

Abstract
Little is known about the functional capability of microbial communities in shallow-sea hydrothermal systems (water depth of <200 m). This study analyzed two high-throughput pyrosequencing metagenomic datasets from the vent and the surface water in the shallow-sea hydrothermal system offshore NE Taiwan. This system exhibited distinct geochemical parameters. Metagenomic data revealed that the vent and the surface water were predominated by Epsilonproteobacteria (Nautiliales-like organisms) and Gammaproteobacteria ( Thiomicrospira -like organisms), respectively. A significant difference in microbial carbon fixation and sulfur metabolism was found between the vent and the surface water. The chemoautotrophic microorganisms in the vent and in the surface water might possess the reverse tricarboxylic acid cycle and the Calvin−Bassham−Benson cycle for carbon fixation in response to carbon dioxide highly enriched in the environment, which is possibly fueled by geochemical energy with sulfur and hydrogen. Comparative analyses of metagenomes showed that the shallow-sea metagenomes contained some genes similar to those present in other extreme environments. This study may serve as a basis for deeply understanding the genetic network and functional capability of the microbial members of shallow-sea hydrothermal systems.
Introduction
The discovery of deep-sea hydrothermal vents in the late 1970s expanded our knowledge of the extent of microhabitats for microorganisms and the possible origins of life on Earth [1,2]. Deep-sea hydrothermal vent chimneys are known to harbor numerous metabolically diverse microorganisms along sharp physical and chemical gradients [2–6]. The geochemistry and microbial communities of deep-sea hydrothermal systems, such as the Lost City chimneys in the Mid-Atlantic Ridge [3–5] and Alvinellapompejana , a polychaete in the East Pacific Rise hydrothermal vent fields [6], have been studied in significant detail. Shallow-sea hydrothermal vents (at water depths of <200 m) are far more ubiquitous than previously recognized [7,8]. They provide more instant access to investigate the metabolic potential and adaptation of microbial communities to extreme environments. However, comparatively few investigations of microbial communities in shallow-sea hydrothermal fields have been conducted.
Shallow-sea hydrothermal vents usually occur near active coastal or submarine volcanoes [7,8]. Such an area of andesite-hosted shallow-sea hydrothermal venting at pH ~1.5−5.0 (acidic) and 30 °C to 116 °C with slightly sulfidic fluids is located 1 km east of Kueishantao Island, near the southern end of the Okinawa Trough [9,10]. Elemental sulfur (S0) is naturally enriched in shallow-sea hydrothermal fluids [9], and gas discharging from these vents is dominated by carbon dioxide (CO2). Among previously investigated hydrothermal fields [11], fluids in the shallow-sea hydrothermal system near Kueishantao Island contain moderate amounts of hydrogen (H2) and methane (CH4) but very low amount of hydrogen sulfide (H2S) because of short interactions of rock and seawater [9]. By contrast, fluids at the serpentinite-hosted Lost City hydrothermal field are highly enriched in CH4 and H2 with near-zero concentrations of CO2. In addition, the Lost City chimneys are alkaline (pH ~9.0 to 11.0) and contain more abundant H2S than the shallow-sea vents [11]. Deep-sea vent communities are often dominated by symbiotrophic forms, such as microbial communities associated with Alvinellapompejana [6] and Riftiapachyptila [12]. The geochemical conditions of A . pompejana are characterized by venting of fluids at 29 °C to 84 °C and pH ~5.3 to 6.9 (slightly acidic to near neutral) with a similar level of free H2S as the shallow-sea vents [6].
With the development of next-generation sequencing technologies, tag pyrosequencing of 16S rRNA genes has been applied to reveal a new and enormous bacterial diversity in deep-sea or shallow-sea hydrothermal environments [3,4,13]. For example, serpentinite-hosted Lost City chimneys harbor endosymbionts with close phylogenetic relationships to the sulfur-oxidizing bacterium Thiomicrospira of the class Gammaproteobacteria and to CH4-oxidizing bacteria of the order Methanosarcinales [3–5]. Bacteria affiliated within Gammaproteobacteria and Epsilonproteobacteria are dominant in 4143-1 chimney at the Fuca Ridge hydrothermal vent [14]. Among the most active and abundant microorganisms in Guaymas Basin plumes are sulfur-oxidizing bacteria of the SUP05 group of Gammaproteobacteria [15,16]. The most prevalent microorganisms in the A . pompejana episymbiont community belonged to Epsilonproteobacteria [6]. Previous studies showed that chemolithoautotrophs from Epsilonproteobacteria and Gammaproteobacteria are predominant primary producers in shallow-sea hydrothermal systems [13,17]. However, the 16S rRNA gene survey offered only limited information on the biogeographic patterns of microbial consortia within an environment. Functional genes rather than species may be the appropriate parameter for understanding biological patterns of bacterial communities [18]. Functional gene analyses showed that species belonging to Gammaproteobacteria and Epsilonproteobacteria in deep-sea hydrothermal vent ecosystems have the potential to grow chemoautotrophically through the Calvin-Benson-Bassham (CBB) cycle and the reductive tricarboxylic acid (rTCA) cycle, respectively [5,19]. Such species can also gain energy by oxidizing reduced sulfur compounds or hydrogen [5,19]. For example, the SUP05 group of Gammaproteobacteria can oxidize reduced sulfur compounds using the energy of hydrogen oxidation in Guaymas Basin plumes [16]. In addition, microbial populations are metabolically active in deep-sea hydrothermal systems with genes encoding the oxidation of methane and ammonia [15,20]. Riftia symbionts might use the rTCA and CBB cycle pathways for carbon fixation, which is possibly an adaptation to the dynamic vent environment [12].
Previous studies conducted functional metagenomic investigations of deep-sea hydrothermal vent systems [5,6,14,21–23]. However, data on the metabolic capacity of microbes in shallow-sea hydrothermal vent systems remain insufficient. This study is the first to describe in detail the functional potential of free-living microbes from the Kueishantao shallow-sea hydrothermal field using high-throughput sequencing technology. Our comparative metagenomic analysis provided insights into the metabolic processes potentially associated with the adaptation of microbial communities into extreme environments.
Results and Discussion
Overview of the geochemical context of sampling sites
Compared with deep-sea vents, shallow hydrothermal systems are characterized by the presence of a gas phase and the enrichment of O2 [7]. Furthermore, the extensive mixing of thermal fluids with oxygenated seawater generates micro-scale redox gradients within shallow-sea hydrothermal systems, thereby affecting various ecosystems [7]. Thus, sampling sites denoted as G1 (vent) and G2 (surface water immediately over the vent) were selected to represent the distinct environment conditions of the Kueishantao shallow-sea hydrothermal system. Dissolved inorganic carbon, ammonium, and phosphate in the vent were all higher than those in the surface water. Nitrate, nitrite, CH4, salinity, and pH were lower at the vent than at the surface water, whereas temperature was higher at the vent than at the surface water (49 °C) (Supplementary Table S1).
Characteristics of Kueishantao Shallow-Sea Hydrothermal System Metagenomes
After removing artificial replicates, 266,487 (G1) and 299,124 (G2) sequence reads with average read lengths of 348 and 401 bp, respectively, were used for the analysis. Approximately 62.5% and 66.1% of the total predicted proteins (146,873 and 161,383, respectively) from the G1 and G2 metagenomes show matches against the M5NR database of the MetaGenome Rapid Annotation with Subsystem Technology (MG-RAST) server [24], with 84,209 and 101,639 matches to functional categories, respectively. The unassembled sequences were used for comparative metagenomic analysis. The assembly of single reads from G1 and G2 datasets resulted in 41 and 95 contigs (>5 kbp in length), respectively.
Taxonomic distribution of metagenomic sequences
The taxonomic classification of protein-coding genes was assigned to the IMG annotation source using the best hit classification of MG-RAST [24]. Bacterial sequences dominated both samples with 97.0% (G1) and 98.7% (G2) of all annotated sequences. Meanwhile, a low number of eukaryotic or other sequences were found. Some sequences were related to proteins affiliated with archaeal members of the class Thermococci, accounting for 1.8% of annotated sequences in the G1 metagenome. Approximately 1.4% of the total gene sequences were assigned to Cyanobacteria in the G2 metagenome.
The bacterial community was diverse with representatives of more than 40 classes. Epsilonproteobacteria was the most dominant class in the vent, accounting for 79.5% of the total assigned sequences of G1. In the surface water, Gammaproteobacteria was the most abundant class, accounting for 61.6% of the total sequences of G2, followed by Alphaproteobacteria (16.2%) and Betaproteobacteria (9.5%). As significant difference in microbial community composition was found between the vent and the surface water. Further statistical analysis indicated that a significant overrepresentation of the sulfur-reducing genera Nautilia and Caminibacter within the order Nautiliales of the class Epsilonproteobacteria was observed in the vent and that a significant overrepresentation of the sulfur-oxidizing genus Thiomicrospira was found in the surface water (q < 0.05, normalized based on metagenome and effect size; Figure 1). Among the sequences assembled from the G1 metagenomic dataset, the majority of the predicted genes show the highest similarity to Nautilia profundicola strain Am-H [19] or Caminibacter mediatlanticus TB-2T [25] (Figure 2). Most of the large contigs in the G2 metagenomic dataset contain open reading frames (ORFs) with significant sequence similarity to the completed genome sequence of Thiomicrospira crunogena XCL-2 [26] (Figure 2). The assembly of these contigs from the metagenomic data provided direct evidence that these communities were dominated by a few populations and may not be very diverse. Bacteria in the Thiomicrospira and Nautiliales lineages were originally found and isolated from deep-sea hydrothermal vents [19,25,26]. Thus, these organisms could inhabit geochemically distinct habitats. Remarkably, these contigs contained the important genes for ecological implications (Figure 2).
Figure 1. Comparison of the taxonomic profiles at the genus level of two samples from the shallow-sea hydrothermal system.

The taxonomic profiles for the vent (G1 colored blue) and surface water immediately above the vent (G2 colored orange) metagenomic datasets were computed using MG-RAST and STAMP v2.0. Corrected P-values (q-values) were calculated based on Fisher’s exact test using Storey’s FDR approach. Dots on either side of the dashed trend line were enriched in one of the two samples. Labeled dots at greater distances from the dashed trend line indicate that these subsystems had greater proportional differences (%) between two metagenomes. A filter was applied to remove features with q value >0.05.
Figure 2. Representative open reading frames encoding carbon fixation, and sulfur, nitrogen and phosphorus utilization-associated functions present in the G1 and the G2 contigs.

The % identities of the homologs to the genes from the reference genomes ( N . profundicola or C . mediatlanticus as references for the G1 dataset analysis and T . crunogena as a reference for the G1 dataset analysis) are listed in the colored boxes. #N/A indicates that no best matches to T . crunogena were found.
Other significantly different bacterial groups included the genera Campylobacter , Nitratriuptor, Sulfurovum , Lebetimonas , Sulfurimonas , Arcobacter , Hydrogenimonas , Nitratifractor , Sulfurospirillum , and Helicobacter in the vent and Shewanella , Vibrio, Nitrosococcus , and Marinobacter in the surface water (q < 0.05). Epsilonproteobacteria at the deep-sea hydrothermal vents could be considered an evolutionary source of human/animal pathogens [6]. Microbial groups ( Campylobacter and Helicobacter ) phylogenetically related to important pathogens were found at the shallow-sea hydrothermal vent. Candidatus Pelagibacter (SAR11 clade) was the most abundant among the Alphaproteobacteria species in the surface water. Overall, the taxonomic affiliation of protein sequences displayed a similar pattern of bacterial diversity to taxonomic analysis based on the identified 16S rRNA genes using the M5NRA database (data not shown).
Metabolic profiling of Kueishantao shallow-sea hydrothermal systems
Sulfur metabolism
Major differences in the enrichment of specific gene families within the sulfur pathway were observed (Figure 3). The genes encoding sulfate adenylyltransferase and adenylylsulfate kinase were overrepresented in the vent (Figure 3). However, the sulfate reduction pathway involving dissimilatory sulfite reductase, adenylylsulfate reductase, and sulfate adenylyltransferase is incomplete [27]. However, genes encoding for polysulfide reductase (Psr) were present in a contig from the G1 dataset (Figure 2), resulting the reduction of polysulfide derived from elemental sulfur to sulfide [27]. Thus, sulfur reduction and not sulfate reduction could be performed by the microorganisms in the vent. Psr gene sequences in the vent were annotated to species of Nautilia or Caminibacter . Meanwhile, the contig present in the G1 dataset contained an ORF with an 83% amino acid identity to a sulfide-quinone reductase (Sqr) encoded by C . mediatlanticus , resulting in the catalysis of the oxidation of sulfide to elemental sulfur (Figure 2) [27]. In addition, 309 and 227 Sqr sequences were detected in the G1 and G2 datasets, respectively, indicating that sulfide oxidation might be an important process in the shallow-sea hydrothermal system. The majority of retrieved Sqr gene sequences from the G1 and G2 datasets were assigned to Nautiliales-like organisms and Thiomicrospira -like organisms, respectively. An overabundance of the other genes associated with sulfur oxidation was present in the G2 dataset (Figure 3). The bacterial community in the surface water possesses genes encoding for key enzymes of three pathways involved in sulfur oxidation [27]: genes encoding adenylylsulfate reductase, sulfide dehydrogenase, and Sox enzyme complex (Figure 3). Most of the sox gene sequences were affiliated with Thiomicrospira -like organisms. The sox operons (soxXYZA, soxCD, soxB, and soxH) encoding enzymes for the oxidation of inorganic sulfur compounds [27] were also observed in the G2 contigs (Figure 2). Other genes involved in the oxidation of inorganic sulfur compounds, including sulfite oxidase and thiosulfate sulfurtransferase, were also found in the G2 dataset (Figure 3), which is mainly contributed by sequences related to those in Gammaproteobacteria. Therefore, the microbial community in the surface water was probably capable of oxidizing a wide range of reduced sulfur compounds. Moreover, sulfur assimilation in the surface water may involve the metabolism of other sulfur-containing compounds, such as dimethylsulfoniopropionate (DMSP) and cysteine (Figure 3). Genes encoding demethylase for DMSP degradation were found only in the G2 dataset (Figure 3), and the sequences were affiliated with those found in the Roseobacter and SAR11 clade.
Figure 3. Metagenomic profile comparisons of genes associated with sulfur metabolic pathways determined using STAMP analysis.
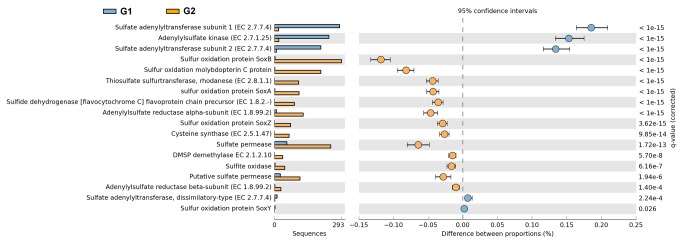
Positive differences between proportions denote greater abundances in the G1 dataset (blue), whereas negative differences between proportions show greater abundances in the G2 dataset (orange) for the given genes. Corrected P-values (q-values) were calculated based on Fisher’s exact test using Storey’s FDR approach. Features with q value <0.05 were considered significant and were thus retained.
Carbohydrate metabolism and carbon fixation
Approximately 10.5% and 8.8% of the annotated reads from the G1 and G2 metagenomes were categorized within the carbohydrate subsystem. The majority of these sequences were linked to the serine−glyoxylate cycle, central carbohydrate, and CO2 fixation subsystems (Supplementary Figure S1). In both metagenomes, the single most abundant component of the carbohydrate subsystems was the serine−glyoxylate cycle followed by the presence of functions involved in the TCA cycle, glycolysis and gluconeogenesis, the CBB cycle, Entner−Doudoroff pathway, pentose phosphate pathway, and pyruvate metabolism (Supplementary Figure S1). However, distinctive differences between the metagenomes in the SEED subsystems were found (q < 0.05, Supplementary Figure S1).
As shown in Figure 4A, the genes for the rTCA cycle [28] were significantly more abundant in the G1 dataset (ATP-dependent citrate lyase, pyruvate: ferredoxin oxidoreductase, and 2-oxoglutarate: ferredoxin oxidoreductase) than in the G2 dataset (q < 0.05). This result suggested that microorganisms might utilize the rTCA cycle for CO2 fixation in the vent. To identify the likely taxonomic source of specific genes in the metagenomes, all matches obtained by MG-RAST could BLASTX against the National Center for Biotechnology Information (NCBI) nr database. The majority of the annotated sequences involved in the rTCA cycle were related to proteins affiliated with the order Nautiliales. Genes involved in CO2 uptake (carboxysome) and photorespiration (oxidative C2 cycle) were overrepresented in the G2 dataset compared with the G1 dataset (q < 0.05, Supplementary Figure S1). This finding suggested that microbes in the surface water possessed a distinguishable carbon fixation pathway. Genes encoding ribulose-1,5-bisphosphate carboxylase (RuBisCO) and phosphoribulokinase that mediate the CBB cycle [28] were enriched in the G2 dataset (Figure 4B). Moreover, one contig in the G2 metagenomic dataset contained a carboxysome operon, including genes for RuBisCO, carboxysome shell proteins, and carbonic anhydrase (Figure 2). Carboxysomes are metabolic modules that enhance the fixation of CO2 by RuBisCO [29]. An ORF that is highly similar to the phosphoribulokinase of T . crunogena was found in the G1 contigs (Figure 2). Genes involved in the CBB cycle in the G1 dataset were mainly affiliated to sequences from Thiomicrospira . Thus, organisms in the system might utilize the rTCA and CBB cycles for CO2 fixation, similar to what is observed for deep-sea vents [4–6]. Microbial communities in the surface water might have possessed two other CO2 fixation pathways, namely, the 3-hydroxypropionate cycle and the reductive acetyl-coenzyme-A pathway [28]. This finding may be attributed to the presence of genes encoding key enzymes involved in these pathways [Figure 4(C) and 4(D)].
Figure 4. Comparison of genes encoding for key enzymes in the carbon fixation pathways determined using STAMP analysis, including (A) the reverse tricarboxylic acid cycle, (B) the Calvin-Bassham-Benson cycle, (C) the 3-hydroxypropionate cycle, and (D) the reductive acetyl-CoA pathway.

Genes encoding enzymes for the G1 (blue) and G2 (orange) datasets were identified based on KEGG functions within the MG-RAST system.
Nitrogen metabolism
Genes encoding nitrate reductase and the nitrite reductase operon were found in the G2 contig of T . crunogena (Figure 2), but the nitrate/nitrite transporter was absent in the genome. Homologs of nitrite reductase sequences were identified in the G1 metagenomic dataset. Genes encoding periplasmic nitrate reductase and dissimilatory nitrite reductase were more prevalent in the G1 dataset than in the G2 dataset, especially within the members of Epsilonproteobacteria. Genes encoding nitrate reductase and ferredoxin-nitrite reductase were enriched in the G2 dataset (q < 0.05, Supplementary Figure S2) and were affiliated with Thiomicrospira -like organisms. These data suggested that the microbes in both sites might utilize nitrate as their electron acceptor or nitrogen source. Furthermore, the enrichment of genes encoding hydroxylamine reductase in the G2 dataset suggested that nitrate might be reduced through a nitrate assimilation pathway with hydroxylamine as a key intermediate [30] (Supplementary Figure S2). The gene encoding for ammonia monooxygenase, a key enzyme for ammonia oxidation [20], was not detected in either metagenome.
Phosphorus metabolism
Both metagenomes contained high-affinity inorganic phosphate transporters (PstB and PstC). However, the significant enrichment of the low-affinity inorganic phosphate transporter was mostly found in the G1 dataset (q < 0.05, Supplementary Figure S3). The phosphonate operon encoding proteins for organic phosphate utilization in T . crunogena was syntenic with one contig in the G2 dataset (Supplementary Figure S3). The overrepresented Phn genes encoding proteins for phosphonate utilization in the surface water [31] might enhance bacterial phosphorus uptake (Supplementary Figure S3).
Energy resource
As shown in Figure 2, various metagenomic sequences obtained from shallow-sea hydrothermal systems were highly similar to the genome of the isolates from deep-sea hydrothermal systems [19,25,26]. Based on the identified genes, the dominant organisms in the shallow-sea hydrothermal system near Kueishantao Island with a geochemically dynamic environment can perform different types of metabolism using various abundant potential electron donors (H2, elemental sulfur) and acceptors (elemental sulfur, CO2, nitrate, O2) as well as potentially lethal levels of heavy metals, such as Fe. Chemoautotrophic bacteria inhabiting the Lost City chimneys [5] and A . pompejana [6] can obtain their metabolic energy by catalyzing the oxidation and reduction of sulfur, respectively. Similar sulfur reduction or oxidation pathways were found in the Kueishantao shallow-sea hydrothermal field. Under reducing conditions in the vent, both sulfur and polysulfides can serve as terminal electron acceptors [27]. One contig in the G1 dataset appeared to contain a complete Ni–Fe hydrogenase operon similar to the one in N . profundicola , enabling bacteria to use H2 as an energy source in the vents [32]. Bacteria in the surface water can potentially obtain their energy by oxidizing reduced sulfur compounds in the presence of sox genes. In general, the oxidation of reduced sulfur compounds can be coupled to the reduction of electron acceptors, including oxygen and nitrate [27]. Furthermore, one contig in the G2 dataset contained genes encoding for cbb3-type cytochrome c oxidases with the potential to mediate aerobic respiration or to act as an electron acceptor even under oxygen-limited conditions [33].
Our 16S rRNA clone data suggested that a shift readily occurred in the predominant microbial population from Epsilonproteobacteria to Gammaproteobacteria across the redox gradients from the vents to the surface water (data not shown). The co-occurrence of sulfur-oxidizing and sulfur-reducing activities could couple their distinct biogeochemical processes based on reciprocal exchange of sulfur compounds and thereby increase the overall energy efficiency of the shallow-sea hydrothermal community even under a relative lack of sulfide conditions. This result is in agreement with the results from the deep-sea hydrothermal microbial symbiosis [6].
According to 16S rRNA gene analysis, archaea related to methanogens and methanotrophs contribute to a much greater percentage of the total sequences in the deep-sea hydrothermal vent than in the shallow-sea hydrothermal system [13,17]. Deep-sea CH4-rich hydrothermal fluids support the growth of large methanogenic and methanotrophic communities, such as those in the Lost City chimneys [5,11]. No genes encoding the key enzymes in methanogenesis/methanotrophy (mcrA and pmoA) [15] were found in the shallow-sea hydrothermal system. This result suggested that these microbial processes are not dominant, although their fluids contained abundant CH4.
Overall, the metabolic profiles of the chemoautotrophic members in the Kueishantao shallow-sea hydrothermal field were similar to those in deep-sea hydrothermal fields, with sulfur metabolism and carbon fixation being of particular importance. Varying concentrations of chlorophyll a in the Kueishantao shallow-sea hydrothermal field indicated that phytoplankton likely contributed to carbon fixation using light as the alternative energy source (Supplementary Table S1). Genes encoding for a light-driven proton pump (proteorhodopsin) involving phototrophy were detected in the surface water and were related to those in the SAR11 clade (details in annotation tables for a metagenome at the MG-RAST Website). Furthermore, the higher concentrations of dissolved organic carbon in the deep-sea hydrothermal fields provided energy support for the heterotrophic activity in the Kueishantao shallow-sea hydrothermal system (Supplementary Table S1). The taxes associated with heterotrophy are more prevalent in our metagenomes than those typically found in deep-sea hydrothermal systems [5,6].
Stress genes
Genes associated with stress response, resistance, and virulence contributed by different bacterial groups were identified in the shallow-sea hydrothermal system (Supplementary Figure S4). Among these genes, the G1 dataset was overrepresented in some subsystems, involving bacterial hemoglobins, periplasmic stress, acid resistance mechanisms, pathogenicity, multidrug resistance efflux pump, and arsenic resistance (q < 0.05, Supplementary Figure S4). Genes assigned to oxidative stress and cobalt−zinc−cadmium resistance subsystems, heat shock dnaK gene cluster, and copper homeostasis were statistically overrepresented in the G2 dataset (q < 0.05, Supplementary Figure S4). Thiomicrospira species were identified as important sources of these genes.
Functional comparisons of the hydrothermal system metagenomic datasets
Based on the relative abundance of Clusters of Orthologous Group (COG) categories and SEED subsystems, multidimensional scaling (MDS) plots showed that most of the samples from the open ocean to the coast, as well as biofilm samples (Supplementary Table S2), clustered closely together at the functional level, apart from hydrothermal field samples (H1: Lost City chimneys [5], H2: A . pompejana episymbiont community [6]) (Figure 5). This result suggested that hydrothermal systems had functional community profiles distinct from other samples. COG analysis also indicated that the sample from H2 was closely related to the G1 samples on the first ordination axis (Figure 5B). Species belonging to Epsilonproteobacteria were also predominant [6].
Figure 5. Multidimensional scaling (MDS) plots of samples using Bray–Curtis similarity according to (A) SEED subsystem and (B) Clusters of Orthologous Groups of protein functional annotations.
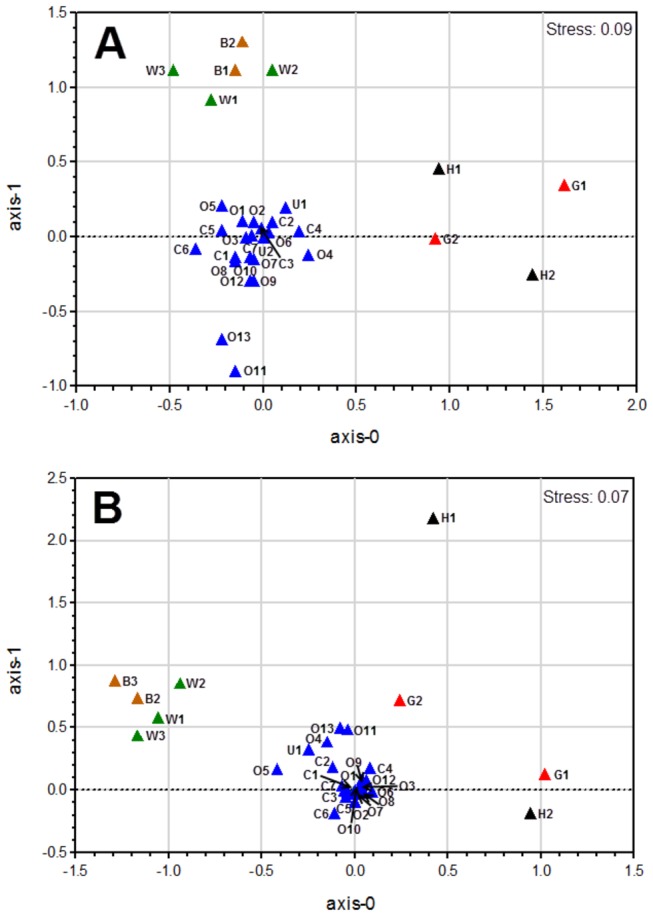
Color represents different sampling areas and each habitat label type (n) is indicated [G: shallow-sea hydrothermal field (red), H: deep-sea hydrothermal field (black), O: open sea (blue), C: coastal and estuary (blue), U: other habitats in common ocean (blue), B: biofilm (orange), W: whale biofilm (green). For details, see Supplementary Table S2]. Samples from each of the respective environments clustered together based on their functional profile. The stress values are reported in the top right corner of the figures and represent the goodness-of-fit.
The two-way functional comparisons revealed some dissimilarity in the SEED subsystems of the samples (Supplementary Table S3). A relatively higher abundance of cobalt−zinc−cadmium resistance genes and virulence genes (e.g. phage integration and excision genes) were found in the deep-sea vents (H1, H2) than in the shallow-sea vent (G1). The genes with significant overrepresentation in the shallow-sea vent relative to deep-sea vents included those for bacterial mobility and chemotaxis (q < 0.05). Campylobacter and Helicobacter species accounted for most of these genes (details in annotation tables for a metagenome at the MG-RAST Website). In submerged environments, motility and chemotaxis-related functions enable bacteria to respond rapidly to environmental changes [34].
Similarity percentage (SIMPER) analysis further revealed the main contributors to the dissimilarity between the two datasets (Figure 6). More genes associated with motility (COG1868, COG1344) and chemotaxis (COG0643) were identified in the G1 dataset than in the other datasets (Figure 6). Type IV secretion systems were more abundant in G1 than in the other sites that were used for both DNA and protein transfer between bacteria or between bacteria and hosts [35] (Figure 6). The type IV secretion system and transposases have potentially important functions in the horizontal gene transfer for deep-sea microbes [21]. Among the vents, H1 contained the most genes encoding transposases (COG2826, COG3039, COG3436) (Figure 6). The G1 dataset also contained abundant genes encoding transposases (COG3676). This finding suggested that the horizontal gene transfer in the shallow-sea hydrothermal fields and in the deep-sea vent chimney biosphere might be a common occurrence. Two-component systems are commonly used by prokaryotes to sense and respond to changing environmental conditions [36]. As shown in Figure 6, the shallow-sea hydrothermal system contains genes for signal transduction EAL domain proteins (COG5001) and FOG: CheY-like receiver protein (COG0784) through which microorganisms can respond to chemical composition changes outside the cell [36]. The H1 metagenome was enriched in genes associated with signal transduction functions, particularly the EAL domain protein (COG5001) and the FOG: GGDEF and FOG: EAL domain protein (COG2199, COG2200) sequences (Figure 6). These domains are involved in the regulation of bacterial growth and survival phenotypes such as the biofilm [36]. In addition, genes encoding PAS/PAC domain proteins (COG2202) were found in the H1 dataset; these genes function as internal sensors of redox potential and oxygen [37]. The H2 metagenome had more genes encoding MutS proteins (COG0249) for DNA mismatch repair than the other metagenomes (Figure 6).
Figure 6. Selected COGs in the order of their contribution to the difference between two samples as assessed using similarity percentage analysis.

Each row represents the relative frequency of genes among samples (A: G1, B: G2, C: H1, D: H2). The value of normalized abundance of genes (relative to the single-copy gene RecA) is assigned with a color relative to the maximum value among all comparisons of each COG, from white to red. The colors represent 0 (white) to maximum (MAX, red) increments of MAX/10. The higher values indicate greater gene abundance. COG descriptions are listed along the row. Among these COG categories, the maximum RecA-normalized gene abundance is shown in a bracket.
Genes encoding the key enzyme for rTCA cycle-pyruvate: ferredoxin oxidoreductase (COG0674, COG1013) were more abundant in the shallow-sea vent than in the deep-sea vent (Figure 6), reflecting a possible adaptation of the organisms to the high CO2 present within the shallow-sea hydrothermal system. In addition, greater abundance of phosphoenolpyruvate carboxykinase and phosphoenolpyruvate carboxylase (COG1866, COG2352) was found in the shallow-sea vent than in the deep-sea vent (Figure 6). These enzymes elicit anaplerotic activities by replenishing oxaloacetate to the rTCA/TCA cycle [38]. Genes encoding nitrate reductase (COG5013) and nitric oxide reductase (COG4263) involved in nitrogen metabolism were enriched in the H2 dataset. Ni–Fe hydrogenases (COG0374) for H2 oxidation were observed primarily in the G1 and H2 datasets. Glycosyltransferases (COG0438, COG0436) and the phosphotransferase system (COG1263, COG1264) were more abundant in the H2 metagenome than in the G1 metagenome and were involved in catalyzing the attachment of sugars to protein and transporting sugar [39], respectively. Moreover, glycosyltransferases might have important functions in biofilm development in several bacteria [40]. Vent fluids from the shallow-sea hydrothermal system contain extremely low concentrations of trace metals, such as Fe [7]. Transporter for Fe3+ (COG1629, COG0609) genes were enriched in the H1 dataset, whereas Fe2+ transporter genes (COG0370) were identified in all datasets (Figure 6).
All these functions could contribute to community adaptations to the hydrothermal environment. The functional genes varied among the metagenomes. Moreover, the microbial communities in the shallow-sea hydrothermal system contained some apparent functional features (Figure 6) that are not found in other environments [5,6]. However, special functional categories, such as transposase and carbon fixation, were found in several datasets. This result indicated that a core of genes was shared by the vent-associated bacterial community.
Materials and Methods
Sampling
Samples were collected in July and August 2010 from two depths in the hydrothermal vent system (121°57′E, 24°50′N): one located in the vent (G1: 17.2 m) and one immediately located above the vent (G2: 0 m). The vents were identified by scuba divers, and their positions were located by the global positioning system. Geochemical features of each sampling site were obtained, as shown in Supplementary Table S1. An enriched content of elemental sulfur was observed during sampling. All necessary permits were obtained for the described field studies. Two permits were required and obtained, one from the Coast Guard Administration of Taiwan and the other from the Fisheries Management Office of the Yilan County, Taiwan.
A total of 20 L of seawater was filtered onto 3 µm of GF/C filters (PALL Corporation) and then collected in 0.22 µm of Sterivex filter units (millipore) [41]. After filtration, the Sterivex units were filled with 1.8 mL of lysis buffer (50 mM Tris-HCl, 40 mM EDTA, 0.75 M sucrose, pH 8.3) and were stored at −80 °C until DNA extraction.
DNA extraction and sequencing
Nucleic acid extraction was performed on a Sterivex filter unit as previously described [42]. Briefly, the samples were added with 100 µL of lysozyme (final concentration of 6.25 mg/mL) and RNase A (final concentration of 100 µg/mL) and then incubated for 1 h. Subsequently, the samples were added with 100 µL of Proteinase K (QIAGEN) and 100 µL of 20% (w/v) sodium dodecyl sulfate and then incubated at 55 °C for 2 h in a hybridization oven. After separation of this lysate, nucleic acids were extracted with an equal volume of phenol: chloroform: isoamyl alcohol (25:24:1, v: v: v). DNA samples were further concentrated by centrifugation (3500×g) with Amicon Ultra-15 30K Centrifugal Filter Units (millipore) and washed several times in TE buffer (pH 8.0). DNA concentration was estimated using a NanoDrop 2000 spectrophotometer (Thermo, Fisher, USA) and analyzed through gel electrophoresis. Approximately, 5 µg of DNA per sample was sent for pyrosequencing to the Chinese National Human Genome Center (Shanghai, China). Shotgun sequencing runs were performed on libraries prepared from environmental samples of community DNA using the 454 GS FLX Titanium protocols.
Metagenome sequencing, assembly, and annotation
The 454 sequencing reads were filtered using an in-house developed program to remove low-quality reads. A total of ~92.82 (118.79) Mbp (G1) and ~120.22 (172.79) Mbp (G2) of unique sequence data was generated from the G1 and G2 samples. Raw sequencing reads from both datasets were submitted to the MG-RAST server (version 3.0) for gene annotation (http://metagenomics.anl.gov/) [24]. The artificially created duplicate reads were removed automatically by MG-RAST [24]. The putative ORFs were identified, and their corresponding protein sequences were searched with BLAST against the M5NR non-redundant protein database in the MG-RAST (an E-value cutoff of less than 1 × 10-3). M5NR is an integration of many sequence databases [including the NCBI GenBank, COG, Kyoto Encyclopedia of Genes and Genomes (KEGG), and SEED] into a single, searchable database [24]. Raw sequencing reads were assembled into contigs employing de novo assembler software Newbler (454 Life Sciences, Roche Applied Sciences, Branford, CT, USA). Newbler could be superior to some assemblers for merging 454 sequencing reads into longer contig, but the amount of contigs produced by Newbler was possibly less [43,44].
Comparative metagenome analyses
Taxonomic and functional profiles within MG-RAST (hits to IMG, M5NRA, SEED, COG, and KEGG databases) were extracted (an E-value cutoff of less than 1 × 10-5 and a minimum read length of 50 bp) to compare functional attributes across metagenomes. For all subsequent analyses, gene counts were normalized against the total number of hits in their respective databases to remove bias in different sequencing efforts as described previously [45,46]. To explore functional differences between two metagenomic datasets, Statistical Analysis of Metagenomic Profiles (STAMP) v2.0 software package [47] was employed to test for significant differences in both taxonomic and functional distribution between metagenomes. Statistical significance of differences between samples (q value) was assessed using the two-sided Fisher’s exact test with Storey’s false discovery rate method of multiple test correction within STAMP [47]. The confidence intervals were determined using the Newcombe−Wilson method. Features with a q value of <0.05 were deemed significant.
To extend comparative metagenomic analyses, other marine environmental metagenomic datasets publicly available in MG-RAST were selected (Supplementary Table S2). This E-value (1 × 10-3) was used for the comparative analyses of metagenomic datasets with different read lengths [45,46]. The abundance of COG categories or SEED subsystems per metagenome was transformed using square root, and Bray−Curtis similarities were calculated on the data matrices. Non-metric MDS was used to determine the similarity among datasets with the PRIMER-E ecological software package [48,49]. The similarities are presented in a multidimensional space by plotting more similar samples closer together using the Ginkgo software [50]. SIMPER analysis in PRIMER-E was used to determine the similarity or difference between the COG abundance distributions among representative metagenomic datasets from the hydrothermal fields [49]. To remove the bias of average genome size on the sampling of gene from a given metagenomic community, the abundance of gene in each COG was normalized against the number of single-copy RecA gene per metagenome [51,52]. The top 50 COG values representing relatively more contributions to the differences between the two samples were selected for heatmap visualization.
Data availability
The sequence data are available under “Kueishantao metagenomes project” in the MG-RAST database (http://metagenomics.anl.gov/) (ID 4487624.3 for vent, and ID 4487625.3 for surface water above the vent). All individual sequence reads have been deposited at the NCBI Short Read Archive (SRA) under the accessions SRX202013 for the vent and SRX202014 for surface water above the vent datasets.
Supporting Information
Comparisons of SEED carbohydrate subsystem for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Classification of a pathway is based on SEED subsystem hierarchy 3 of the MG-RAST.
(TIF)
Metagenomic profile comparisons of genes involved in nitrogen metabolism for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Enzyme identification was based on KEGG functions within the MG-RAST system.
(TIF)
Metagenomic profile comparisons of genes associated with phosphorus utilization pathways for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Enzyme identification was based on KEGG functions within the MG-RAST system.
(TIF)
Comparison of genes associated with stress and virulence determined using STAMP analysis. Gene identifications were based on subsystem hierarchy 4 of the MG-RAST system.
(TIF)
Geochemical data of sampling sites.
(XLS)
Information on publicly available metagenomes used in this study.
(XLS)
Selected pair-wise comparisons of SEED subsystems for the shallow-sea and deep-sea hydrothermal fields’ metagenomic datasets.
(XLS)
Acknowledgments
We thank Bing-Jye Wang and Rui Zong for their assistance in collecting samples in the cruise. Professor John Hodgkiss of the University of Hong Kong is thanked for polishing the English. We gratefully acknowledge the thoughtful comments, valuable suggestions, and helpful criticisms of the reviewers.
Funding Statement
This work was supported by National Basic Research Program of China (2011CB808800/2013CB955700), NSFC (41276131/91028001) Public Science and Technology Research Funds Projects of Ocean 201105021&MELRI1005 *LMSR 7.26.13. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Corliss JB, Dymond J, Gordon LI, Edmond JM, Von Herzen RP et al. (1979) Submarine thermal sprirngs on the galapagos rift. Science 203: 1073-1083. doi:10.1126/science.203.4385.1073. PubMed: 17776033. [DOI] [PubMed] [Google Scholar]
- 2. Reysenbach AL, Shock E (2002) Merging genomes with geochemistry in hydrothermal ecosystems. Science 296: 1077-1082. doi:10.1126/science.1072483. PubMed: 12004120. [DOI] [PubMed] [Google Scholar]
- 3. Flores GE, Campbell JH, Kirshtein JD, Meneghin J, Podar M et al. (2011) Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ Microbiol 13: 2158-2171. doi:10.1111/j.1462-2920.2011.02463.x. PubMed: 21418499. [DOI] [PubMed] [Google Scholar]
- 4. Roussel EG, Konn C, Charlou JL, Donval JP, Fouquet Y et al. (2011) Comparison of microbial communities associated with three Atlantic ultramafic hydrothermal systems. FEMS Microbiol Ecol 77: 647-665. doi:10.1111/j.1574-6941.2011.01161.x. PubMed: 21707671. [DOI] [PubMed] [Google Scholar]
- 5. Brazelton WJ, Baross JA (2010) Metagenomic comparison of two Thiomicrospira lineages inhabiting contrasting deep-sea hydrothermal environments. PLOS ONE 5: e13530. doi:10.1371/journal.pone.0013530. PubMed: 20975831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grzymski JJ, Murray AE, Campbell BJ, Kaplarevic M, Gao GR et al. (2008) Metagenome analysis of an extreme microbial symbiosis reveals eurythermal adaptation and metabolic flexibility. Proc Natl Acad Sci U S A 105: 17516-17521. doi:10.1073/pnas.0802782105. PubMed: 18987310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tarasov VG, Gebruk AV, Mironov AN, Moskalev LI (2005) Deep-sea and shallow-water hydrothermal vent communities: Two different phenomena? Chem Geol 224: 5-39. doi:10.1016/j.chemgeo.2005.07.021. [Google Scholar]
- 8. Maugeri TL, Bianconi G, Canganella F, Danovaro R, Gugliandolo C et al. (2010) Shallow hydrothermal vents in the southern Tyrrhenian Sea. Chem Ecol 26: 285-298. doi:10.1080/02757541003693250. [Google Scholar]
- 9. Chen CTA, Zeng Z, Kuo FW, Yang TF, Wang BJ et al. (2005) Tide-influenced acidic hydrothermal system offshore NE Taiwan. Chem Geol 224: 69-81. doi:10.1016/j.chemgeo.2005.07.022. [Google Scholar]
- 10. Yang L, Hong H, Guo W, Chen CTA, Pan PI et al. (2012) Absorption and fluorescence of dissolved organic matter in submarine hydrothermal vents off NE Taiwan. Mar Chem 128-129: 64-71. doi:10.1016/j.marchem.2011.10.003. [Google Scholar]
- 11. Kelley DS, Karson JA, Früh-Green GL, Yoerger DR, Shank TM et al. (2005) A serpentinite-hosted ecosystem: the Lost City hydrothermal field. Science 307: 1428-1434. doi:10.1126/science.1102556. PubMed: 15746419. [DOI] [PubMed] [Google Scholar]
- 12. Robidart JC, Bench SR, Feldman RA, Novoradovsky A, Podell SB et al. (2008) Metabolic versatility of the Riftia pachyptila endosymbiont revealed through metagenomics. Environ Microbiol 10: 727-737. doi:10.1111/j.1462-2920.2007.01496.x. PubMed: 18237306. [DOI] [PubMed] [Google Scholar]
- 13. Maugeri TL, Lentini V, Gugliandolo C, Italiano F, Cousin S et al. (2009) Bacterial and archaeal populations at two shallow hydrothermal vents off Panarea Island (Eolian Islands, Italy). Extremophiles 13: 199-212. doi:10.1007/s00792-008-0210-6. PubMed: 19050821. [DOI] [PubMed] [Google Scholar]
- 14. Xie W, Wang F, Guo L, Chen Z, Sievert SM et al. (2011) Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J 5: 414-426. PubMed: 20927138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lesniewski RA, Jain S, Anantharaman K, Schloss PD, Dick GJ (2012) The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME J 6: 2257-2268. doi:10.1038/ismej.2012.63. PubMed: 22695860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anantharaman K, Breier JA, Sheik CS, Dick GJ (2013) Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc Natl Acad Sci U S A 110: 330-335. doi:10.1073/pnas.1215340110. PubMed: 23263870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Zhao Z, Chen CT, Tang K, Su J et al. (2012) Sulfur metabolizing microbes dominate microbial communities in andesite-Hosted shallow-sea hydrothermal systems. PLOS ONE 7: e44593. doi:10.1371/journal.pone.0044593. PubMed: 22970260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T (2011) Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci U S A 108: 14288-14293. doi:10.1073/pnas.1101591108. PubMed: 21825123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campbell BJ, Smith JL, Hanson TE, Klotz MG, Stein LY et al. (2009) Adaptations to submarine hydrothermal environments exemplified by the genome of Nautilia profundicola . PLOS Genet 5: e1000362 PubMed: 19197347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baker BJ, Lesniewski RA, Dick GJ (2012) Genome-enabled transcriptomics reveals archaeal populations that drive nitrification in a deep-sea hydrothermal plume. ISME J 6: 2269-2279. doi:10.1038/ismej.2012.64. PubMed: 22695863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brazelton WJ, Baross JA (2009) Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J 3: 1420-1424. doi:10.1038/ismej.2009.79. PubMed: 19571895. [DOI] [PubMed] [Google Scholar]
- 22. Hügler M, Petersen JM, Dubilier N, Imhoff JF, Sievert SM (2011) Pathways of carbon and energy metabolism of the epibiotic community associated with the deep-sea hydrothermal vent shrimp Rimicaris exoculata . PLOS ONE 6: e16018. doi:10.1371/journal.pone.0016018. PubMed: 21249205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang F, Zhou H, Meng J, Peng X, Jiang L et al. (2009) GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc Natl Acad Sci U S A 106: 4840-4845. doi:10.1073/pnas.0810418106. PubMed: 19273854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM et al. (2008) The metagenomics RAST server-A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf 9: 386. doi:10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giovannelli D, Ferriera S, Johnson J, Kravitz S, Pérez-Rodríguez I et al. (2011) Draft genome sequence of Caminibacter mediatlanticus strain TB-2T, an epsilonproteobacterium isolated from a deep-sea hydrothermal vent. Stand Genomics Sci 5: 135-143. doi:10.4056/sigs.2094859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scott KM, Sievert SM, Abril FN, Ball LA, Barrett CJ et al. (2006) The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PLOS Biol 4: e383. doi:10.1371/journal.pbio.0040383. PubMed: 17105352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamamoto M, Takai K (2011) Sulfur metabolisms in epsilon- and gamma-proteobacteria in deep-sea hydrothermal fields. Front Microbiol 2: 192 PubMed: 21960986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sievert Hügler M (2011) Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Annu. Rev Mar Sci 3: 261-289. doi:10.1146/annurev-marine-120709-142712. [DOI] [PubMed] [Google Scholar]
- 29. Yoshizawa Y, Toyoda K, Arai H, Ishii M, Igarashi Y (2004) CO2-responsive expression and gene organization of three ribulose-1,5-bisphosphate carboxylase/oxygenase enzymes and carboxysomes in Hydrogenovibrio marinus strain MH-110. J Bacteriol 186: 5685–5691. doi:10.1128/JB.186.17.5685-5691.2004. PubMed: 15317772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Canfield DE, Glazer AN, Falkowski PG (2010) The evolution and future of Earth’s nitrogen cycle. Science 330: 192-196. doi:10.1126/science.1186120. PubMed: 20929768. [DOI] [PubMed] [Google Scholar]
- 31. Villarreal-Chiu JF, Quinn JP, McGrath JW (2012) The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front Microbiol 3: 19 PubMed: 22303297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petersen JM, Zielinski FU, Pape T, Seifert R, Moraru C et al. (2011) Hydrogen is an energy source for hydrothermal vent symbioses. Nature 476: 176-180. doi:10.1038/nature10325. PubMed: 21833083. [DOI] [PubMed] [Google Scholar]
- 33. Pitcher RS, Brittain T, Watmough NJ (2002) Cytochrome cbb(3) oxidase and bacterial microaerobic metabolism. Biochem Soc Trans 30: 653-658. PubMed: 12196157. [DOI] [PubMed] [Google Scholar]
- 34. Lertsethtakarn P, Ottemann KM, Hendrixson DR (2011) Motility and chemotaxis in Campylobacter and Helicobacter . Annu Rev Microbiol 65: 389-410. doi:10.1146/annurev-micro-090110-102908. PubMed: 21939377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alvarez-Martinez CE, Christie PJ (2009) Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev 73: 775-808. doi:10.1128/MMBR.00023-09. PubMed: 19946141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stock AM, Robinson VL, Goudreau PN (2000) Two-component signal transduction. Annu Rev Biochem 69: 183-215. doi:10.1146/annurev.biochem.69.1.183. PubMed: 10966457. [DOI] [PubMed] [Google Scholar]
- 37. Taylor BL, Zhulin IB (1999) PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev 63: 479-506. PubMed: 10357859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chao YP, Liao JC (1993) Alteration of growth yield by overexpression of phosphoenolpyruvate carboxylase and phosphoenolpyruvate carboxykinase in Escherichia coli . Appl Environ Microbiol 59: 4261-4265. PubMed: 8285716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jervis AJ, Langdon R, Hitchen P, Lawson AJ, Wood A et al. (2010) Characterization of N-linked protein glycosylation in Helicobacter pullorum . J Bacteriol 192: 5228-5236. doi:10.1128/JB.00211-10. PubMed: 20581208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou M, Zhu F, Dong S, Pritchard DG, Wu H (2010) A novel glucosyltransferase is required for glycosylation of a serine-rich adhesin and biofilm formation by Streptococcus parasanguinis . J Biol Chem 285: 12140-12148. doi:10.1074/jbc.M109.066928. PubMed: 20164186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walsh DA, Zaikova E, Hallam SJ (2009) Large Volume (20L+) Filtration of coastal seawater samples. J Vis Exp 28: e1161 PubMed: 19543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wright JJ, Lee S, Zaikova E, Walsh DA, Hallam SJ (2009) DNA Extraction from 0.22 µM Sterivex filters and cesium chloride density gradient centrifugation. J Vis Exp 31: e1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mundry M, Bornberg-bauer E, Sammeth M, Feulner PG (2012) Evaluating characteristics of De Novo assembly software on 454 transcriptome data: a simulation approach. PLOS ONE 7: e31410. doi:10.1371/journal.pone.0031410. PubMed: 22384018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pignatelli M, Moya A (2011) Evaluating the fidelity of de novo short read metagenomic assembly using simulated data. PLOS ONE 6: e19984. doi:10.1371/journal.pone.0019984. PubMed: 21625384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith RJ, Jeffries TC, Roudnew B, Fitch AJ, Seymour JR et al. (2012) Metagenomic comparison of microbial communities inhabiting confined and unconfined aquifer ecosystems. Environ Microbiol 14: 240-253. doi:10.1111/j.1462-2920.2011.02614.x. PubMed: 22004107. [DOI] [PubMed] [Google Scholar]
- 46. Jeffries TC, Seymour JR, Gilbert JA, Dinsdale EA, Newton K et al. (2011) Substrate type determines metagenomic profiles from diverse chemical habitats. PLOS ONE 6: e25173. doi:10.1371/journal.pone.0025173. PubMed: 21966446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parks DH, Beiko RG (2010) Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26: 715-721. doi:10.1093/bioinformatics/btq041. PubMed: 20130030. [DOI] [PubMed] [Google Scholar]
- 48. Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18: 117-143. doi:10.1111/j.1442-9993.1993.tb00438.x. [Google Scholar]
- 49. Clarke KR, Gorley RN (2001) PRIMER v5. User manual/tutorial. UK: Plymouth. [Google Scholar]
- 50. Bouxin G (2005) Ginkgo, a multivariate analysis package. J Veg Sci 16: 355-359. doi:10.1111/j.1654-1103.2005.tb02374.x. [Google Scholar]
- 51. Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB et al. (2004) Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432: 910-913. doi:10.1038/nature03170. PubMed: 15602564. [DOI] [PubMed] [Google Scholar]
- 52. Toulza E, Tagliabue A, Blain S, Piganeau G (2012) Analysis of the global ocean sampling (GOS) project for trends in iron uptake by surface ocean microbes. PLOS ONE 7: e30931. doi:10.1371/journal.pone.0030931. PubMed: 22363520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparisons of SEED carbohydrate subsystem for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Classification of a pathway is based on SEED subsystem hierarchy 3 of the MG-RAST.
(TIF)
Metagenomic profile comparisons of genes involved in nitrogen metabolism for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Enzyme identification was based on KEGG functions within the MG-RAST system.
(TIF)
Metagenomic profile comparisons of genes associated with phosphorus utilization pathways for the G1 (blue) and G2 (orange) datasets determined using STAMP analysis. Enzyme identification was based on KEGG functions within the MG-RAST system.
(TIF)
Comparison of genes associated with stress and virulence determined using STAMP analysis. Gene identifications were based on subsystem hierarchy 4 of the MG-RAST system.
(TIF)
Geochemical data of sampling sites.
(XLS)
Information on publicly available metagenomes used in this study.
(XLS)
Selected pair-wise comparisons of SEED subsystems for the shallow-sea and deep-sea hydrothermal fields’ metagenomic datasets.
(XLS)
Data Availability Statement
The sequence data are available under “Kueishantao metagenomes project” in the MG-RAST database (http://metagenomics.anl.gov/) (ID 4487624.3 for vent, and ID 4487625.3 for surface water above the vent). All individual sequence reads have been deposited at the NCBI Short Read Archive (SRA) under the accessions SRX202013 for the vent and SRX202014 for surface water above the vent datasets.