

Abstract
The ultimate goal of this research is to construct a new direct CO2 fixation system using photosystems in living algae. Here, we report light-driven formate production from CO2 by using cyanobacterial photosystem I (PS I). Formate, a chemical hydrogen carrier and important industrial material, can be produced from CO2 by using the reducing power and the catalytic function of formate dehydrogenase (FDH). We created a bacterial FDH mutant that experimentally switched the cofactor specificity from NADH to NADPH, and combined it with an in vitro-reconstituted cyanobacterial light-driven NADPH production system consisting of PS I, ferredoxin (Fd), and ferredoxin-NADP+-reductase (FNR). Consequently, light-dependent formate production under a CO2 atmosphere was successfully achieved. In addition, we introduced the NADPH-dependent FDH mutant into heterocysts of the cyanobacterium Anabaena sp. PCC 7120 and demonstrated an increased formate concentration in the cells. These results provide a new possibility for photo-biological CO2 fixation.
Introduction
Formate is an important energy carrier in the bacterial kingdom. Biological formate is produced by the degradation of pyruvate, amino acids, l-(+)-tartaric acid, oxalate, and hypoxanthine [1,2]. It can also be produced by the direct reduction of CO2 catalyzed by formate dehydrogenase (FDH), and this approach is increasingly gaining attention [3].
FDHs consist of several groups of enzymes that vary significantly in their quaternary structure, occurrence, and type of prosthetic groups. One such group is the NADH-dependent FDH (EC1.2.1.2) (NADH-FDH), which catalyzes the oxidation of formate to CO2 and the reduction of CO2 to formate coupled with the oxidation-reduction of NAD+/NADH (HCOO- + NAD+ ↔ NADH + CO2) [4]. NADH-FDH has previously been applied to light-driven formate production systems by using Zn-porphyrin or TiO2 particles as a photosensitizer [5–7]. In these systems, electrons are supplied by photosensitizers and transported via mediators such as methyl viologen, to diaphorase reducing NAD+ to NADH. The NADH-FDH then catalyzes the CO2 reduction by using the resulting NADH to produce formate.
Based on the results of previous studies, we designed a new biological system (Figure 1). In our system, the ability of cyanobacteria, algae, and plants to effectively produce NADPH using a photosynthetic light reaction [8,9] was taken as the basis for the design, in order to produce formate by using NADPH as a substrate for FDH. All the components are biologically producible molecules and can, therefore, be regenerated within the cell, enabling the creation of a sustainable formate production mechanism.
Figure 1. The proposed system for light-driven formate.
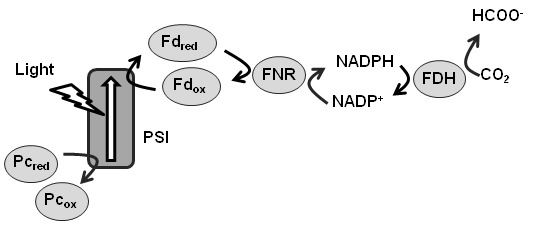
production.
In oxygenic photosynthesis, photochemical reactions are carried out in 2 separate photosystems. Photosystem II (PS II) catalyzes the anodic half-cell reaction: H2O + 2hν → 1/2O2 + 2e- + 2H+, whereas photosystem I (PS I) catalyzes the cathodic half-cell reaction: oxidized Fd (Fdox) + reduced plastocyanin (PCred) + hν → reduced Fd (Fdred) + oxidized PC (PCox). Fdred then transfers electrons to NADP+ via ferredoxin-NADP+-reductase (FNR), thereby reducing it to NADPH: 2Fdred + H+ + NADP+ → 2Fdox + NADPH. NADPH can be used as a reducing agent in various biosynthetic processes, including CO2 fixation via the Calvin–Benson cycle. However, direct formate production by using NADPH and CO2 does not progress in oxygen-evolving phototrophs because of the oxygen-sensitivity of FDH.
To create a light-driven formate production system in vivo, we focused on cyanobacterial heterocysts, which carry out a strictly anaerobic reaction, i.e., nitrogen fixation [10]. Heterocysts differentiate from vegetative cells, which carry out oxygen-evolving photosynthesis, and are deficient in PS II activity. Their internal environment is, therefore, micro-oxic and oxygen-sensitive nitrogenase is expressed in the cells. The electrons necessary for the nitrogen fixation are supplied from neighboring vegetative cells in the form of sucrose. Sucrose is degraded into CO2, protons, and electrons, and the electrons are photo-excited by PS I and transferred to nitrogenase to reduce nitrogen [10]. The micro-oxic and reductive environment makes heterocysts an ideal reaction vessel for light-driven formate production by FDH.
We first created NADPH-dependent FDH (NADPH-FDH) because all the known NADH-FDHs are specific for NADH. In a previous study, site-saturation mutagenesis at the amino acid residues determining the cofactor specificity of NADH-FDH were examined and some mutants, in which the cofactor preference in the degradation of formate was shifted from NAD+ to NADP+, were found [11]. However, these studies did not report any data on the productivity of formate and its stability. Here, we prepared several mutants of FDHs from Pseudomonas sp. 101 ( Pseudomonas ), Candida boidinii (Candida), potato, Arabidopsis thaliana ( Arabidopsis ), and Thiobacillus sp. KNK65MA ( Thiobacillus ), and investigated their formate productivity from NADPH and CO2. We then verified our formate production system in vitro and in heterocysts by using the NADPH-FDH with the highest formate productivity.
Materials and Methods
Bacterial strains and Culture conditions
Escherichia coli (E. coli) DH5α (RBC Bioscience, New Taipei City, Taiwan) and E. coli BL21 (DE3) (Merck, Rahway, New Jersey) were used for the genetic manipulation and expression of recombinant FDH, respectively. For protein expression, 0.5 mM (as a final concentration) of isopropyl-l-d-thiogalactopyranoside (IPTG; Nacalai tesque, Kyoto, Japan) was added to each Luria-Bertani (LB) medium. The filamentous heterocystous cyanobacterium Nostoc sp. strain PCC 7120 (also called Anabaena sp. strain PCC 7120) (hereinafter, Anabaena ) was purchased from the Pasteur Culture Collection of Cyanobacteria (France). Anabaena and the transformed cells were grown at 30◦C under a light intensity of 30 µE∙m-2∙s-1 in the BG11 medium as previously described [12]. Liquid cultures were bubbled with air. For nitrogen deprivation experiments, cells grown in the BG11 medium until they reached an OD600 of 0.4–0.5 were washed with the nitrogen-free medium (BG110) and then re-suspended in the BG110 medium. Neomycin was added to the medium at a final concentration of 30 µg/mL, when required.
Plasmids
The synthesized genes coding for FDHs from Pseudomonas , Candida , potato, Arabidopsis , and Thiobacillus were subcloned using NdeI and NotI sites in pET22b (Merck) as C-terminal His-tagged proteins. According to previous studies [11], the amino acid residues determining the cofactor specificity of NADH-FDH were suggested to be at 222 and 224 in Pseudomonas , 197 and 199 in potato, 227 and 229 in Arabidopsis , and 222 and 224 in Thiobacillus . The nucleotides corresponding to the above pairs of amino acids were substituted with those corresponding to Glu/Ser/Ala/Gln and Asn/His, respectively, by means of PCR site-directed mutagenesis using mixed primers, 5′-GTTCATCTGCACTATACC (C or G or T) (A or C) GCGT(A or C) AC CGCCTGCCGGAAAGCGTG-3′ for Pseudomonas , 5′-CTGCAATCTGCTGTACCAC (C or G or T)(A or C)GCGT(A or C) A CAAAATGGATTCTGAACTG-3′ for potato, 5′- GTTGCAACCTGCTGTACCAT (C or G or T)(A or C) GCGT(A or C) A CCAGATGGCACCGGAACTG -3′ for Arabidopsis , and 5′-GTTAAACTGCATTATACC (C or G or T)(A or C) GCGT(A or C) A CCGTCTGCCGGAAGCAGTG-3′for Thiobacillus , and their complementary primers (underlines indicate codons corresponding to amino acid residues determining the cofactor specificity). The introduction of mutations into Candida gene was performed using non-mixed primers, 5′-CGAAAGAACTGCTGTATTACC A G C G T A A TGCCCTGCCGAAAGAAGCAG-3′ and its complementary primer (underlines indicate codons corresponding to mutation sites (Asp195 → Gln, Tyr196→Arg and Gln197→Asn)).
The DNA fragments coding for plastocyanin (Pc), ferredoxin (Fd) and ferredoxin–NADP+ reductase (FNR) were amplified from Synechocystis sp. PCC6803 genome using primer pairs; 5′-AGATATACATATGTCTAAAAAGTTTTTAAC-3′ and 5′-ATATAGTGCGGCCGCCTCAACGACAACTTTGCC-3′ for Pc, 5′-AGATATACATATGGCATCCTATACCGTTA-3′ and 5′-ATATAGTGCGGCCGCGTAGAGGTCTTCTTCTTTG-3′ for Fd, and 5′-AGATATACATATGTACAGTCCCGGTTAC-3′ and 5′-ATATAGTGCGGCCGCGTAGGTTTCCACGTGCCAG-3′ for FNR, respectively. The fragments were digested with NdeI and NotI, and inserted into pET22b treated with the same enzymes.
For the expression of NADPH-FDH in Anabaena heterocysts, we used a hetR promoter and a shuttle vector, pAM505, which contained replicons of E. coli and Anabaena [12,13]. A fragment coding for the NADPH-FDH gene was amplified from pET22b harboring a gene coding for a Pseudomonas FDH mutant, PsFDH[QN] (please see Results and Discussion), using primers, 5′-AAACTGGAGACTCATAATGGCGAAAGTGCTGTG-3′, and 5′-AATAATTGAGCTCTCAGTGGTGGTGGTGGTGGTGTTCGAGTGCGGCCGC-3′. The hetR promoter (approximately 1000 bp upstream of hetR coding region) was amplified from Anabaena genome according to Wang et al. [14]. The fragments were then digested and inserted into pAM505 using SalI and SacI sites, resulting in pAM505-fdh. Furthermore, the FNR gene was inserted into the downstream of the FDH, generating a vector for co-expression of FDH and FNR, pAM505-fdhfnr.
The helper plasmid pRL623, which harbors a mob gene and methylase genes [15] and a self-transmissible plasmid RP4 [16] were provided from Prof. Wolk (Michigan State University).
Introduction of the shuttle vector into Anabaena
Plasmid pAM505-fdh was first introduced into E. coli HB101 carrying a helper plasmid pRL623 [15]. The resultant transformed cells were mixed with Anabaena and E. coli J53 carrying a self-transmissible plasmid RP4 [16–18], and the exoconjugant Anabaena carrying pAM505-fdh ( Anabaena -fdh) was selected on a BG11 agar plate containing neomycin. For co-expression of PsFDH(QN) and FNR, we introduced pAM505-fdhfnr into Anabaena and isolated the exoconjugant Anabaena -fdhfnr similarly to that described above.
Purification of proteins
The FDH proteins, Pc, Fd and FNR were purified under aerobic conditions using a His-Accept Ni-chelating resin (Nacalai tesque). The 50 mM Tris buffer (pH 7.6) containing 500 mM NaCl was used as binding and wash buffer. The binding buffer containing 500 mM imidazole was used for the elution of His-tagged proteins.
Purification of PS I was conducted essentially as previously described by Nakamoto and Hasegawa [19] except for the solubilization condition. To solubilize PS I from thylakoid membrane, the membrane was suspended in the buffer containing 1.6% dodecyl-β-d-maltoside, 50 mM Tris-HCl (pH8.0), 10 mM NaCl and 10% sucrose.
SDS-PAGE and western blot analyses
For SDS-PAGE, gradient gels (5–20%, SuperSepTMAce, Wako, Osaka, Japan) and the Rapid Stain CBB Kit (Nacalai Tesque) were used, according to manufacturer’s instructions. For western blot analysis, proteins in SDS-PAGE gel were transferred onto polyvinylidene difluoride membrane (Immobilon, Millipore) for 1 h under 80 mA constant-current conditions. Blots were blocked with 0.5% (w/v) skim milk in TBST buffer (25 mM Tris-HCl pH7.4, 150 mM NaCl, 0.1% (v/v) Tween 20) and then incubated with a mouse biotin-conjugated anti-histidine-tag antibody (Penta-His Biotin Conjugate, Qiagen, Venlo, Netherlands) and Streptavidin-Horseradish Peroxidase conjugate (Prozyme, Hayward, California) diluted in blocking solution. Blots were washed with TBST buffer. Immunoreaction of HRP was carried out using Pierce Western Blotting Substrate (Thermo Scientific, Headquarters, Waltham, Massachusetts). The resulting chemiluminescence was detected using the ImageQuant LAS 4000 mini (GE Healthcare, Little Chalfont, United Kingdom).
Assays
The formate degradation activities of the FDH proteins were measured spectroscopically in 2 mL of phosphate-buffered saline (PBS) containing 1 mM NADP+ or NAD+, 100 mM formate, 0.05% Triton X-100, and 5 µg/mL FDH. The reaction was initialized by the addition of a FDH sample and the change in the absorption of NADPH (or NADH) was monitored at 340 nm (the extinction coefficient is 6.22 mM-1cm-1). The analysis was performed in triplicate. The Michaelis-Menten parameters were determined from the NAD(P)+ dependence of initial rates of NAD(P) H production in PBS containing 0.05–2 mM NAD(P) D+, 500 mM formate, 0.05% Triton X-100, and 5 µg/mL FDH, and from the formate dependence obtained in the same conditions, except for the NAD(P) D+ and formate concentrations being constant (2 mM) and variable (10–500 mM), respectively.
The formate formation reaction was carried out under strictly anaerobic conditions. A tightly sealed branched flask was used with FDH placed in one arm and PBS, NADPH, and Triton X-100 in the other. The 2 solutions were degassed by repeated cycles of evacuation and argon gas purging, followed by charging with 100% CO2 for 1 min at the final cycle. The reaction was initiated by mixing the 2 solutions at 25° C. The final concentrations of FDH, NAD(P) H, and Triton X-100 were 1.7 mg/ml (38 µM), 1 mM, and 0.05%, respectively. The concentration of the dissolved CO2 in the buffer was measured using a CO2 sensor, CGP-31 (TOA DKK, Tokyo, Japan), and it was observed to be between 1.0 and 1.3 mg/ml. The concentration of formate was determined by withdrawing 100-µl aliquots from the solution without exposure to O2 at intervals of 60 min, and subsequently analyzing them with ion chromatography by using a HPLC system equipped with a Shim-pack SCR-102H column (300 mm length × 8.0 mm φ; mobile phase, 4 mM p-toluenesulfonic acid; column temperature, 40° C) and a conductivity detector, CDD-10AVP (Shimadzu, Kyoto, Japan). The analysis was performed in triplicate.
Light-dependent formate production was performed in 3 mL PBS by using isolated PS I at a concentration of 100 µg chlorophyll/mL, 10 mM ascorbate, 10 µM PC, 2 µM Fd, 0.5 µM FNR, 1 mM NADP+, 4 mM NADPH, 2.5 mg/mL (56 µM) FDH mutant (PsFDH[QN]), 0.05% Triton X-100, and 10% sucrose. The solution containing all the components was degassed by repeating cycles of evacuation and argon gas purging, followed by charging with 100% CO2 for 1 min during the final cycle. The reaction was initiated by illuminating with visible light (<420 nm) at an intensity of 1000 µmol (photon) m-2 s-1 and monitored by sampling at regular intervals without exposure to O2. The analysis was performed in triplicate.
Intracellular formate levels were estimated using the extracts from Anabaena -WT, Anabaena -fdh and Anabaena -fdhfnr cell pellets. Anabaena -WT, Anabaena -fdh and Anabaena -fdhfnr were initially grown in BG11 medium by bubbling with air under a fluorescent lamp at an intensity of 15 µmol photon m-2 s-1, and the cells in the exponential growth phase (OD700 ~3) were washed and transferred into BG110 medium and cultured for a further 36 h. The formation of heterocysts was confirmed microscopically. The media were then degassed and bubbled with air, air + 10% CO2, N2 + 10% CO2, or argon + 10% CO2, for 12 h under relatively strong light (150 µmol photon m-2 s-1). The cultures were transferred anaerobically to the centrifuge tubes in a globe box, centrifuged at 20,000 × g and lysed by grinding with glass beads in 0.1N HCl. The lysates were adjusted to pH 3, centrifuged, and subjected to HPLC analysis. The cultivation and analysis were performed in triplicate.
Results and Discussion
Conversion of the cofactor specificities of FDH mutants
To engineer NADH-FDHs, the FDH genes from Pseudomonas , Candida , potato, Arabidopsis , and Thiobacillus were subcloned into pET22b and mutated at the positions corresponding to the amino acid residues determining the cofactor specificity (i.e., 222 and 224 in Pseudomonas , 195–197 in Candida , 197 and 199 in potato, 227 and 229 in Arabidopsis , and 222 and 224 in Thiobacillus ). The resulting plasmids were transformed into E. coli BL21 (DE3). Cells were grown in LB medium at 37° C and induced with 0.5 mM IPTG. FDH mutants were expressed as a soluble and apo form and purified under aerobic conditions. SDS-PAGE analyses indicated that purified FDH samples from Arabidopsis and Thiobacillus contained additional bands at a slightly lower molecular weight besides an expected band, indicating the partial digestion during the purification; however, those from Pseudomonas , Candida and potato were purified to homogeneity.
Cofactor specificities of FDH mutants were first evaluated by measuring the NAD+ or NADP+ reduction rate. FDH mutants from Arabidopsis and Thiobacillus tended to aggregate and become inactivated during the reaction; however, those from Pseudomonas , Candida , and potato were relatively stable. Several mutants switched the cofactor specificity from NADH to NADPH (Table 1), and among them, Pseudomonas FDH mutations (222→Gln/224→Asn, PsFDH[QN]), Candida FDH mutant (195→Gln/196Arg/197→Asn, CbFDH[QRN]), Arabidopsis FDH mutant (227→Gln/197→Asn, AtFDH[QN]) and potato FDH mutant (195→Gln/197 → His, PoFDH[QH]) exhibited a relatively high NADP+ reduction activity. Using these selected mutants, we next determined the Michaelis-Menten parameters. As shown in Table 2, PsFDH[QN] and CbFDH[QRN] exhibited the highest Vmax values compared with the other 2 mutants, AtFDH[QN] and PoFDH[QH]. PsFDH[QN] showed relatively low K m values in both substrates, NADP and formate, implying a relatively small perturbation by the mutations. K m values for NAD+ of the selected mutants, except for PsFDH[QN], could not be determined because of the low affinity for NAD+ in the concentration range examined (0–20 mM).
Table 1. Cofactor specificities of the WT and mutant FDH proteins.
| NADP+ reduction (µM/h-1) | NAD+ reduction (µM/h-1) | NADP+/NAD+ | |
|---|---|---|---|
| Pseudomonas | |||
| WT (DH) | 24.0 ± 1.7 | 1010 ± 44 | 0.024 |
| EN | 30.0 ± 0.5 | 417 ± 45 | 0.072 |
| SN | 279 ± 1 | 579 ± 30 | 0.48 |
| SH | 184 ± 2 | 429 ± 10 | 0.43 |
| AN | 141 ± 6 | 138 ± 2 | 1.0 |
| AH | 153 ± 2 | 203 ± 10 | 0.75 |
| QN | 293 ± 5 | 30.9 ± 4.2 | 9.5 |
| QH | 299 ± 13 | 31.0 ± 3.9 | 9.6 |
| Candida | |||
| WT (DYH) | 16.8 ± 0.6 | 302 ± 4 | 0.056 |
| QRN | 265 ± 0.3 | 51.0 ± 9.6 | 5.2 |
| Thiobacillus | |||
| WT (DH) | 18.9 ± 0.4 | 926 ± 6 | 0.020 |
| SN | 0.4 ± 0.2 | 3.9 ± 2.9 | 0.10 |
| SH | 3.0 ± 1.2 | 2.6 ± 1.2 | 1.15 |
| AN | 0 ± 0 | 3.1 ± 2.6 | 0 |
| AH | 0.2 ± 0.1 | 3.2 ± 2.6 | 0.063 |
| QN | 0.8 ± 0.1 | 0.3 ± 0.1 | 2.6 |
| QH | 0.6 ± 0.4 | 0.4 ± 0.2 | 1.5 |
| Arabidopsis | |||
| WT (DL) | 1.3 ± 0.2 | 416 ± 61 | 0.0031 |
| SN | 6.8 ± 3.0 | 5.4 ± 4.0 | 1.3 |
| SH | 4.5 ± 2.6 | 4.9 ± 4.4 | 0.92 |
| AN | 5.8 ± 3.0 | 3.1 ± 2.3 | 1.9 |
| AH | 14.6 ± 1.7 | 3.3 ± 0.9 | 4.4 |
| QN | 3.4 ± 2.4 | 5.1 ± 1.6 | 0.67 |
| QH | 70.5 ± 13.9 | 4.2 ± 2.7 | 17 |
| Potato | |||
| WT (DL) | 6.7 ± 0.2 | 803 ± 3 | 0.0083 |
| EN | 5.5 ± 1.5 | 112 ± 1 | 0.049 |
| SN | 53.1 ± 0.7 | 8.6 ± 3.1 | 6.2 |
| SH | 91.7 ± 5.5 | 75.5 ± 1.8 | 1.2 |
| AH | 116 ± 2 | 11.1 ± 3.8 | 10 |
| QN | 93.4 ± 1.4 | 3.7 ± 2.3 | 25.2 |
| QH | 149 ± 11 | 3.4 ± 1.0 | 44 |
FDHs were added up to a final concentration of 5 µg/mL into PBS containing 0.8 mM NAD+ or NADP+, 50 mM sodium formate, and 0.05% Triton X-100 at 30° C. NADH or NADPH production was monitored at 340 nm. All the measurements were performed in triplicate. The two-letter or three-letter descriptions in the left column indicate 2 or 3 one-letter codes of amino acid residue at the position of 222 and 224 in Pseudomonas , 195, 196 and 197 in Candida , 197 and 199 in potato, 227 and 229 in Arabidopsis , and 222 and 224 in Thiobacillus .
Table 2. Michaelis-Menten parameters of the mutant FDH proteins.
|
NADP+ reduction
|
NAD+ reduction
|
||||
|---|---|---|---|---|---|
| Vmax | K m(NADP+) | K m(formate) | Vmax | K m(NAD+) | |
| (µM/h-1) | (mM) | (mM) | (µM/h-1) | (mM) | |
| PsFDH[QN] | 590 ± 20 | 0.35 ± 0.01 | 63 ± 3 | 33 ± 23 | 1.0 ± 0.6 |
| CbFDH[QRN] | 500 ± 200 | 0.18 ± 0.01 | 150 ± 28 | ND | ND |
| AtFDH[QN] | 260 ± 34 | 0.91 ± 0.07 | 96 ± 14 | ND | ND |
| PoFDH[QH] | 320 ± 19 | 0.62 ± 0.05 | 120 ±12 | ND | ND |
The reactions were carried out in PBS containing 0.05–2 mM NAD(P)+ 500 mM formate, 0.05% Triton X-100, and 5 µg/mL FDH or in the same conditions, except for the NAD(P) D+ and formate concentrations being constant (2 mM) and variable (10–500 mM), respectively. Michaelis-Menten parameters were determined by Lineweaver-Burk plots. ND indicates the case in which the data could not be fitted to Lineweaver-Burk plots.
Cofactor specificities of PsFDH(QN), CbFDH(QRN), and PoFDH(QH) for formate production were determined for the following reaction: NAD(P)H + CO2 → HCOO- + NAD(P)+. Because this reaction is inhibited by gaseous oxygen, it was performed under strict anaerobic conditions (100% CO2 atmosphere). Consequently, we observed that the reactivity of PsFDH(QN) with NADPH was 5 times that with NADH, and produced formate at 32 µM/h, corresponding to a specific activity of 19 nmol (mg protein)-1 h-1, which was twice the amount produced in the reaction of the wild-type protein with NADPH (Figure 2). The reaction of PsFDH(QN) was also carried out in the presence of a high concentration of NADPH (10 mM), but it resulted in only a 1.3-fold activity compared to when 1 mM NADPH was used (closed diamonds in Figure 3). This suggested that the cofactor binding sites were almost saturated with NADPH at 1 mM concentration.
Figure 2. Formate production activities of NADPH-FDHs.
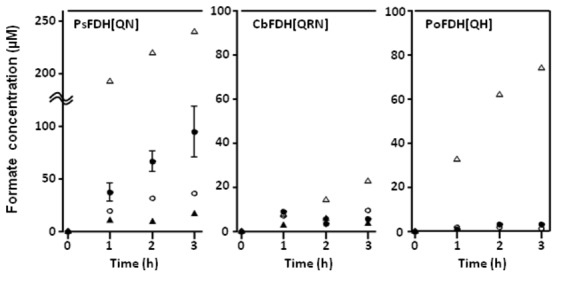
The reaction solution contained 1.67 mg/mL of FDH, 1 mM NAD(P) H, 0.05% Triton X-100 and 1.1–1.3 mg/ml dissolved CO2. The concentration of formate was determined by a HPLC equipped with a Shim-pack SCR-102H column and a conductivity detector, CDD-10AVP (Shimadzu, Kyoto, Japan). Open circles, closed circles, open triangles and closed triangles represent data of combinations of WTs with NADPH, mutants with NADPH, WTs with NADH, and mutants with NADH, respectively. All measurements were performed in triplicate.
Figure 3. Substrate dependence and light sensitivity of PsFDH(QN).
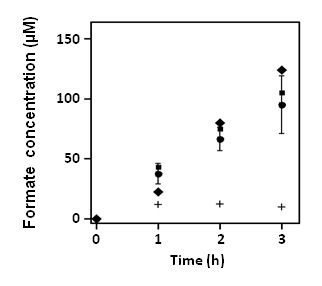
The formate production in the presence of 1 mM NADPH (circles) were compared with that in the presence of 10 mM NADPH (diamonds), that under visible light at an intensity of 1000 µmol photon m-2 s-1 (squares), and that in which NaHCO3 was used instead of CO2 (crosses). These experiments were performed as mentioned in Figure 2, except as described above.
To confirm the photosensitivity of PsFDH(QN), the reaction was conducted under visible light (<420 nm) at an intensity of 1000 µmol photon m-2 s-1. No significant change was observed, demonstrating an insensitivity to light (closed squares in Figure 3). When instead of charging with CO2, NaHCO3 was added to the phosphate buffer (final concentration 50 mM) with NADPH, and Triton X-100, followed by degassing and mixing with the FDH solution, the rate of formate production was significantly lower (crosses in Figure 3). The observed concentration of dissolved CO2 in the buffer (0.040–0.060 mg/mL) was significantly lower than that in the solution charged with CO2 (1.1–1.3 mg/mL), which explains why no significant formate production from NaHCO3 was observed. The significant low CO2 concentration in the buffer added with NaHCO3 was mainly attributed to the degassing process.
CbFDH(QRN) and PoFDH(QH) showed much lower formate production activities than PsFDH(QN) (2.4 µM/h and 1.3 µM/h [1.4 and 0.77 nmol (mg protein)-1 h-1], respectively), likely due to their tendency to aggregate and become inactive (Figure 2).
In vitro light-driven formate production
Using PsFDH(QN), a light-driven formate production system was constructed in vitro. The system consisted of PS I (100 µg chlorophyll/mL) as photosensitizer; 10 mM ascorbate as a sacrificial electron donor; 10 µM PC as an electron carrier at the donor side of PS I; 2 µM Fd, 0.5 µM FNR, 1 mM NADP+, and 4 mM NADPH as an electron relay system at the acceptor side of PS I; and 2.5 mg/mL (~56 µM) PsFDH(QN) as the catalyst for formate production. The formate concentration linearly increased with illumination time after about a 30-min lag time (closed squares in Figure 4). The maximum rate of formate production achieved was 26 µM/h. When expressed in units of formate per mg chlorophyll per hour, the rate is equivalent to 0.26 µmol (mg chlorophyll)-1 h-1. In the control reaction under dark condition (open circles in Figure 4), formate production was observed at a rate of 5 µM/h, which was significantly lower than that under illumination. In the reaction under a 10% O2 and 90% CO2 gas atmosphere (open triangles in Figure 4), formate was not detected, demonstrating the O2-sensitivity of the system.
Figure 4. In vitro light-driven CO2 fixation by PS I and NADPH-FDH.
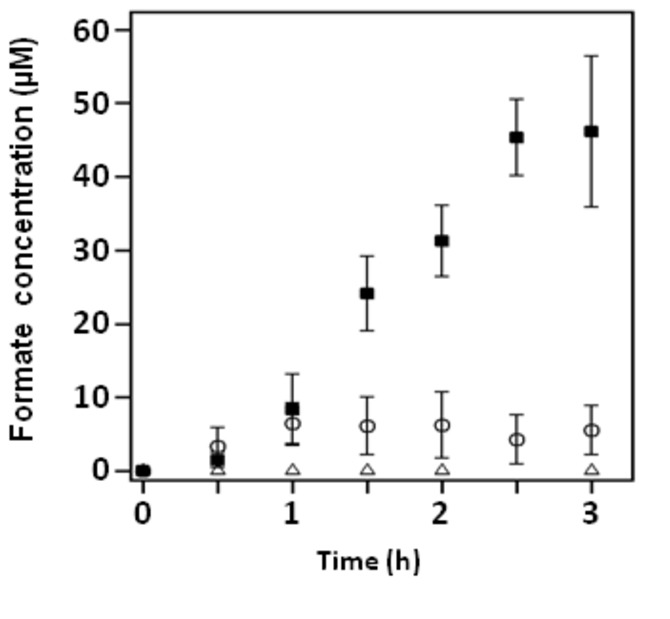
The reaction solutions contained PS I (100 µg chlorophyll/mL), 10 mM ascorbate, 10 µM PC, 2 µM Fd, 0.5 µM FNR, 1 mM NADP+, and 4 mM NADPH, 2.5 mg/mL (~56 µM) PsFDH(QN), 0.05% Triton X-100 and 10% sucrose in PBS. The solution were degassed and then charging with 100% CO2 for 1 min. The reaction was initiated by illuminating with visible light (<420 nm) at an intensity of 1000 µmol photon m-2 s-1, and monitored by sampling at regular intervals without exposure to O2 (closed square). The control reactions were performed without the illumination (open circles) and in the presence of 10% oxygen gas (open triangles).
The rate-determining factor was then assessed using the total activity of PsFDH(QN) and the rate of supply of NADPH. The total activity of 7.5 mg of PsFDH(QN) in 3 mL of reaction solution was estimated to produce 47 µM formate/h using its specific activity of 19 nmol (mg protein)-1 h-1 as described above. This rate was in the same range as the maximum rate of formate production at 30-60 min after initiation. On the other hand, the total activity of the 0.3 mg chlorophyll PS I in 3 mL solution was estimated to be 15 mM NADPH/h using its specific activity of 150 µmol (mg chlorophyll)-1 h-1 which was determined in the same conditions except for no FDH protein and no NADP+ being added and the concentration of PS I being 5 µg chlorophyll/mL. However, because the total NADP(H) concentration was 5 mM, almost all NADP+ was expected to be reduced to NADPH. In fact, the NADPH concentration in the control reaction solution without PsFDH(QN) was analyzed spectrophotometrically and estimated to reach 4.5 mM (NADPH/NADP+ ~ 9), which is high enough to push the equilibrium towards formate production, within 60 min after the initiation of the reaction under anaerobic conditions. The time required for all NADP+ to be converted to NADPH was consistent with the lag time observed in Figure 4. Therefore, the rate-determining factor was identified in the accumulation of NADPH from 0 min to 30 min, and in the activity of FDH from 30 min onward.
In vivo light-driven formate production
To recreate the light-driven formate production system in vivo, we attempted to express PsFDH(QN) in Anabaena heterocysts, which differentiate in order to carry out nitrogen fixation during nitrogen starvation [10]. Heterocysts are deficient in PS II, and their internal environment is micro-oxic. The cells receive carbohydrates from neighboring vegetative cells and use them as electron donors to PS I. The photo-excited electrons derived from PS I are used for the reduction of N2 gas to ammonium by nitrogenase [20]. The micro-oxic and reductive environment makes heterocysts an ideal reaction vessel for light-driven formate production by PsFDH(QN). For the expression of PsFDH(QN) in heterocysts, we constructed two shuttle vectors, pAM505-fdh and pAM505-fdhfnr, and introduced them into Anabaena ( Anabaena -fdh and Anabaena -fdhfnr).
To confirm the expression of PsFDH(QN), western blot analysis was carried out. Anabaena -fdh and Anabaena -fdhfnr were cultivated in nitrogen-deficient BG11 medium (BG110), in which the differentiation to heterocysts and the expression of PsFDH(QN) was to be induced. Cell extracts were separated on 10% polyacrylamide gels and then blotted onto polyvinylidene difluoride membranes. Chemiluminescence images were obtained after blocking, incubation with anti-His tag antibody-biotin conjugate and streptavidin-Horseradish Peroxidase conjugate and reaction with chemiluminescence reagent. The bands were successfully detected at the expected molecular weight of PsFDH(QN) in the samples from both exoconjugants, but the intensity of the sample from BG110 was approximately 2-fold that from BG11 (Figure 5). Although it is suggested that the expression of the PsFDH(QN) was enhanced in response to nitrogen deprivation, it remains to be solved whether the increase occurred mainly in the heterocysts. Assuming the heterocyst-specific response, taken together with the fact that approximately 1 heterocyst develops for every 10–20 vegetative cells; it is speculated that the PsFDH(QN) concentration in the heterocysts was 10–20 times that in the vegetative cells.
Figure 5. Expression of PsFDH[QN] in Anabaena -fdh.
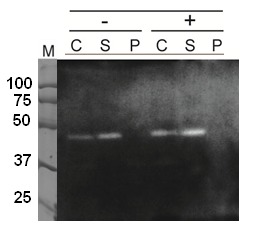
Anabaena -fdh cells were cultured in non-inducible ((-), BG11) or inducible ((+), BG110) medium and their whole lysates (C), lysate supernatants (S) and lysate precipitates (P) were analyzed by Western blotting using anti-Histag antibody HRP conjugate. Chemiluminescent signals were detected at approximately 44 kDa as expected.
Anabaena -WT, Anabaena -fdh and Anabaena -fdhfnr were initially grown in BG110 medium by bubbling with air, air + 10% CO2, N2 + 10% CO2, or argon + 10% CO2. We observed that only when Anabaena -fdh was incubated under argon + 10% CO2, a peak was detected at the same retention time (12.6 min) as formate with an intensity corresponding to a concentration of 28 µM (Figure 6). To confirm whether the peak could be assigned to formate, the lysate was neutralized and treated with 10 µM Candida boidinii FDH (Sigma-Aldrich, St. Louis, Missouri) and 10 mM NAD+ to advance the reaction: HCOO- + NAD+ → CO2 + NADH. Thereupon, only the peak at 12.6 min among the observed peaks disappeared (Figure 5), demonstrating that it was derived from formate. Therefore, we concluded that introducing PsFDH(QN) into heterocysts accelerated in vivo formate production.
Figure 6. Formate production by pAM505-fdh exoconjugant cyanobacteria.
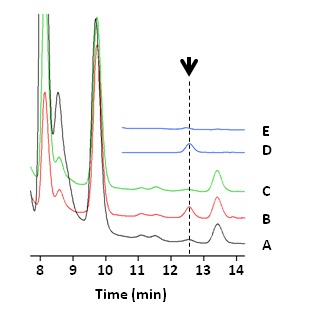
The wild-type and exoconjugant Anabaena cultures (BG11, OD700 ~3) were inoculated into BG110 and cultivated for 36 h under a light of 15 µmol photon m-2 s-1 and under air atmosphere. The media were then degassed and bubbled with argon + 10% CO2 for 12 h under a light of 150 µmol photon m-2 s-1. The cells anaerobically harvested were homogenized in 1N HCl. The resulting lysate was analyzed by ion-chromatography and the chromatograms of Anabaena -WT (A) and Anabaena -fdh (B) are shown. The lysate of Anabaena -fdh was also treated with Candida boidinii FDH (Sigma-Aldrich, Missouri, USA) and NAD+ overnight and was then analyzed (C). The chromatograms of formate solutions without and with the treatment with Candida boidinii FDH and NAD+ are shown as controls (D and E, respectively).
One of the possible interpretations of this result is that under argon + 10% CO2, the electron flux toward N2 fixation in heterocysts is decreased and the NADPH concentration is increased, facilitating the reaction: CO2 + NADPH → HCOO- + NADP+ by PsFDH(QN). Another interpretation is that nitrogenase can accept electrons in the absence of N2 and produce H2 [20], which reacts with endogenous hydrogenase, producing NAD(P) H, which is used as a substrate for PsFDH(QN) to produce formate. In this study, no significant amounts of formate were detected in the lysates of Anabaena -fdhfnr. Although the level of expression of PsFDH(QN) in Anabaena -fdhfnr was almost the same as that in Anabaena -fdh, the growth rate of Anabaena -fdhfnr was lower than Anabaena -WT and Anabaena -fdh (see Figure S1), and a large quantities of cell debris appeared in the BG110 medium, suggesting the occurrence of cell lysis and the outflow of cell contents. Notably, by over repeated subculture, the ability of Anabaena -fdh to produce formate sometime disappeared because of the appearance of plasmids lacking FDH gene. In addition to this problem, the analysis of these phenomena at molecular level becomes difficult because of the low frequency of heterocysts. The development of stable expression system and a quantitative understanding of the process in heterocysts are currently under investigation.
Summary
Here we have shown a novel example of direct CO2 reduction using the photo-reducing power of PS I. The system proposed here consisted of proteins (PS I, Fd, FNR, and PsFDH[QN]) and a natural electron carrier (NADP[H]). We have demonstrated the feasibility of creating a CO2 reduction system in vivo by introducing PsFDH(QN) into heterocysts and have successfully enhanced the level of formate production. Further work investigating the detailed electron flux in the cells needs to be carried out, in addition to the optimization of the electron supply to the CO2 reduction system, the suppression of the N2 fixation and the design of the system for formate secretion.
Supporting Information
Growth curves of Anabaena wild-type (open circles), Anabaena -fdh (closed squares) and Anabaena -fdhfnr (closed triangles). xmlns:xlink="http://www.w3.org/1999/xlink" xmlns:mml="http://www.w3.org/1998/Math/MathML">Culture conditions were: at 30◦C, light intensity of 30 mE∙m-2∙ s-1 and bubbling with air in BG11 medium.
(TIF)
Acknowledgments
The authors gratefully thank Prof. C. Peter Wolk (Michigan State University) for gifting us plasmids, pRL623 and RP4, and E. coli strain, J53. The authors are also grateful to Dr. Shigeki Ehira (Chuo University) and Prof. Yasuhiro Kashino (University of Hyogo) for helpful discussions.
Funding Statement
This research was supported by PRESTO (Precursory Research for Embryonic Science and Technology) in JST (Japan Science and Technology Agency) program. The URL of any funder's website is "http://www.jst.go.jp/kisoken/presto/en/index.html". The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hornsey DJ (1971) An investigation of formate metabolism in bacteria using radiorespirometry. Int J Appl Radiat Isot 22: 381-383. doi:10.1016/0020-708X(71)90066-4. PubMed: 4933058. [DOI] [PubMed] [Google Scholar]
- 2. Leonhartsberger S, Korsa I, Böck A (2002) The molecular biology of formate metabolism in enterobacteria. J Mol Microbiol Biotechnol 4: 269-276. PubMed: 11931558. [PubMed] [Google Scholar]
- 3. Crable BR, Plugge CM, McInerney MJ, Stams AJ (2011) Formate formation and formate conversion in biological fuels production. Enzyme Res, 2011: 2011: Article ID 532536. PubMed: 21687599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferry JG (1990) Formate dehydrogenase. FEMS Microbiol Rev 7: 377-382. PubMed: 2094290. [DOI] [PubMed] [Google Scholar]
- 5. Tsujisho I, Toyoda M, Amao Y (2006) hotochemical and enzymatic synthesis of formic acid from CO 2 with chlorophyll and dehydrogenase system 7. Cat Co. pp. 173-176 [Google Scholar]
- 6. Miyatani R, Amao Y (2004) Photochemical synthesis of formic acid from CO2 with formate dehydrogenase and water-soluble zinc porphyrin. J Mol Cat B 27: 121-125. doi:10.1016/j.molcatb.2003.11.003. [Google Scholar]
- 7. Kurayama F, Matsuyama T, Yamamoto H (2004) A feasibility study of a new photosynthesis bioreactor design using TiO2 particles combined with enzymes. Ad Powder Tec 15: 51-61. doi:10.1163/15685520460740061. [Google Scholar]
- 8. Moal G, Lagoutte B (2012) Photo-induced electron transfer from photosystem I to NADP(+): Characterization and tentative simulation of the in vivo environment. Biochim Biophys Acta 1817: 1635-1645. doi:10.1016/j.bbabio.2012.05.015. PubMed: 22683536. [DOI] [PubMed] [Google Scholar]
- 9. Bengis C, Nelson N (1975) Purification and properties of the photosystem I reaction center from chloroplasts. J Biol Chem 250: 2783-2788. PubMed: 804481. [PubMed] [Google Scholar]
- 10. Kumar K, Mella-Herrera RA, Golden JW (2010) Cyanobacterial heterocysts. Cold Spring Harb Perspect Biol 2: a000315. doi:10.1101/cshperspect.a000315. PubMed: 20452939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu W, Zhu D, Hua L (2009) Site-saturation mutagenesis of formate dehydrogenase from Candida bodinii creating effective NADP+-dependent FDH enzymes. J Mol Cat B 6: 157-161. [Google Scholar]
- 12. Wei TF, Ramasubramanian TS, Golden JW (1994) Anabaena sp. strain PCC 7120 ntcA gene required for growth on nitrate and heterocyst development. J Bacteriol 176: 4473-4482. PubMed: 7913926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoon HS, Golden JW (1998) Heterocyst pattern formation controlled by a diffusible peptide. Science 282: 935-938. doi:10.1126/science.282.5390.935. PubMed: 9794762. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Xu X (2005) Regulation by hetC of genes required for heterocyst differentiation and cell division in Anabaena sp. strain PCC 7120. J Bacteriol 187: 8489-8493. doi:10.1128/JB.187.24.8489-8493.2005. PubMed: 16321953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elhai J, Vepritskiy A, Muro-Pastor AM, Flores E, Wolk CP (1997) Reduction of conjugal transfer efficiency by three restriction activities of Anabaena sp. strain PCC 7120. J Bacteriol 179: 1998-2005. PubMed: 9068647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas CM, Smith CA (1987) Incompatibility group P plasmids: genetics, evolution, and use in genetic manipulation. Annu Rev Microbiol 41: 77-101. doi:10.1146/annurev.mi.41.100187.000453. PubMed: 3318684. [DOI] [PubMed] [Google Scholar]
- 17. Elhai J, Wolk CP (1988) Conjugal transfer of DNA to cyanobacteria. Methods Enzymol 167: 747-754. doi:10.1016/0076-6879(88)67086-8. PubMed: 3148842. [DOI] [PubMed] [Google Scholar]
- 18. Ehira S, Ohmori M (2012) The pknH gene restrictively expressed in heterocysts is required for diazotrophic growth in the cyanobacterium Anabaena sp. strain PCC 7120. Microbiology 158: 1437-1443. doi:10.1099/mic.0.057729-0. PubMed: 22383473. [DOI] [PubMed] [Google Scholar]
- 19. Nakamoto H, Hasegawa M (1999) Targeted inactivation of the gene psaK encoding a subunit of photosystem I from the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol 40: 9–16. doi:10.1093/oxfordjournals.pcp.a029479. PubMed: 10189699. [DOI] [PubMed] [Google Scholar]
- 20. Bothe H, Schmitz O, Yates MG, Newton WE (2010) Nitrogen fixation and hydrogen metabolism in cyanobacteria. Microbiol Mol Biol Rev 74: 529-551. doi:10.1128/MMBR.00033-10. PubMed: 21119016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth curves of Anabaena wild-type (open circles), Anabaena -fdh (closed squares) and Anabaena -fdhfnr (closed triangles). xmlns:xlink="http://www.w3.org/1999/xlink" xmlns:mml="http://www.w3.org/1998/Math/MathML">Culture conditions were: at 30◦C, light intensity of 30 mE∙m-2∙ s-1 and bubbling with air in BG11 medium.
(TIF)