

It is widely accepted that the transcription factor STAT5 has a key role in many hematological malignancies and mediates anti-apoptotic as well as growth-stimulatory functions. In BCR-ABL1-driven disease, STAT5 has an even more privileged position, as it supports the leukemic cells in counteracting therapeutic challenges posed by tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib and dasatinib.1-3
In contrast to the two-faced god Janus, after whom the STAT upstream Janus kinases are named, STAT5 appears to have at least four “faces” that support chronic myeloid leukemia (CML) triggered by the BCR-ABL1 oncoprotein. Two faces have been known for a long time and are consequently well documented: STAT5 is capable of stimulating cell proliferation and enhances cell viability by upregulation of anti-apoptotic genes such as BCLXL and MCL1.1,2 The two additional faces were discovered only recently: STAT5 is able to counteract TKI-induced cell death and to increase the probability of acquiring BCR-ABL1 mutations.3,4
CML used to be extremely deadly. Although the development of BCR-ABL1 TKIs has lessened the threat posed by the disease, the success is tempered by the development of resistance to the drugs, especially resulting from mutations in the BCR-ABL1 gene itself. Increased levels of STAT5 protein counteract TKI therapy in two ways: the high STAT5 protein levels observed in human patients after prolonged disease not only shield cells from TKI attack, but also favor BCR-ABL1 mutations.4 Interestingly, the loss of responsiveness does not extend to conventional chemotherapeutic drugs but is limited to TKIs.3
Our experiments have uncovered a clear and statistically highly significant correlation between the expression levels of STAT5 and the occurrence of BCR-ABL1 mutations. We propose that production of reactive oxygen species (ROS) triggered by STAT5 mediates the effect.4 STAT5 and BCR-ABL1 act in tandem—the BCR-ABL1 oncoprotein is required to enable STAT5 to enhance intracellular levels of ROS. In non-transformed cells, enforced STAT5 expression does not trigger ROS. Accordingly, the Janus kinase JAK2 is of no importance for STAT5-mediated ROS production. JAK2 is the main upstream kinase of STAT5 in hematopoietic cells, but we have provided evidence that a pronounced signal rewiring takes place in BCR-ABL1+ cells, placing STAT5 under the direct control of the BCR-ABL1 oncoprotein.5 BCR-ABL1 itself phosphorylates a critical tyrosine residue that drives dimerization/oligomerization of STAT5, which is a prerequisite for nuclear translocation and DNA binding. The formation of STAT5 oligomers is important for the regulation of ROS levels and indicates that a set of oligomer-dependent STAT5 target genes critically regulates BCR-ABL1-driven ROS production.
As a consequence of STAT5 upregulation, the rate of double-strand breaks (DSBs) increases, representing a first step toward an enhanced mutation rate.4 The group of Thomas Skorski has pioneered studies on the role of ROS in BCR-ABL1-driven leukemia. The work has convincingly revealed the link between BCR-ABL1, ROS production and increased mutations rates.6,7 We now add STAT5 to the landscape. The role of ROS may go beyond increasing mutation rates: in a recent paper that investigated JAK2V617F-driven disease, inhibiting ROS was shown to have major consequences, inhibiting and significantly impairing disease development.8 JAK2V617F-driven disease also critically depends on activation of STAT5, so STAT5 is somehow involved in the production of ROS driven by JAK2V617, although the mechanistic details remain a matter of speculation.
It is conceivable that the development of third-generation TKIs such as ponatinib will take care of mutation-related resistance, as ponatinib potently inhibits the most feared T315I “gatekeeper” mutation, the single mutation that has sounded the death knell for all treatment options based on TKIs. This optimistic point of view does not take into account the fact that cancers generally win the evolutionary race between targeted drugs and drug escape. Unless we manage to solve the difficulties posed by the high resistance of CML stem cells to TKIs, the problem of resistance will persist in the future. Even sequential therapy with TKIs will select for BCR-ABL1 compound mutations that confer resistance to multiple inhibitors. A further concern is the occurrence of BCR-ABL1-independent resistance to targeted therapy. Based on its critical multifaceted role in CML, STAT5 remains an attractive therapeutic target for two reasons. First, CML cells depend on an active STAT5 pathway irrespective of their BCR-ABL1 mutation status. Second, inhibition of STAT5 activity decreases levels of ROS and thereby reduces the probability of acquiring additional disease-driving mutations. The discovery that STAT5 acts as a trigger of ROS thus provides a further rationale for targeting STAT5 in leukemia (Fig.1).
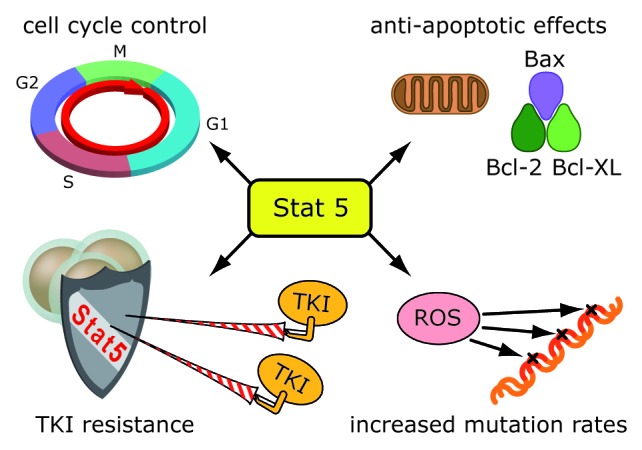
Figure 1. Multiple functions of STAT5 in BCR-ABL transformed cells. Activation of STATS (1) impacts on cell cycle control (2) enhances anti-apoptotic effects (3) and results in an up-regulation of ROS (4) thereby contributing to an increased mutation rate that contributes to TKI resistance. The enhanced mutation rate is counterbalanced by STATS effects on pro-survival genes such as Bcl-2 and Bcl-XL.
Acknowledgments
This work was supported by the Austrian Science Fund (FWF): [SFB 2810].
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25116
References
- 1.Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, et al. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107:4898–906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117:3409–20. doi: 10.1182/blood-2009-10-248211. [DOI] [PubMed] [Google Scholar]
- 4.Warsch W, Grundschober E, Berger A, Gille L, Cerny-Reiterer S, Tigan AS, et al. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012;3:1669–87. doi: 10.18632/oncotarget.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, et al. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8:285–93. doi: 10.1038/nchembio.775. [DOI] [PubMed] [Google Scholar]
- 6.Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–27. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–53. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 8.Marty C, Lacout C, Droin N, Le Couédic JP, Ribrag V, Solary E, et al. A role for reactive oxygen species in JAK2(V617F) myeloproliferative neoplasm progression. Leukemia. 2013 doi: 10.1038/leu.2013.102. [DOI] [PubMed] [Google Scholar]