66:8058–8064. doi: 10.1158/0008-5472.CAN-06-1364. [DOI] [PubMed] [Google Scholar]
- 31.Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, Li W, Guo Z, Xu K, Kim O, Kong X, Melamed J, Qiu S, Chen H, Qiu Y. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70:5587–5596. doi: 10.1158/0008-5472.CAN-09-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie Y, Xu K, Dai B, Guo Z, Jiang T, Chen H, Qiu Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene. 2006;25:70–78. doi: 10.1038/sj.onc.1209058. [DOI] [PubMed] [Google Scholar]
- 33.Saharinen P, Ekman N, Sarvas K, Parker P, Alitalo K, Silvennoinen O. The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cδ. Blood. 1997;90:4341–4353. [PubMed] [Google Scholar]
- 34.Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS, Shih HM, Kung HJ, Chen RH. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol. Cell. Biol. 2000;20:2043–2054. doi: 10.1128/mcb.20.6.2043-2054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, MacSwords J, Eyler CE, McLendon RE, Heddleston JM, Shou W, Hambardzumyan D, Lee J, Hjelmeland AB, Sloan AE, Bredel M, Stark GR, Rich JN, Bao S. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19:498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 37.Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP. Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J. Biol. Chem. 2007;282:32689–32698. doi: 10.1074/jbc.M703412200. [DOI] [PubMed] [Google Scholar]
- 38.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell. Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciccimaro E, Hevko J, Blair IA. Analysis of phosphorylation sites on focal adhesion kinase using nanospray liquid chromatography/multiple reaction monitoring mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20:3681–3692. doi: 10.1002/rcm.2783. [DOI] [PubMed] [Google Scholar]
- 40.Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001;8:1058–1063. doi: 10.1038/nsb721. [DOI] [PubMed] [Google Scholar]
- 41.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bae JH, Lew ED, Yuzawa S, Tomé F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, Nelson PS, Liu XS, Brown M, Balk SP. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White MF, Shoelson SE, Keutmann H, Kahn CR. A cascade of tyrosine autophosphorylation in the b-subunit activates the phosphotransferase of the insulin receptor. J. Biol. Chem. 1998;263:2969–2980. [PubMed] [Google Scholar]
- 45.Lopaczynski W, Terry C, Nissley P. Autophosphorylation of the insulin-like growth factor I receptor cytoplasmic domain. Biochem. Biophys. Res. Commun. 2000;279:955–960. doi: 10.1006/bbrc.2000.4046. [DOI] [PubMed] [Google Scholar]
- 46.Baserga R. The IGF-I receptor in cancer research. Exp. Cell Res. 1999;253:1–6. doi: 10.1006/excr.1999.4667. [DOI] [PubMed] [Google Scholar]
- 47.Gao T, Furnari F, Newton AC. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He H, Hirokawa Y, Gazit A, Yamashita Y, Mano H, Kawakami Y, Kawakami, Hsieh CY, Kung HJ, Lessene G, Baell J, Levitzki A, Maruta H. The Tyr-kinase inhibitor AG879, that blocks the ETK-PAK1 interaction, suppresses the RAS-induced PAK1 activation and malignant transformation. Cancer Biol. Ther. 2004;3:96–101. doi: 10.4161/cbt.3.1.643. [DOI] [PubMed] [Google Scholar]
- 50.Turk BE, Hutti JE, Cantley LC. Determining protein kinase substrate specificity by parallel solution-phase assay of large numbers of peptide substrates. Nat. Protoc. 2006;1:375–379. doi: 10.1038/nprot.2006.57. [DOI] [PubMed] [Google Scholar]
- 51.Breitkopf SB, Asara JM. Determining in vivo phosphorylation sites using mass spectrometry. Curr. Prot. Mol. Biol. 2012;Chapter 18(Unit18.19):1–27. doi: 10.1002/0471142727.mb1819s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ, Akey CW. The holo-apoptosome: Activation of procaspase-9 and interactions with caspase-3. Structure. 2011;19:1084–1096. doi: 10.1016/j.str.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang X, Turke AB, Qi J, Song Y, Rexer BN, Miller TW, Jänne PA, Arteaga CL, Cantley LC, Engelman JA, Asara JM. Using tandem mass spectrometry in targeted mode to identify activators of class IA PI3K in cancer. Cancer Res. 2011;71:5965–5975. doi: 10.1158/0008-5472.CAN-11-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang X, Chen S, Asara JM, Balk SP. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110β and p110δ catalytic subunits. J. Biol. Chem. 2010;285:14980–14989. doi: 10.1074/jbc.M109.085696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8:994–999. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.