

Abstract
The Nod-like receptor, Nlrp3, has been linked to inflammatory diseases and adjuvant-mediated immune responses. A wide array of structurally diverse agents does not interact directly with Nlrp3, but is thought to activate the Nlrp3 inflammasome by inducing a common upstream signal, such as lysosome rupture. To test the connection between lysosome integrity and Nlrp3 signaling, we analyzed inflammasome activation following stimulation of murine macrophages with lysosome-destabilizing agents and pyroptosis inducers. Here we provide evidence that lysosomal rupture and the corresponding release of lysosomal hydrolases is an early event in macrophages exposed to the lysosome-destabilizing adjuvants LLOMe and alum. Lysosome rupture preceded cell death induction mediated by these agents and was associated with the degradation of low-molecular weight proteins, including the inflammasome component caspase-1. Proteolysis of caspase-1 was controlled by specific cathepsins, but was independent of autocatalytic processes and Nlrp3 signaling. Consistent with these findings, lysosome-disrupting agents triggered only minimal caspase-1 activation and failed to cause caspase-1-dependent cell death (pyroptosis), generally associated with Nlrp3 signaling. In contrast, lysosome rupture was a late event in macrophages exposed to prototypical pyroptosis inducers. These agents triggered extensive Nlrp3 signaling prior to lysosome rupture with only minimal impact on the cellular proteome. Taken together, our findings suggest that lysosome impairment triggers a cascade of events culminating in cell death but is not crucial for Nlrp3 signaling. The significant differences observed between lysosome-disrupting agents and pyroptosis inducers might explain the distinct immunologic responses associated with these compounds.
Keywords: Nlrp3 inflammasome, caspase-1, lysosome rupture, necrosis, pyroptosis
Introduction
The Nod-like receptors (NLRs) are intracellular surveillance receptors that have been associated with a wide variety of proinflammatory processes, including septic shock, chronic inflammatory disease and adjuvant activities.1 The best-studied NLR is Nlrp3, which triggers the formation of a high-molecular weight inflammasome complex containing the downstream proteins Asc and caspase-1 upon activation.2-4 Upon activation, the Nlrp3 inflammasome mediates two caspase-1-dependent inflammatory processes: (1) necrotic cell death (pyroptosis) and (2) processing of IL-1β and IL-18.
A wide range of chemically and structurally unrelated agents activates the Nlrp3 inflammasome (reviewed in ref. 5). Nlrp3 inducers include not only compounds derived from bacterial, fungal and viral pathogens, but also a range of non-infectious agents, such as inorganic crystalline particles.5 It is unclear how these structurally unrelated compounds are able to activate the Nlrp3 inflammasome. It has been postulated that Nlrp3 ligands do not activate the Nlrp3 inflammasome directly, but rather trigger Nlrp3 activation through induction of an upstream stimulus.6 Potassium efflux and disruption of lysosomes or mitochondria have been suggested as upstream signals for Nlrp3 signaling.6,7 However, the exact mechanism and contribution of these potential stress signals to Nlrp3 signaling remains to be elucidated.
Recent studies have shown that insoluble particulates, such as the adjuvant alum, silica, monosodium urate and calcium pyrophosphate dehydrate, activate the Nlrp3 inflammasome.8-10 Endocytosis of these crystalline particles triggers the disintegration of endolysosomal organelles and the release of lysosomal contents into the cytosol.8-10 Based on these studies, it has been postulated that lysosome rupture could trigger Nlrp3 signaling.8,11,12 This theory was further supported by the fact that inhibitors of lysosomal cathepsins block caspase-1 activation by all Nlrp3 inducers tested, including non-particulate agents.8,13 In addition, cathepsin inhibitors also block caspase-1 activation mediated by Nalp1b and Ipaf inducers, suggesting that lysosome rupture might be a common upstream event necessary for inflammasome activation. Identifying the relationship between lysosome integrity and inflammasome signaling is the focus of this study.
To analyze the role of lysosomal impairment in Nlrp3 signaling, we used the lysosome-destabilizing agents Leu-Leu-OMe (LLOMe) and alum, as model systems. Here we demonstrate that these agents trigger very different cellular and inflammatory responses than the prototypical Nlrp3 inducers, ATP and nigericin. We found that alum- and LLOMe-mediated lysosome rupture was a primary event that triggered caspase-1-independent cell death and cathepsin-dependent proteolysis of cytosolic proteins. In contrast, lysosome rupture occurred only after Nlrp3 signaling and induction of caspase-1-dependent necrosis (pyroptosis) in cells treated with the prototypical Nlrp3 inducers, nigericin and ATP. Taken together, our findings indicate that lysosome rupture is not a common signal for Nlrp3 activation. The differences observed between lysosome-disrupting agents and prototypical pyroptosis inducers may also explain the distinct immune responses associated with these agents.
Results
Lysosome-disrupting adjuvants trigger caspase-1 activation, but not caspase-1-mediated cell death
The ability of lysosome-disrupting agents to activate the Nlrp3 inflammasome suggested that lysosome rupture might act as a secondary signal in Nlrp3 activation.8,11 This was further supported by the fact that inhibitors of lysosomal cathepsins block Nlrp3 signaling and caspase-1 activation by all Nlrp3 inducers tested.8,13,14 We have previously shown that lysosome-disrupting agents trigger a caspase-1-independent cell death.15 This is surprising, as activated caspase-1 is generally highly cytotoxic and triggers pyroptotic cell death.16,17 This caspase-1-mediated cell death is a hallmark of inflammasome signaling mediated by the NLRs, Nlrp3 and Nlrp1b.16-18 It is unclear why lysosome-disrupting agents activate Nlrp3 but fail to trigger pyroptotic cell death. This conundrum suggested that lysosome-disrupting agents and prototypical pyroptosis inducers trigger distinct cellular responses. We therefore performed a comparative study of caspase-1-induced cellular events mediated by lysosome-disrupting agents and by pyroptosis inducers.
As our model for lysosome-disrupting agents, we used the insoluble adjuvant, alum, and the soluble agent, Leu-Leu-OMe (LLOMe).15,19,20 LLOMe is converted in lysosomes into a lytic polymer, causing synchronized and complete rupture of these organelles.19,20 As positive controls for inflammasome-activating agents, we used the pyroptosis inducers, ATP, nigericin and anthrax lethal toxin (LT). To analyze the ability of lysosome-disrupting agents and pyroptosis inducers to activate caspase-1, we challenged LPS-primed bone marrow-derived murine macrophages with increasing concentrations of these agents.
Consistent with previous studies, we found that lysosome-disrupting agents and pyroptosis inducers triggered both necrotic cell death and IL-1β release. While lysosome-disrupting agents and pyroptosis inducers triggered necrotic cell death with high efficiency, they responded differently to the caspase-1 inhibitor, Boc-D-CMK (Fig. 1A). As expected, Boc-D-CMK efficiently blocked cell death mediated by the pyroptosis inducers ATP and nigericin, but not cell death mediated by lysosome-disrupting agents (Fig. 1A). Boc-D-CMK also blocked cell death mediated by the pyroptosis inducer, anthrax lethal toxin (LT), which activates the Nalp1b inflammasome.18,21 In contrast, Boc-D-CMK did not block cell death mediated by the lysosome-disrupting agents, alum and LLOMe (Fig. 1A). These results suggested that, in contrast to pyroptosis inducers, lysosome-disrupting agents trigger caspase-1-independent cell death. To test the involvement of the Nlrp3 inflammasome in this process, we challenged macrophages lacking Nlrp3 components with lysosome-disrupting agents and pyroptosis inducers. We found that Nlrp3 and Asc deficiency did not block cell death mediated by the lysosome-disrupting agents, alum and LLOMe, which indicated caspase-1-independent cell death (Fig. 1C). In contrast, Nlrp3- and Asc-deficiency prevented cell death by the pyroptosis inducers ATP and nigericin (Fig. 1C). Taken together, these findings indicated that lysosome-destabilizing agents trigger Nlrp3 and caspase-1-independent cell death.

Figure 1. Alum and LLOMe trigger Nlrp3-independent cell death and are poor inducers of IL-1β release. (A and B) BALB/c macrophages were primed with 250 ng/ml LPS in the presence or absence of 100 μM CA-074-Me for 2 h. Macrophages were then exposed to increasing concentrations of LLOMe, ATP or nigericin for 4 h or alum for 6 h, in the presence or absence of 40 μM Boc-D-CMK. IL-1β release was assessed by ELISA (A), and cell death by CytotoxOne LDH activity assays (B) from supernatants. (C) Wild type and Nalp3-, Ipaf- and Asc-deficient C57BL/6 macrophages were primed for 2 h with 250 ng/ml of LPS in the presence or absence of 100 μM CA-074-Me. Cells were then treated with 150 μg/ml alum, 2.5 mM LLOMe or with varying doses of ATP and nigericin for 3 h. Plasma membrane impairment was determined by propidium iodide exclusion assays. All measurements were taken in triplicate from representative experiments.
As lysosome-disrupting agents failed to trigger caspase-1-mediated cell death, we analyzed their ability to trigger IL-1β release, the second activity associated with activated caspase-1. We found that the IL-1β release mediated by lysosome-disrupting agents was strikingly dissimilar to that triggered by pyroptosis inducers (Fig. 1B). We found that alum and LLOMe triggered distinctly less IL-1β release in murine macrophages than did the prototypical pyroptosis inducers, ATP and nigericin (Fig. 1B). While IL-1β release (and cell death) increased with higher ATP or nigericin concentrations (Fig. 1B), IL-1β decreased after an initial peak, with increasing LLOMe concentrations (Fig. 1B). These results are consistent with LLOMe- and alum-induced Nlrp3 signaling, though with lower efficiency than prototypical pyroptosis inducers.
While the caspase-1 inhibitor, Boc-D-CMK, failed to block cell death mediated by lysosome-disrupting agents, Boc-D-CMK blocked IL-1β release mediated by both lysosome-disrupting agents and pyroptosis inducers (Fig. 1B). It is conceivable that low levels of caspase-1 activation mediated by lysosome-disrupting agents are not sufficient for pyroptosis induction. These results indicate that lysosome-disrupting agents trigger caspase-1-dependent IL-1β release, but not caspase-1-dependent cell death. In contrast to caspase-1 inhibitors, the cathepsin inhibitor CA-074-Me blocked both cell death and IL-1β release mediated by lysosome-disrupting agents (Fig. 1A–C). Taken together, our results highlight the distinct Nlrp3 responses mediated by lysosome-disrupting agents and pyroptosis inducers.
Cathepsin-dependent lysosome rupture precedes plasma membrane impairment in alum and LLOMe-treated macrophages
The ability of insoluble particles to trigger IL-1β release suggested that lysosomal impairment is a critical step in inflammasome signaling and caspase-1 activation. To determine whether lysosome rupture is a common upstream signal for Nlrp3 inflammasome signaling, we determined the kinetics of lysosome rupture and cell death induction following challenge with lysosome-disrupting agents and pyroptosis inducers.
To this end, we labeled endolysosomal vesicles with fluorescent dextran and determined the kinetics of dextran release from lysosomes in LLOMe-, alum- and nigericin-treated macrophages by confocal microscopy. Consistent with rapid lysosome rupture, fluorescent dextran progressed from a punctate lysosomal to a diffuse cytosolic staining 30 min after LLOMe exposure (Fig. 2A). Notably, plasma membrane impairment, a hallmark of necrotic cell death, occurred 30 to 60 min after lysosome rupture in LLOMe-treated macrophages (Fig. 2A). Lysosome rupture also preceded necrotic cell death in alum-treated macrophages, though with slower kinetics (Fig. 2A). These results indicated that lysosome rupture precedes necrotic cell death in macrophages with the lysosome-disrupting agents, alum and LLOMe. In contrast, the pyroptosis inducer nigericin did not trigger any discernable lysosome rupture prior to a loss of plasma membrane integrity. In fact, necrotic macrophages showed no discernable signs of lysosome rupture 2 h after nigericin exposure (Fig. 2A). Importantly, the cathepsin inhibitor CA-074-Me blocked lysosome rupture mediated by alum and LLOMe (Fig. 2A). As CA-074-Me also blocks alum and LLOMe-induced cell death, our findings suggested that lysosome rupture drives alum and LLOMe-induced cell death.

Figure 2. Lysosome rupture precedes cell death mediated by alum and LLOMe, but not by pyroptosis inducers. (A) Confocal microscopy of lysosome rupture in macrophages exposed to 250 ng/ml LPS in the presence or absence of 100 μM CA-074-Me for 2 h. Cells were then treated with 150 μg/ml alum, 2.5 mM LLOMe or 15 μM nigericin, and stained with Hoechst (blue), 20 MW FITC-Dextran (green), CellMask Orange (red), TO-PRO-3 (white). 2D-fluorescent intensity plots of FITC-dextran-labeled representative cells following alum, LLOMe and nigericin exposure. (B) Flow cytometry analysis of LLOMe and LT-treated cells. Murine macrophages were exposed to 2.5 mM LLOMe or 500 ng/ml LT, and lysosome and membrane integrity were measured at different time points using flow cytometry using LysoTracker and PI, respectively.
To quantify lysosomal integrity and cell death induction, we used the LysoTracker, a marker for lysosomes with low pH, and the vital stain propidium iodide. Consistent with our studies using fluorescent dextran, we observed a loss of lysosome integrity 30 min, and necrotic cell death (PI-positive cells) 90 min after LLOMe exposure (Fig. 2B). Notably, we found a complete shift in the LysoTracker signal 30 min post LLOMe exposure, consistent with rapid and synchronized lysosome rupture. As nigericin non-specifically quenches the LysoTracker signal even in the absence of lysosomal impairment,22,23 we used the NLR (Nlrp1b) inducer anthrax lethal toxin (LT) as a control for a pyroptosis inducer. In contrast to LLOMe, LT destabilized lysosomes only after cells showed signs of necrotic cell death. As observed with nigericin-treated macrophages, lysosome rupture was a late event in LT-treated cells, and occurred only after induction of cell death (Figs. 2A and B).
Our findings suggested that lysosome rupture drives necrotic cell death in cells treated with LLOMe and alum, but not in cells exposed to pyroptosis inducers. The kinetics further indicated that lysosomal rupture occurred in cells with intact plasma membranes, suggesting that released proteolytic enzymes are retained within the cytoplasm in this process. As pyroptosis inducers triggered lysosome rupture only after cells already showed signs of necrotic cell death, it appears that lysosome rupture is an epiphenomenon in pyroptosis induction and inflammasome activation.
Lysosome-disrupting adjuvants trigger Nlrp3-independent degradation of proinflammatory proteins
To further assess the ability of lysosome-disrupting agents to activate the Nlrp3 inflammasome, we analyzed processing of Nlrp3-associated proteins by immunoblotting. We challenged murine macrophages with the lysosome-disrupting agents, alum and LLOMe and with the pyroptosis inducers, ATP and nigericin. The pyroptosis inducer ATP triggered cleavage and release of IL-1β, consistent with Nlrp3 activation (Fig. 3A). The activated inflammasome generally recruits and activates only a fraction of cellular caspase-1. Accordingly, we found only limited processing of pro-caspase-1 in cells exposed to the pyroptosis inducers ATP and nigericin (Figs. 3A and B). Intriguingly, the lysosome-disrupting agents, alum and LLOMe, triggered a more pronounced drop in levels of pro-caspase-1, and of the cytokines IL-1β and IL-18 than did the pyroptosis inducers (Fig. 3A). The drop of pro-IL-1β levels, however, was associated with only minimal IL-1β release in alum-treated cells, and no release at all in LLOMe-treated cells (Fig. 3A).

Figure 3. LLOMe and alum trigger the depletion of caspase-1 and NLR-associated cytokines. (A) BALB/C macrophages were primed with 250 ng/ml LPS in the presence or absence of 100 μM CA-074-Me for 2 h. Cells were subsequently challenged with 2.5 mM LLOMe, 5 mM ATP or 150 μg/ml alum for various times. Lysates were probed for pro-caspase-1, IL-18 and IL-1β, and supernatants for mature IL-1β by immunoblotting. (B) C57BL/6-derived macrophages were exposed to 250 ng/ml LPS in the absence and presence of 100 μM CA-074-Me for 2 h. Cells were subsequently challenged with 2.5 mM LLOMe or 20 mM nigericin. Cell death (top panel) was measured by PI exchange assays, and caspase-1 and actin levels (bottom panel) were determined from cellular lysates by immunoblotting at different time points post LLOMe/nigericin exposure.
We initially hypothesized that the depletion of intracellular caspase-1 was due to autocatalytic processing of inflammasome components. Upon inflammasome activation, the 45 kDa pro-form of capase-1 is generally auto-catalytically processed into 20 and 10 kDa subunits.11,24 Intriguingly, the LLOMe-induced drop in pro-caspase-1 was not accompanied by generation of the p20 subunit, as seen in nigericin-treated macrophages (Fig. 3B). To further investigate involvement of autocatalytic processes in the reduction of caspase-1 levels, we challenged wild type, Nlrp3- and Asc-deficient macrophages with LLOMe and the pyroptosis inducer, nigericin. We found that Nlrp3 and Asc deficiency did not block the LLOMe-induced drop in caspase-1 levels and necrotic cell death (Fig. 4). In contrast, Nlrp3 and Asc deficiency prevented nigericin-induced drop in caspase-1 levels and pyroptosis induction, consistent with Nlrp3 signaling (Fig. 4). Taken together, these results further underscore the fundamentally different cellular responses mediated by lysosome-disrupting agents and pyroptosis inducers.
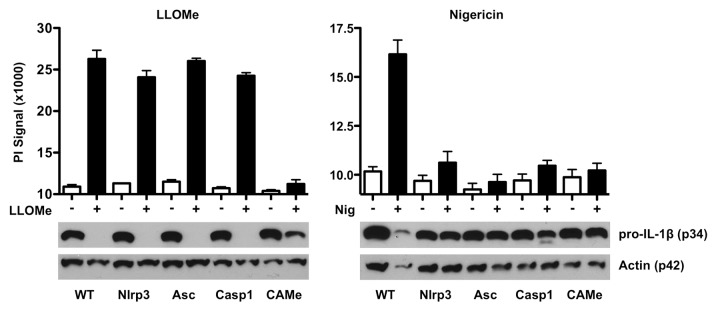
Figure 4. LLOMe-mediated caspase-1 depletion is independent of Nlrp3 signaling. Wild-type and Nalp3/Asc/caspase-1-deficient C57BL/6 macrophages were primed with 250 ng/ml LPS in the presence or absence of 100 μM CA-074-Me (CAMe) for 2 h. Cells were then challenged with 2.5 mM LLOMe or 10 mM nigericin for 2 h, and cell death was measured using PI exchange assays (top row). Cellular lysates were examined for levels of pro-caspase-1 and actin by immunoblotting (bottom row).
Alum and LLOMe-mediated inflammasome degradation is dependent on the activity of specific cathepsins
The drop in caspase-1 levels observed in LLOMe-treated cells, which was independent of Nlrp3 signaling, was correlated with induction of necrotic cell death (Figs. 3B and 4). Caspase-1 levels decreased with increasing levels of necrotic cell death in LLOMe-treated macrophages (Fig. 3B). Moreover, the cathepsin B inhibitor, CA-074-Me, blocked both necrotic cell death and the drop in caspase-1 levels observed in LLOMe-treated cells (Fig. 3B). These results indicated a close correlation between necrotic cell death and proteolysis of proinflammatory proteins in LLOMe-treated cells. These studies further indicated that both processes were cathepsin-dependent. Previous work by our lab and others has identified specific cathepsins controlling cell death mediated by alum and LLOMe.15 As cathepsin C is critical for LLOMe-mediated lysosome rupture and necrotic cell death,15,19,20 we asked whether cathepsin C is also critical for the LLOMe-induced degradation of inflammasome components. We therefore challenged a panel of cathepsin-deficient macrophages with LLOMe. As predicted, cathepsin C deficiency prevented both IL-1β degradation and necrotic cell death in LLOMe-treated macrophages (Fig. 5). This process was highly specific, as deficiency of cathepsins B, L and S had no impact on LLOMe-induced protein degradation and cell death (Fig. 5). Together, these data suggested that specific cathepsins control cell death and the degradation of inflammatory proteins mediated by lysosome-disrupting agents.
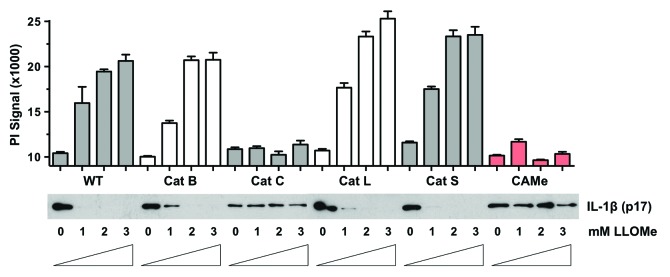
Figure 5. Cathepsin C controls LLOMe-mediated degradation of IL-1β. Wild type, cathepsin B-, C-, L- and S-deficient macrophages were primed with 250 ng/ml LPS for 2 h and then treated with increasing concentrations of LLOMe for 2 h. LLOMe-treated wild type macrophages were also tested in the presence 100 μM CA-074-Me. Cell death was measured by propidium iodide exclusion assay, and IL-1β levels were determined by immunoblotting from lysates. Representative experiment is shown, and PI measurements were performed in triplicates.
Lysosome-disrupting agents trigger broad protein degradation
We demonstrated that the cathepsin inhibitor CA-074-Me and cathepsin C deficiency prevents LLOMe-induced lysosome rupture, cell death and protein degradation. We also showed that lysosome rupture is an early event preceding cell death in LLOMe and alum-treated macrophages. It is therefore reasonable to assume that lysosome rupture triggers both necrotic cell death and protein degradation mediated by LLOMe. Lysosomes are the digestive organelles of the cell, containing many promiscuous and constituently active hydrolases, whose destructive potential is limited by being contained within lysosomes.25,26 Indeed the endopeptidase activity of many cathepsins is at least partially retained at cytosolic pH.27 Therefore it stands to reason that lysosomal release of these promiscuous and constitutively active hydrolases into the cytosol might trigger cell death and protein degradation in alum- and LLOMe-treated macrophages. As lysosomal proteases are highly promiscuous, we reasoned that alum and LLOMe may trigger not only the degradation of pro-inflammatory proteins, but also other cytosolic proteins.
To examine effects of lysosome disruption on the cellular proteome, we challenged LPS-treated macrophages with LLOMe or nigericin and analyzed lysates from these cells by differential 2D electrophoresis (DIGE). As predicted, LLOMe triggered a broad degradation of proteins within these cells, visible on a macro-level by DIGE (Fig. 6A). We found that proteins of lower molecular weight and of more acidic pI were preferentially degraded (Fig. 6A). Intriguingly, this subset of the cellular proteome contains inflammasome-associated proteins (Fig. 6B). By contrast, nigericin triggered only minimal changes in the proteome, though the cells were similarly necrotic, with minimal degradation of low molecular weight and of acidic pI proteins (Fig. 6B).
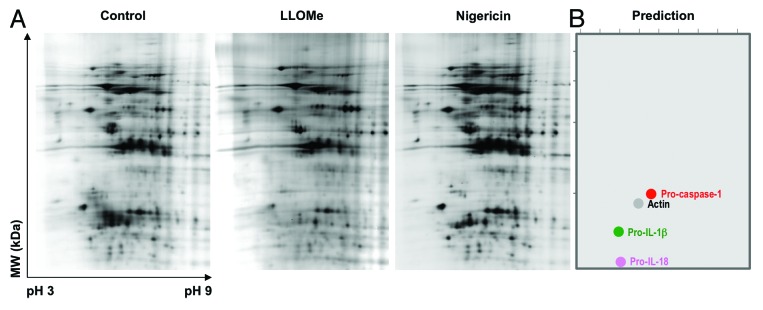
Figure 6. LLOMe triggers broad degradation of cellular proteins in macrophages. (A) 2D electrophoresis gel of proteins isolated from untreated, LLOMe- and nigericin-treated macrophages. C57BL/6-derived macrophages were primed with 250 ng/ml for 2 h, and then challenged with 2.5 mM LLOMe or 10 mM nigericin. Protein lysates were isolated from untreated, nigericin and LLOMe-treated macrophages 90 min post nigericin/LLOMe exposure. Protein samples were labeled with Cy2, Cy3 and Cy5, respectively, and separated on a 2D-DIGE gel. The red box on each gel highlights the low PI and low molecular weight region where the greatest variability is observed. (B) Bioinformatic prediction of the location of murine actin, pro-caspase-1, pro-IL-18 and pro-IL-1β on a 2D PAGE gel.
In summary, here we demonstrated the fundamental differences between lysosome-destabilizing agents and pyroptosis inducers in terms of proteolytic and necrotic events induced by these agents. Our findings indicated that alum- and LLOMe-induced lysosome rupture is a catastrophic event, resulting in the degradation of proinflammatory proteins and necrotic cell death. We demonstrated that these proinflammatory events are dependent on specific cathepsins and independent of Nlrp3 signaling.
Discussion
Lysosome-disrupting agents and pyroptosis inducers trigger distinct cellular responses
The structural diversity of Nlrp3 inducers suggests that these agents do not interact with Nlrp3 directly, but that they activate Nlrp3 by inducing a common upstream signal.6,28,29 As lysosome-disrupting agents trigger Nlrp3, lysosome rupture has been suggested to be such an upstream signal in Nlrp3 activation.6 As unequivocal evidence for this theory was lacking, we used the lysosome-disrupting agents alum and LLOMe as a model system to analyze the role of lysosome rupture in Nlrp3 signaling. Here we present evidence that lysosome rupture triggers a range of cellular responses, including cell death and the degradation of proinflammatory proteins in alum- and LLOMe-treated macrophages. A side-by-side comparison revealed that lysosome-disrupting agents and pyroptosis inducers trigger very different cellular responses (Table 1). These differences might explain the distinct immunologic responses associated with lysosome-disrupting agents and pyroptosis inducers.
Table 1. Activation of distinct cellular pathways by lysosome-disrupting agents and pyroptosis inducers.
| Lysosome-destabilizing agents | Pyroptosis inducers | |
|---|---|---|
|
Lysosome rupture precedes cell death |
Yes |
No |
|
Lysosome-mediated cell death |
Yes |
No |
|
Degradation of inflammatory proteins |
Yes |
No |
|
Caspase-1-mediated cell death (pyroptosis) |
No |
Yes |
|
Nlrp3 signaling |
Weak |
Strong |
| IL-1β release | Weak | Strong |
Lysosome rupture triggers cathepsin-dependent cell death
Our findings and a recent study by our group indicate that alum and LLOMe trigger a novel form of necrotic cell death that is distinct from the two established forms of programmed necrotic cell death: pyroptosis and necroptosis.13,15,30 We demonstrated that lysosome-disrupting agents, and in particular the dimethylester LLOMe, trigger rapid, complete and synchronized lysosome rupture. Lysosomes contain a number of enzymes, including lipases, proteases, amylases and nucleases, that digest foreign bodies and recycle cellular components.26,31 Many of these enzymes are highly promiscuous and are primarily regulated by being compartmentalized within lysosomes.32
While hydrolases generally perform optimally at an acidic pH, many lysosomal hydrolases, including specific cathepsins, retain their activity even after release into the neutral pH of the cytosol.33-35 It is very conceivable that these constitutively active hydrolases contribute directly to cell death and the broad degradation of cytosolic proteins (see below) we observed in cells treated with lysosome-disrupting agents. Based on our studies, lysosome rupture appears to be an early catastrophic event driving a series of downstream events, including necrotic cell death.15 We call this novel form of necrotic cell death lysosome-mediated necrosis (LMN).
Lysosome rupture and caspase-1-mediated cell death
As lysosome rupture has been implicated as a secondary signal in Nlrp3 signaling, we correlated lysosome rupture and inflammasome signaling mediated by a range of Nlrp3 inducers. Traditional inflammasome inducers regulate both cytokine signaling and cell death through caspase-1 in a process known as pyroptosis. While the lysosome-disrupting agents, alum and LLOMe, trigger IL-1β release in a caspase-1-dependent fashion, they induce cell death in a caspase-1-independent manner. Similar results were obtained with the lysosome-disrupting agent, silica, as well.15 This discrepancy between caspase-1-dependent IL-1β release and caspase-1-independent cell death appears to be specific to lysosome-disrupting agents. These findings are surprising, as caspase-1 activation is a highly cytotoxic event when potassium ionophores (Nlrp3 inducers) or anthrax lethal toxin (Nlrp1b inducer) are used.16,17,21
Role of lysosome rupture in Nlrp3 signaling
Consistent with previous studies,8-10 we found that lysosome-disrupting agents activate the Nlrp3 inflammasome, albeit much less efficiently than the prototypical Nlrp3 inducers, ATP and nigericin. While lysosome rupture was an early event in alum and LLOMe-treated cells, it was a late event in cells challenged with the pyroptosis inducers, nigericin and LT. In these cells, lysosome rupture occurred after caspase-1 activation and induction of necrotic cell death. It is therefore reasonable to assume that lysosome rupture is not an upstream signal for inflammasome activation mediated by pyroptosis inducers. Our findings indicate that the lysosome-destabilizing agent, LLOMe, triggers synchronized and complete lysosome rupture with very pronounced downstream effects. This might explain why LLOMe consistently yielded lower levels of IL-1β release and caspase-1 activation than alum, which triggers more measured lysosome rupture. Taken together, our results suggest lysosome rupture is a weak inducer of caspase-1 and appears to be sufficient for promoting IL-1β release but not pyroptotic cell death. Minimal Nlrp3 and caspase-1 activation might explain why lysosome-disrupting agents trigger caspase-1-dependent IL-1β release and fail to trigger caspase-1-dependent cell death.
Lysosomal rupture triggers cathepsin-dependent protein degradation
Here we provide evidence that alum and LLOMe trigger a sharp drop in the proforms of IL-1β and caspase-1. We found that this drop was independent of autocatalytic processes and Nlrp3 signaling. The degradation of pro-caspase-1, IL-18 and IL-1β, however, coincided with lysosome rupture, and was blocked by cathepsin inhibitors. Using genetic approaches, we have demonstrated that a specific cathepsin (cathepsin C) not only controls cytopathic effects, but also the adaptive immune response mediated by LLOMe.16 Here we report that cathepsin C is also critical for LLOMe-induced degradation of low-molecular proteins. We found more pronounced degradation of proinflammatory proteins in cells exposed to LLOMe compared with those treated with alum. This might be due to the fact that LLOMe triggers more rapid, synchronized and complete lysosome rupture than alum in murine macrophages.
In contrast, lysosomal rupture occurred late, and only after plasma membrane impairment in cells exposed to pyroptosis inducers, consistent with a lack of discernible protein degradation associated with these inducers. This might explain the higher Nlrp3 signaling efficiency of pyroptosis inducers compared with lysosome-disrupting agents. However, our findings do not rule out the possibility that a tempered release of lysosomal cathepsins might activate the Nlrp3 inflammasome. It is conceivable that low-level lysosome permeabilization might still activate the inflammasome before lysosome disruption causes cell death and degrades the components of the complex.
Protein degradation might be further enhanced by the fact that lysosome rupture occurred significantly before a loss of plasma membrane integrity in alum- and LLOMe-treated macrophages. Because of this, the released lysosomal enzymes were not immediately released from these cells. This transient containment might result in higher cytosolic concentrations of hydrolases and cathepsins, and presumably enhances their destructive potential in LLOMe and alum-treated cells. Taken together, our findings suggest that lysosome rupture is critical for the degradation of proinflammatory proteins mediated by lysosome-destabilizing agents.
Role of lysosome rupture in adjuvant activities
It is well established that the lysosome-disrupting agent alum is an effective adjuvant. We have recently demonstrated that the lysosome-disrupting agent, LLOMe, is an even more powerful enhancer of the adaptive immune response.15 We found that cell death is critical for the Th2-biased immune response mediated by LLOMe.15 These findings are consistent with studies implicating the necrotic release of cellular components, such as uric acid and DNA, in alum-induced adjuvant responses.36-40
Initial studies have also implicated Nlrp3 signaling in alum-mediated adjuvant effects.11 However, recent studies suggested that caspase-1 and Nlrp3 are dispensable for alum-enhanced immune responses.36,37,40-46 These studies are consistent with our findings that Nlrp3 activation by alum and LLOMe is too weak to drive an Nlrp3-dependent necrotic cell death and immune responses. Taken together, these studies suggest that lysosome rupture is a primary event that antagonizes Nlrp3 signaling and promotes necrotic cell death, which, in return, appears to result in a Th2-biased immune response. As many insoluble particles, including alum, silica, asbestos, cholesterol plaques and ultra-high molecular weight polyethylene also trigger lysosomal rupture and necrotic cell death,8,9,15,47-49 it is conceivable that lysosome-mediated cell death and proteolysis contribute to the inflammatory responses and diseases associated with these lysosome-destabilizing agents.
Here we present evidence that lysosome rupture mediated by alum and LLOMe triggers a cascade of events starting with (1) the release of proteolytic enzymes into the cytosol; (2) degradation of proinflammatory proteins; (3) a loss of plasma membrane integrity and (4) finally a systemic immune response.50 In the case of LLOMe, we demonstrated that cathepsin C is critical for lysosome rupture and all the mentioned downstream events. We also demonstrated that lysosome-disrupting agents and pyroptosis inducers trigger significantly different cellular events. In contrast to lysosome-destabilizing agents, pyroptosis inducers triggered caspase-1-mediated cell death, lysosome rupture only after cell death induction and no caspase-1-independent processing of inflammatory proteins. Taken together, our findings illustrate significant differences between the inflammatory processes mediated by lysosome-disrupting agents and prototypical inflammasome-inducing agents. It remains to be shown how these fundamental different cellular responses affect systemic immune responses.
Materials and Methods
Chemicals and reagents
Imject Alum was purchased from Thermo Scientific. Cell culture reagents were purchased from Fisher Scientific. Boc-D-CMK and Leu-Leu-OMe were purchased from Bachem. Propidium iodide, ATP and LPS (0111:B4) were purchased from Sigma-Aldrich. Nigericin was purchased from EMD Chemical. LT was purchased from Wadsworth Laboratories, and Cytotox One was from Promega. Precast gels and Coomassie solutions were from Biorad and low-endotoxin fetal calf serum was from Atlanta Biologicals.
Generation of primary cell lines and cell culture
Wild-type BALB/c and C57BL/6 mice were purchased from Jackson Labs. Inflammasome-deficient mice were provided by Dr Fayyaz Sutterwala,51 and cathepsin-deficient mice were provided by Drs Johanna Joyce and Thomas Reinheckel.52 All mice were euthanized humanly using CO2 in compliance with standard protocols. Primary macrophages were generated from bone marrow from femurs and tibias as described previously.21,52 In short, bone marrow was grown for one week in complete DMEM containing 20% L929 preconditioned media. Marrow was grown for 6 d, at which point adherent cells were stripped and replated in DMEM containing 10% FCS and 10% L929 preconditioned media.
Cell death assays and ELISAs
Necrotic cell death was determined propidium iodide (PI) exclusion and LDH release assays. PI exclusion assays were performed in phenol-red-free DMEM and analyzed with a Victor 2 plate reader from Perkin Elmer. LDH release was determined using the CytotoxOne kit from Promega, according to the manufacturer’s instructions. IL-1β Ready-Set-Go ELISA kits were purchased from eBioscience and were performed according to manufacturer’s recommendations. ELISA measurements were performed in triplicate and analyzed with a Victor 2 plate reader from Perkin Elmer.
Western blotting
Western blotting was performed as described previously.21 Membranes were probed with the following antibodies: anti-caspase-1 (Santa Cruz Biotechnologies), anti-actin (Sigma-Aldrich), anti-IL 18 (BioVision), anti-IL-1β (R and D Systems Antibodies), anti-cathepsin B (R and D Systems Antibodies) and anti-cytochrome C (BD Biosciences). The anti-caspase-1 p20 antibody was a kind gift from Dr Peter Vandanbeele (Ghent University). All secondary antibodies were HRP conjugated. Antibodies against goat, rabbit and donkey were obtained from Santa Cruz, and anti-mouse secondary antibodies were from Amersham Biosciences.
Acknowledgments
We thank R. Kitsis and F. Sutterwala for the Nlrp3−/−, Asc, caspase-1−/− and Ipaf−/− mice; and J. Joyce for the cathepsin B−/−, C−/−, L−/− and S−/− mice. This work was supported, in whole or in part, by the US National Institutes of Health grant 1R56AI092497-01A1, by the Martin Turkish Foundation and the M-O’Connor-Foundation. Dr Michael Lisanti and his laboratory were supported by the resources of Thomas Jefferson University.
Glossary
Abbreviations:
- LLOMe
L-leucyl-L-leucine methyl ester
- IL
interleukin
- LPS
lipopolysaccharide
- TLR
Toll-like receptor
- NLR
nucleotide-binding domain and leucine rich repeat-containing
- ELISA
enzyme-linked immunosorbent assay
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24903
References
- 1.Williams A, Flavell RA, Eisenbarth SC. The role of NOD-like Receptors in shaping adaptive immunity. Curr Opin Immunol. 2010;22:34–40. doi: 10.1016/j.coi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 3.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 4.Nour AM, Yeung YG, Santambrogio L, Boyden ED, Stanley ER, Brojatsch J. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect Immun. 2009;77:1262–71. doi: 10.1128/IAI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin CC, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30:628–31. doi: 10.1007/s10875-010-9440-3. [DOI] [PubMed] [Google Scholar]
- 7.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–20. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci USA. 2009;106:870–5. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40:620–3. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hentze H, Lin XY, Choi MS, Porter AG. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ. 2003;10:956–68. doi: 10.1038/sj.cdd.4401264. [DOI] [PubMed] [Google Scholar]
- 14.Newman ZL, Leppla SH, Moayeri M. CA-074Me protection against anthrax lethal toxin. Infect Immun. 2009;77:4327–36. doi: 10.1128/IAI.00730-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson LS, Lima H, Jr., Goldberg MF, Gocheva V, Tsiperson V, Sutterwala FS, et al. Cathepsin-mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)-associated adjuvants. J Biol Chem. 2013;288:7481–91. doi: 10.1074/jbc.M112.400655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–70. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 18.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105:4312–7. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiele DL, Lipsky PE. The action of leucyl-leucine methyl ester on cytotoxic lymphocytes requires uptake by a novel dipeptide-specific facilitated transport system and dipeptidyl peptidase I-mediated conversion to membranolytic products. J Exp Med. 1990;172:183–94. doi: 10.1084/jem.172.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiele DL, Lipsky PE. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci USA. 1990;87:83–7. doi: 10.1073/pnas.87.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, et al. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle. 2007;6:758–66. doi: 10.4161/cc.6.6.3991. [DOI] [PubMed] [Google Scholar]
- 22.Varnes ME, Glazier KG, Gray C. pH-dependent effects of the ionophore nigericin on response of mammalian cells to radiation and heat treatment. Radiat Res. 1989;117:282–92. doi: 10.2307/3577329. [DOI] [PubMed] [Google Scholar]
- 23.Bevensee MO, Bashi E, Boron WF. Effect of trace levels of nigericin on intracellular pH and acid-base transport in rat renal mesangial cells. J Membr Biol. 1999;169:131–9. doi: 10.1007/s002329900525. [DOI] [PubMed] [Google Scholar]
- 24.Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68:765–83. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guicciardi ME, Leist M, Gores GJ. Lysosomes in cell death. Oncogene. 2004;23:2881–90. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 26.Schröder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics. 2010;10:4053–76. doi: 10.1002/pmic.201000196. [DOI] [PubMed] [Google Scholar]
- 27.Jäättelä M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003;4:416–23. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 28.Griffiths RJ, Stam EJ, Downs JT, Otterness IG. ATP induces the release of IL-1 from LPS-primed cells in vivo. J Immunol. 1995;154:2821–8. [PubMed] [Google Scholar]
- 29.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–9. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 30.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 31.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 32.Cuervo AM, Dice JF. Lysosomes, a meeting point of proteins, chaperones, and proteases. J Mol Med (Berl) 1998;76:6–12. doi: 10.1007/s109-1998-8099-y. [DOI] [PubMed] [Google Scholar]
- 33.Averette KM, Pratt MR, Yang Y, Bassilian S, Whitelegge JP, Loo JA, et al. Anthrax lethal toxin induced lysosomal membrane permeabilization and cytosolic cathepsin release is Nlrp1b/Nalp1b-dependent. PLoS ONE. 2009;4:e7913. doi: 10.1371/journal.pone.0007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U, et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem. 2008;283:19140–50. doi: 10.1074/jbc.M802513200. [DOI] [PubMed] [Google Scholar]
- 35.Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276:3149–57. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 36.Kool M, Soullié T, van Nimwegen M, Willart MA, Muskens F, Jung S, et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–82. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- 38.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kool M, Willart MA, van Nimwegen M, Bergen I, Pouliot P, Virchow JC, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–40. doi: 10.1016/j.immuni.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 41.Spreafico R, Ricciardi-Castagnoli P, Mortellaro A. The controversial relationship between NLRP3, alum, danger signals and the next-generation adjuvants. Eur J Immunol. 2010;40:638–42. doi: 10.1002/eji.200940039. [DOI] [PubMed] [Google Scholar]
- 42.Franchi L, Núñez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. 2008;38:2085–9. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKee AS, Munks MW, MacLeod MK, Fleenor CJ, Van Rooijen N, Kappler JW, et al. Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J Immunol. 2009;183:4403–14. doi: 10.4049/jimmunol.0900164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris J, Sharp FA, Lavelle EC. The role of inflammasomes in the immunostimulatory effects of particulate vaccine adjuvants. Eur J Immunol. 2010;40:634–8. doi: 10.1002/eji.200940172. [DOI] [PubMed] [Google Scholar]
- 45.Kuroda E, Ishii KJ, Uematsu S, Ohata K, Coban C, Akira S, et al. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity. 2011;34:514–26. doi: 10.1016/j.immuni.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 46.Seubert A, Calabro S, Santini L, Galli B, Genovese A, Valentini S, et al. Adjuvanticity of the oil-in-water emulsion MF59 is independent of Nlrp3 inflammasome but requires the adaptor protein MyD88. Proc Natl Acad Sci USA. 2011;108:11169–74. doi: 10.1073/pnas.1107941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Driscoll KE, Lindenschmidt RC, Maurer JK, Higgins JM, Ridder G. Pulmonary response to silica or titanium dioxide: inflammatory cells, alveolar macrophage-derived cytokines, and histopathology. Am J Respir Cell Mol Biol. 1990;2:381–90. doi: 10.1165/ajrcmb/2.4.381. [DOI] [PubMed] [Google Scholar]
- 48.Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunol Cell Biol. 2004;82:488–96. doi: 10.1111/j.0818-9641.2004.01272.x. [DOI] [PubMed] [Google Scholar]
- 49.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muehlbauer SM, Lima H, Jr., Goldman DL, Jacobson LS, Rivera J, Goldberg MF, et al. Proteasome inhibitors prevent caspase-1-mediated disease in rodents challenged with anthrax lethal toxin. Am J Pathol. 2010;177:735–43. doi: 10.2353/ajpath.2010.090828. [DOI] [PMC free article] [PubMed] [Google Scholar]