

Abstract
If life were created by intelligent design, we would indeed age from accumulation of molecular damage. Repair is costly and limited by energetic resources, and we would allocate resources rationally. But, albeit elegant, this design is fictional. Instead, nature blindly selects for short-term benefits of robust developmental growth. “Quasi-programmed” by the blind watchmaker, aging is a wasteful and aimless continuation of developmental growth, driven by nutrient-sensing, growth-promoting signaling pathways such as MTOR (mechanistic target of rapamycin). A continuous post-developmental activity of such gerogenic pathways leads to hyperfunctions (aging), loss of homeostasis, age-related diseases, non-random organ damage and death. This model is consistent with a view that (1) soma is disposable, (2) aging and menopause are not programmed and (3) accumulation of random molecular damage is not a cause of aging as we know it.
Keywords: God, rapamycin, senescence, cancer, diseases, natural selection
Introduction
“Natural selection, the blind, unconscious, automatic process…has no purpose in mind. It has no mind and no mind’s eye. It does not plan for the future. It has no vision, no foresight, no sight at all. If it can be said to play the role of watchmaker in nature, it is the blind watchmaker.”1 Richard Dawkins, The Blind Watchmaker: Why the Evidence of Evolution Reveals a Universe without Design
The View from Intelligent Design
DNA, RNA, proteins, lipids, as well as structures containing these molecules can be damaged. Molecular damage constantly occurs. This must lead to aging. Molecular damage can be repaired. Yet, repair is energy-dependent and “expensive.” And the organism, anyway, does not last forever, because it dies from extrinsic causes, such as predators, infections, starvation and accidents. Resource must be allocated: first to vital functions (such as brain respiration) to avoid immediate death, second to growth and reproduction and, third, to repair molecular damage to avoid “aging.” And a truly intelligent designer will even calculate cost-effectiveness of anti-aging repair and design trade-off allocation between repair and reproduction to maximize reproductive success.
In the protected environment, where accidental causes of death are diminished, humans and laboratory/domesticated animals would die from aging (see “Appendix: Footnote 1”).
Blind Watchmaker
Unlike intelligent designer (ID), blind watchmaker (BW) cannot foresee aging. The BW is concerned with development, growth, reproduction and prevention of death from immediate causes: starvation, accidents, predators, weather, genetic and infectious diseases. Early in life, an animal must be strong and competitive. Activated by nutrients and growth factors, intra-cellular signaling networks including the MTOR (target of rapamycin) pathway stimulate growth,2,3 muscle hypertrophy4 and robustness.5 Provided that nutrients (fuel) are readily available, MTOR is active or, metaphorically, “MoTOR” is running (Fig. 1A).
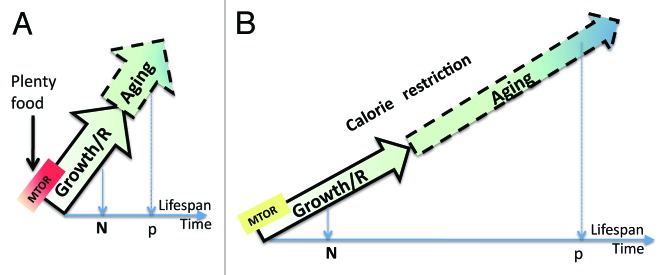
Figure 1. MTOR-driven quasi-programmed aging: plenty of food (overeating) vs. calorie restriction (famine). (A) Plenty of food: activation of the nutrient/MTOR and insulin/MTOR pathway. Calories are used for survival, growth and reproduction (R). Energy-dependent program of developmental growth continues as energy-dependent quasi-program of aging. N (natural), the typical lifespan in the wild; P (protected environment), the typical lifespan in the protected environment (humans, laboratory and domestic animals). In rare cases, protected environments can emerge naturally, especially among large animals. (B) Calorie restriction and famine: Especially during famine, calories/nutrients are used for immediate survival and only “leftovers” for program of developmental growth and reproduction (growth/R). N (natural), the typical lifespan in the wild during famine is short. There is no need to spend any calorie on anti-aging activities, no need to delay aging. P (protected environment), the typical lifespan in the protected environment (humans, laboratory and domestic animals). Lifespan is increased during calorie restriction under protected environment (ideally, no accidental death), exactly because resources are limited to drive energy-dependent quasi-programmed aging.
Because BW cannot design, she is extremely wasteful and messy. But even a small increase in fitness early in life justifies any waste. Furthermore, when development is finished, BW continues to spend energy on “twisted” growth or aging, which is purely harmful, energy-consuming and an unneeded process. But nature is blind. So how does growth convert into aging?
When developmental growth is completed, BW does not care to switch off growth programs driven by MTOR (and other related pathways). What for? Furthermore, MTOR is positively involved in reproduction. Also, the organism is likely to die from extrinsic causes anyway (Fig. 1). So nature is incapable of inhibiting MTOR just to prevent “future” aging. (A hypothetical exception was discussed elsewhere.6,7 One can calculate the cost-effect, but not because anti-aging efforts are limited by resources, but because MTOR is pleiotropic: useful early in life and harmful later. Later in life, MTOR causes cellular aging with inappropriate hyperfunction, an increased production of cytokines, resistance to signals, leading to loss of homeostasis, diseases of aging, organ damage and death, as recently discussed.6
BW is reluctant to constrain mTOR just in order to delay “aging.” Survive right now is more important. The M(o)TOR is running, and the accelerator is stuck. By analogy, a car, running at 75 mph on the highway exits to a parking lot but continues to idle at full speed, as the accelerator is stuck. Importantly, even 50 mph will be too fast. (“Footnote 2”; in theory, either the MTOR activity or synthetic processes may be decreased later in life, but either not sufficiently or too late. For example, RNA/protein synthesis is decreased with aging in model organisms, yet its further inhibition prolongs lifespan,8-13 indicating insufficient natural decrease).
It is not BW, but starvation, scarce resources and stresses that de-activate the nutrient-sensing mTOR pathway, simply for mechanistic reasons not for the purpose of slowing aging. Starvation slows developmental growth, delays reproduction and aging (Fig. 1A), because aging is just driven by the same M(o)TOR, and this is why CR extends lifespan and overeating shortens it.14 (Fig. 1)
Both Models are Disposable Soma Theories (DST)
Soma is disposable by definition. Therefore, regardless of the mechanisms of aging, all models of aging are “disposable soma theories.” For example, soma could be damaged by passive process, such as insufficient repair of molecular damage. As emphasized, “the aging process is caused by the gradual buildup of a huge number of individually tiny faults—some damage to a DNA strand here, a deranged protein molecule there and so on.”15 Or it can be damaged by an active process: myocardial infarction and stroke as a consequence of atherosclerosis, hypertension and thrombosis caused by MTOR-driven aging.6,16
It is misleading to call any one theory “simply” disposable soma theory (DST), implying that other theories are not DSTs.
DST One: Damage/Repair Theory
Known as “disposable soma theory (DST),” this theory postulates that: (1) accumulation of molecular damage causes aging, and (2) repair is costly and limited by resources.15,17,18 Therefore, molecular damage is not repaired completely and eventually causes aging (in a protected environment). This is logical and intelligent. Actually, this is exactly what an intelligent designer would design. However, predictions of this model contradict observations, experiments and medical practice. If repair is limited by resources, the prediction must be the less resources, the shorter lifespan. And vice versa, namely, over-nutrition would provide resources for repair and thereby extend lifespan. However, calorie restriction extends lifespan, whereas overeating shortens it.
To save the theory, it was suggested that during famine/starvation resources are allocated to anti-aging repair (Fig. 1). When starvation is over, then an organism would live a longer life, catching up with reproduction. This is paradoxical.14,19 First, during famine, when the death rate is especially high, it would be sensible to allocate resources to vital functions and fitness, simply to prevent death from starvation and predators. Is it aging that always limits lifespan in the wild according to DST (and even despite famine when external death rate is exceptionally high). This contradicts the evolutionary theory. The DST (repair/damage) theory becomes internally inconsistent, leading to paradoxes as discussed.14 It was even suggested that some harmful conditions such as menopause are programmed and have a purpose.15 And would any rational designer extend life by inflicting damage (hormesis), indicating lack of design, as recently discussed?20 And finally, deletion of numerous genes and processes not only improves an organism, but also extends lifespan.21-36 What is a strange design? Should knockout of springs make clock-watches better? One must agree with Richard Dawkins that an intelligent designer (ID) has never existed. Aging, like life itself, have been shaped by blind watchmaker (BW).
DST Two: Hyperfunction Theory
In the BW scenario, an organism actively causes aging. BW uses energy to foster growth and then for its unneeded continuation, quasi-programmed aging. The long sequence of unforeseen events includes cellular hypertrophy, hyperplasia, hyperfunctions, loss of homeostasis, age-related diseases, organ damage and loss of functions.37,38 At late stages, the process becomes MTOR-independent. Then malfunctions replace hyperfunctions. Since initial hyperfunctions are the most harmful, the model MTOR-driven quasi-programmed aging is becoming known as hyperfunction theory.39
Life Without Food: Eating as Harmful Quasi-Programmed Hyperfunction
The most impressive example is lifespan extension by complete removal of food in C. elegans.40 Whereas nutrients are essential for developmental growth, they are not needed in adult C. elegans, but even hurt them. Still, harmful eating continues. As was discussed in detail, nutrients, insulin and growth factors all activate TOR, driving growth and aging.41 This explains why lack of food extends lifespan. This cannot be easily explained by any trade-offs between maintenance and reproduction.40 The higher the MTOR activity, the faster the aging the way it is blindly “designed.” An adult worm is actively seeking food, thus only accelerating its aging and death.
Gerogenic Conversion (Cellular Aging) and Organismal Aging
One of the most important features of hyperfunction theory is that organismal aging and age-related diseases can be easily explained by cellular aging on molecular mechanisms.38,42,43,38 Cellular senescence is not just cell cycle arrest.44 Senescence is not even necessarily cell cycle arrest (see “Footnote 3”). Cellular senescence develops when signal-transduction pathways, which drive metabolism and mass/size growth (nutrient-sensing, oncogenic, growth-promoting pathway, insulin/mitogen-sensing, gerogenic pathways), are activated, but the cell cycle is nevertheless blocked. Then the cell undergoes MTOR-driven geroconversion or conversion from quiescence/arrest to senescence.45-59 Such gerogenic cells drive age-related diseases and organism aging.38, 60 Because rapamycin suppresses cellular geroconversion, the gerosuppressant rapamycin also suppresses age-related diseases and aging. (See ref. 61 for review and references therein as well as most recent publications.) Interestingly rapamycin can also affect not only geroconversion (physiological aging) but also so-called chronological aging in yeast62 and cancer cells,63 or in other words, acid-induced cell destruction.63-65 Even more intriguingly, rapamycin can prevent replicative aging in certain conditions.55,66-68 Most importantly, it can treat age-related diseases and69-74 extend lifespan in old75 and cancer-prone mice,76 including mice lacking p5377-80 and RB,81 and slow down the aging process.82,83
Why Damage Cannot Cause Aging, As We Know It
Unlike pleotropic MTOR, which is useful early in life, random damage is harmful at any stage of life. Blind watchmaker is especially prone to overdo any necessary task for short-termed benefits. BW repairs it because it is harmful right now, not because BW is concerned with aging later. Broken bone needs to be repaired to save infant life. Molecular damage needs to be repaired too. Or consider progeria,84-87 including Hutchinson-Gilford progeria (HGP) syndrome.88 Infants and children with this syndrome would not be viable in the wild. They would die at a very young age. By a remarkable co-incidence, progeria in animals can be treated by rapamycin.89-91
So BW takes damage repair seriously: damage must be repaired regardless of aging, and this is why it is not a cause of aging. In addition, mutations initiate a sequence of events leading to cancer. Not despite but because of high mutation rate, cancer cells are robust (not weak), and multiple rounds of selection and proliferation non-randomly activate the MTOR pathway in cancer cells (for details, see ref. 92). Finally, BW may blindly repair some rare beneficial mutations too.
But still, damage accumulates. In theory, it will cause a new type of aging (at age of 130, for instance) if MTOR-driven quasi-programmed aging would not terminate life first. By analogy, when in the past people died young from starvation, infections and accidents, then atherosclerosis could not kill them. Some age-related diseases were almost unknown until recently.
Quasi-Program is Not a Program
The evolutionary theory explains that aging cannot be programmed. Still the notion that aging is programmed persists and was even “re-invented.” In my mind, the reason why theories of programmed aging are still “alive” is that aging is so program-like. Human age-related diseases and menopause have distinct molecular, cellular and systemic mechanisms, which are not random at all. How can random damage drive these processes that are so development-like? Even non-programmed random molecular damage theories accept that menopause and death of Pacific salmon are programmed. But, like aging itself, they are not: they are quasi-programmed.
A quasi-program looks like as a program, but it is not. It is a continuation of some useful developmental program. It is a by-product. Unlike an actual program it has no purpose, nor plan. By giving a new application to the Dawkins words, I have the courage to write that, as the blind continuation of developmental growth, a quasi-program of aging “has no purpose in mind. It has no mind and no mind’s eye. It does not plan for the future. It has no vision, no foresight, no sight at all.”1
Amazingly, logical thinking could create non-programmed “damage/repair” theories on one hand, and “programmed aging” theories on the other hand. In contrast, blind watchmaker may unsuspectingly create only quasi-programmed aging. Of course, “DST-one” was not intended as a design theory, but in contrast, like “DST-two,” it was inspired by Darwinian theory of natural selection. But at the end, one may characterize DST-one as a flawed application of BW thinking that, inadvertently, has the character of intelligent invention when viewed through the lens of hyperfunction theory (DST-two).
Appendix
Footnote 1
Many advocates of programmed aging point out that some species of animals (especially, larger animals), live long enough in the wild to die from aging. Yes, some do (by the way, not Pacific Salmon, in which 98% individuals died before spawning (see ref. 14 for explanation); however, it does not mean that aging is either programmed or designed. It is still quasi-programmed. In natural “protected environments,” when extrinsic death rate is temporarily low, nature selects for slow aging and aging tolerance (see aging tolerance in refs. 16 and 20), until most individuals live longer to die from extrinsic causes again. Therefore, aging in the wild may predominate in some species once in a while, because a natural “protected environment” may emerge occasionally. This does not argue for “programmed nature” of aging.
Footnote 2
In theory, either the MTOR activity or synthetic processes may be decreased later in life, but either not sufficiently or too late. For example, RNA/protein synthesis is decreased with aging in model organisms, yet its further inhibition prolongs lifespan,8-13 indicating insufficient natural decrease
Footnote 3
The difference between cell cycle arrest and senescence is unfortunately misunderstood. In the young organism, most cells are arrested but not senescent. They become senescent via gerogenic conversion. In cell culture, cell cycle arrest is a predisposition to senescence. Cell cycle arrest itself does not cause senescence but creates conditions for geroconversion from arrest to senescence.44 MTOR generally causes growth and fosters proliferation, whereas rapamycin slows cell cycle progression and of course does not induce proliferation in the arrested cells.44 What rapamycin and other inhibitors of MTOR do is suppress geroconversion (gerosuppression). Inhibitors of MTOR suppress some markers of senescent phenotype and preserve proliferative/regenerative (PP) potential, which is convenient to measure senescence. The potential to proliferate is not proliferation. This is the potential of normal young cell.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25062
References
- 1.Dawkins R. ISBN Ê 0-393-31570-3. The Blind Watchmaker. New York: W. W. Norton & Company, Inc; 1986. [Google Scholar]
- 2.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mounier R, Lantier L, Leclerc J, Sotiropoulos A, Foretz M, Viollet B. Antagonistic control of muscle cell size by AMPK and mTORC1. Cell Cycle. 2011;10:2640–6. doi: 10.4161/cc.10.16.17102. [DOI] [PubMed] [Google Scholar]
- 5.Dreesen IA, Fussenegger M. Ectopic expression of human mTOR increases viability, robustness, cell size, proliferation, and antibody production of chinese hamster ovary cells. Biotechnol Bioeng. 2011;108:853–66. doi: 10.1002/bit.22990. [DOI] [PubMed] [Google Scholar]
- 6.Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY) 2012;4:861–77. doi: 10.18632/aging.100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blagosklonny MV. Big mice die young but large animals live longer. Aging (Albany NY) 2013;5:227–33. doi: 10.18632/aging.100551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 9.Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, et al. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007;6:111–9. doi: 10.1111/j.1474-9726.2006.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007;445:922–6. doi: 10.1038/nature05603. [DOI] [PubMed] [Google Scholar]
- 11.Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- 12.Tavernarakis N. Protein synthesis and aging: eIF4E and the soma vs. germline distinction. Cell Cycle. 2007;6:1168–71. doi: 10.4161/cc.6.10.4230. [DOI] [PubMed] [Google Scholar]
- 13.Mehta R, Chandler-Brown D, Ramos FJ, Shamieh LS, Kaeberlein M. Regulation of mRNA translation as a conserved mechanism of longevity control. Adv Exp Med Biol. 2010;694:14–29. doi: 10.1007/978-1-4419-7002-2_2. [DOI] [PubMed] [Google Scholar]
- 14.Blagosklonny MV. Paradoxes of aging. Cell Cycle. 2007;6:2997–3003. doi: 10.4161/cc.6.24.5124. [DOI] [PubMed] [Google Scholar]
- 15.Kirkwood T. Why women live longer. Stress alone does not explain the longevity gap. Sci Am. 2010;303:34–5. doi: 10.1038/scientificamerican1110-34. [DOI] [PubMed] [Google Scholar]
- 16.Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8:4055–9. doi: 10.4161/cc.8.24.10310. [DOI] [PubMed] [Google Scholar]
- 17.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–8. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 18.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–47. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 19.Blagosklonny MV. Why the disposable soma theory cannot explain why women live longer and why we age. Aging (Albany NY) 2010;2:884–7. doi: 10.18632/aging.100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY) 2011;3:1051–62. doi: 10.18632/aging.100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 22.Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–62. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- 23.Bartke A, Coschigano K, Kopchick J, Chandrashekar V, Mattison J, Kinney B, et al. Genes that prolong life: relationships of growth hormone and growth to aging and lifespan. J Gerontol A Biol Sci Med Sci. 2001;56:B340–9. doi: 10.1093/gerona/56.8.B340. [DOI] [PubMed] [Google Scholar]
- 24.Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008;7:13–22. doi: 10.1111/j.1474-9726.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 25.Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci USA. 2005;102:3105–10. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borrás C, Monleón D, López-Grueso R, Gambini J, Orlando L, Pallardó FV, et al. RasGrf1 deficiency delays aging in mice. Aging (Albany NY) 2011;3:262–76. doi: 10.18632/aging.100279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shmookler Reis RJ, Xu L, Lee H, Chae M, Thaden JJ, Bharill P, et al. Modulation of lipid biosynthesis contributes to stress resistance and longevity of C. elegans mutants. Aging (Albany NY) 2011;3:125–47. doi: 10.18632/aging.100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the lifespan of C. elegans inhibit tumor growth. Science. 2006;313:971–5. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- 29.Haselton A, Sharmin E, Schrader J, Sah M, Poon P, Fridell YW. Partial ablation of adult Drosophila insulin-producing neurons modulates glucose homeostasis and extends lifespan without insulin resistance. Cell Cycle. 2010;9:3063–71. doi: 10.4161/cc.9.15.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Streeper RS, Grueter CA, Salomonis N, Cases S, Levin MC, Koliwad SK, et al. Deficiency of the lipid synthesis enzyme, DGAT1, extends longevity in mice. Aging (Albany NY) 2012;4:13–27. doi: 10.18632/aging.100424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou KI, Pincus Z, Slack FJ. Longevity and stress in Caenorhabditis elegans. Aging (Albany NY) 2011;3:733–53. doi: 10.18632/aging.100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11:453–65. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katewa SD, Kapahi P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp Gerontol. 2011;46:382–90. doi: 10.1016/j.exger.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharp ZD. Aging and TOR: interwoven in the fabric of life. Cell Mol Life Sci. 2011;68:587–97. doi: 10.1007/s00018-010-0542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian lifespan. Science. 2009;326:140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selman C, Partridge L. A double whammy for aging? Rapamycin extends lifespan and inhibits cancer in inbred female mice. Cell Cycle. 2012;11:17–8. doi: 10.4161/cc.11.1.18736. [DOI] [PubMed] [Google Scholar]
- 37.Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010;9:1859–62. doi: 10.4161/cc.9.10.11872. [DOI] [PubMed] [Google Scholar]
- 38.Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY) 2012;4:861–77. doi: 10.18632/aging.100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gems DH, Guardia YD. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2012 doi: 10.1089/ars.2012.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaeberlein TL, Smith ED, Tsuchiya M, Welton KL, Thomas JH, Fields S, et al. Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging Cell. 2006;5:487–94. doi: 10.1111/j.1474-9726.2006.00238.x. [DOI] [PubMed] [Google Scholar]
- 41.Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY) 2009;1:357–62. doi: 10.18632/aging.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5:2087–102. doi: 10.4161/cc.5.18.3288. [DOI] [PubMed] [Google Scholar]
- 43.Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012;181:1142–6. doi: 10.1016/j.ajpath.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 44.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 46.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 47.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8:4112–8. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, et al. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci Signal. 2009;2:ra11. doi: 10.1126/scisignal.2000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–52. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2010;2:924–35. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci USA. 2012;109:13314–8. doi: 10.1073/pnas.1205690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11:4642–9. doi: 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012;33:597–604. doi: 10.1007/s10059-012-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012;181:278–93. doi: 10.1016/j.ajpath.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, et al. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY) 2012;4:952–65. doi: 10.18632/aging.100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denchi EL. A critical role for TORC1 in cellular senescence. Cell Cycle. 2012;11:2976. doi: 10.4161/cc.21529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iglesias-Bartolome R, Gutkind SJ. Exploiting the mTOR paradox for disease prevention. Oncotarget. 2012;3:1061–3. doi: 10.18632/oncotarget.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darzynkiewicz Z. Running m(o)TOR with the brakes on leads to catastrophe at mitosis. Cell Cycle. 2012;11:4494. doi: 10.4161/cc.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M, Miller RA. Fibroblasts from long-lived mutant mice exhibit increased autophagy and lower TOR activity after nutrient deprivation or oxidative stress. Aging Cell. 2012;11:668–74. doi: 10.1111/j.1474-9726.2012.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological lifespan in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY) 2011;3:1078–91. doi: 10.18632/aging.100402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burtner CR, Murakami CJ, Kennedy BK, Kaeberlein M. A molecular mechanism of chronological aging in yeast. Cell Cycle. 2009;8:1256–70. doi: 10.4161/cc.8.8.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaeberlein M, Kennedy BK. A new chronological survival assay in mammalian cell culture. Cell Cycle. 2012;11:201–2. doi: 10.4161/cc.11.2.18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11:2402–7. doi: 10.4161/cc.20882. [DOI] [PubMed] [Google Scholar]
- 67.Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012;11:2231–2. doi: 10.4161/cc.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polymenis M, Kennedy BK. Chronological and replicative lifespan in yeast: do they meet in the middle? Cell Cycle. 2012;11:3531–2. doi: 10.4161/cc.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolosova NG, Muraleva NA, Zhdankina AA, Stefanova NA, Fursova AZ, Blagosklonny MV. Prevention of age-related macular degeneration-like retinopathy by rapamycin in rats. Am J Pathol. 2012;181:472–7. doi: 10.1016/j.ajpath.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 70.Zheng XF. Chemoprevention of age-related macular regeneration (AMD) with rapamycin. Aging (Albany NY) 2012;4:375–6. doi: 10.18632/aging.100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron. 2012;75:425–36. doi: 10.1016/j.neuron.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Passtoors WM, Beekman M, Deelen J, van der Breggen R, Maier AB, Guigas B, et al. Gene expression analysis of mTOR pathway: association with human longevity. Aging Cell. 2013;12:24–31. doi: 10.1111/acel.12015. [DOI] [PubMed] [Google Scholar]
- 74.Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience. 2012;223:102–13. doi: 10.1016/j.neuroscience.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010;176:2092–7. doi: 10.2353/ajpath.2010.091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, et al. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY) 2012;4:709–14. doi: 10.18632/aging.100498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, et al. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY) 2012;4:715–22. doi: 10.18632/aging.100496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY) 2012;4:660–1. doi: 10.18632/aging.100494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levine AJ, Harris CR, Puzio-Kuter AM. The interfaces between signal transduction pathways: IGF-1/mTor, p53 and the Parkinson Disease pathway. Oncotarget. 2012;3:1301–7. doi: 10.18632/oncotarget.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, et al. Rapamycin extends lifespan of Rb1+/- mice by inhibiting neuroendocrine tumors. Aging (Albany NY) 2013;5:100–10. doi: 10.18632/aging.100533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–82. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spong A, Bartke A. Rapamycin slows aging in mice. Cell Cycle. 2012;11:845. doi: 10.4161/cc.11.5.19607. [DOI] [PubMed] [Google Scholar]
- 84.Dreesen O, Stewart CL. Accelerated aging syndromes, are they relevant to normal human aging? Aging (Albany NY) 2011;3:889–95. doi: 10.18632/aging.100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lachapelle S, Oesterreich S, Lebel M. The Werner syndrome helicase protein is required for cell proliferation, immortalization, and tumorigenesis in Scaffold attachment factor B1 deficient mice. Aging (Albany NY) 2011;3:277–90. doi: 10.18632/aging.100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging (Albany NY) 2011;3:311–8. doi: 10.18632/aging.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Damerla RR, Knickelbein KE, Strutt S, Liu FJ, Wang H, Opresko PL. Werner syndrome protein suppresses the formation of large deletions during the replication of human telomeric sequences. Cell Cycle. 2012;11:3036–44. doi: 10.4161/cc.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824–33. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- 89.Blagosklonny MV. Progeria, rapamycin and normal aging: recent breakthrough. Aging (Albany NY) 2011;3:685–91. doi: 10.18632/aging.100352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4:ra103. doi: 10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, et al. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3:89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- 92.Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY) 2011;3:1130–41. doi: 10.18632/aging.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]