

Abrogation of DNA damage-induced cell cycle checkpoints represents a promising concept for chemosensitization during anticancer treatment. A prominent example is the Chk1 inhibitor UCN-01 that is currently used in clinical trials in combination with various DNA damaging or DNA replication inhibiting drugs. It is well-established that inhibition of Chk1 in the presence of DNA damage leads to an override of the G2 cell cycle checkpoint, resulting in a premature mitotic entry.1 Damaged cells that are forced to enter mitosis show a transient mitotic arrest mediated by the spindle assembly checkpoint (SAC). At the same time, massive chromosome fragmentation is commonly observed, and this seems to be a prerequisite for the subsequent mitotic catastrophe, leading to the induction of apoptosis during mitosis, which involves a Mad2-dependent proapoptotic pathway2 (Fig. 1). However, the exact nature of mitotic defects triggering mitotic catastrophe remains unclear, but it is investigated in the work presented by Beeharry et al.3
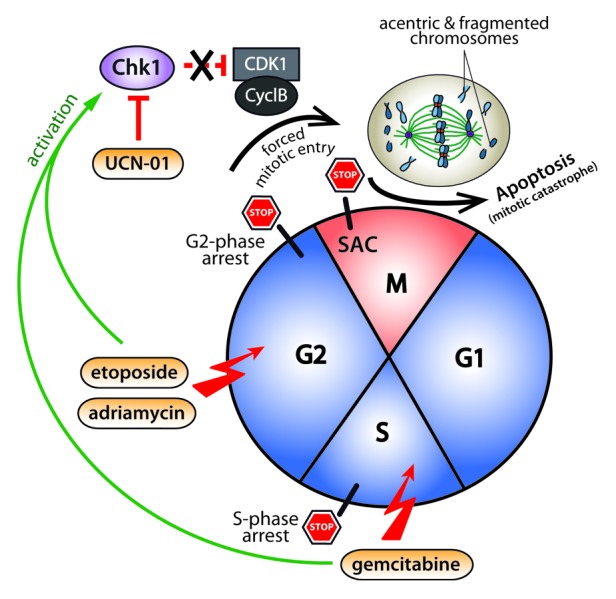
Figure 1. Model illustrating mitotic centromere fragmentation after UCN-01-mediated S and G2 checkpoint override. Replication-stress (gemcitabine treatment) or inhibition of topoisomerase II (etoposide or Adriamycin treatment) leads to cell cycle arrest in S- and G2-phase, respectively. Abrogation of the damage-induced cell cycle checkpoints by treatment with the Chk1 inhibitor UCN-01, forces cells to enter mitosis with incompletely replicated DNA leading to the formation of acentric and fragmented chromosomes. As a consequence of MUGing (mitosis with unreplicated genomes), the cells transiently arrest in mitosis in a spindle assembly checkpoint (SAC)-dependent manner before undergoing mitotic catastrophe and subsequent apoptosis.
In the June 1, 2013 issue of Cell Cycle, Beeharry and colleagues provide interesting new insights into the mitotic defects that result from abrogation of a DNA damage-induced S- or G2-phase checkpoint arrest. Immunofluorescence and electron microscopy analysis of a panel of different pancreatic cancer cell lines revealed that sequential treatment with the DNA replication inhibitor gemcitabine or with topoisomerase II inhibitors such as etoposide and adriamycin in combination with UCN-01 leads to the generation of MUGs (mitosis with unreplicated genomes). Here, MUGs are characterized by severe fragmented mitotic chromosomes with unreplicated centromeres and kinetochores detached from the bulk chromosomes. MUGs were first described by Brinkley et al. some 30 years ago,4 but Beeharry and colleagues now provide the first evidence that MUGs might be the primary outcome of G2 checkpoint override after treatment with chemotherapeutic DNA replication inhibitors. In contrast, G2 checkpoint override after treatment of cells with alkylating agents such as methyl methanesulfonate (MMS), albeit inducing DNA damage and arresting cells in G2 phase of the cell cycle, do not induce MUGs, and accordingly, cells progress though mitosis without further delay. Interestingly, inhibition of topoisomerase II by etoposide and adriamycin, although not grossly inhibiting DNA replication, still causes MUGing. By using FISH analysis with a centromere-specific probe hybridizing to the centromere region of chromosome 7, the authors found that inhibition of topoisomerase II, like inhibition of DNA replication by gemcitabine, results in unreplicated centromeres.3 It was previously shown that replication of centromeric DNA proceeds until metaphase,5 supporting the notion that the replication of centromeres might be sensitive toward topoisomerase II inhibition. However, it remains an open question how topoisomerase II inhibition blocks centromere replication.
The work presented by Beeharry et al. suggests that the underlying cause for mitotic catastrophe observed after G2 checkpoint override might be a lack of replication and subsequent fragmentation of the centromere, resulting in acentric genomes. These interesting results might also explain why cells cannot satisfy the SAC after forced entry into mitosis and arrest in a metaphase-like state.2 Furthermore, it would be interesting to address the question of whether this mitotic arrest or the centromere fragmentation is directly related to the massive chromosome fragmentation that is commonly seen in mitotic cells after checkpoint override. In fact, it might be possible that chromosome condensation that occurs on chromosomes after forced entry into mitosis can generate forces that tear chromosomes apart.
From the clinical point of view, this study provides insights on the choice of chemotherapeutic drugs that are to be combined with G2 checkpoint inhibitors. At least in pancreatic cancer cells, as shown here, the combination of inhibitors of centromere replication and G2 checkpoint inhibitors might be the most powerful combination, in particular for cancer cells lacking functional p53. Appropriate clinical trials will show whether this holds true in a clinical setting. Also, additional studies investigating other cancer entities are needed to provide a more general view on the efficacy of chemotherapeutic drugs that affect centromere replication and S or G2 checkpoint inhibitors.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25175
References
- 1.Tse AN, Schwartz GK. Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–44. doi: 10.1158/0008-5472.CAN-04-0841. [DOI] [PubMed] [Google Scholar]
- 2.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007;67:339–45. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 3.Beeharry N, Rattner JB, Caviston JP, Yen T. Centromere fragmentation is a common mitotic defect of S and G 2 checkpoint override. Cell Cycle. 2013;12:1588–97. doi: 10.4161/cc.24740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkley BR, Zinkowski RP, Mollon WL, Davis FM, Pisegna MA, Pershouse M, et al. Movement and segregation of kinetochores experimentally detached from mammalian chromosomes. Nature. 1988;336:251–4. doi: 10.1038/336251a0. [DOI] [PubMed] [Google Scholar]
- 5.Madan K, Allen JW, Gerald PS, Latt SA. Fluorescence analysis of late DNA replication in mouse metaphase chromosomes using BUdR and 33258 Hoechst. Exp Cell Res. 1976;99:438–44. doi: 10.1016/0014-4827(76)90604-2. [DOI] [PubMed] [Google Scholar]