

Abstract
Phosphatidylinositol-3-kinase (PI3K) signaling is constitutive in most human cancers. Selective inhibition of PI3Kδ (p110δ) by GS-1101 has emerged as a promising therapy in chronic lymphocytic leukemia and indolent lymphomas. In aggressive non-Hodgkin lymphomas such as mantle cell lymphoma (MCL), however, efficacy has been observed, but the extent and duration of tumor control is modest. To determine if tumor killing by GS-1101 is cell cycle-dependent, we show in primary MCL cells by whole-transcriptome sequencing that, despite aberrant expression and recurrent mutations in Cyclin D1, mutations are rare in coding regions of CDK4, RB1 and other genes that control G1-S cell cycle progression or PI3K/AKT signaling. PI3Kδ is the predominant PI3K catalytic subunit expressed, and inhibition by GS-1101 transiently inhibits AKT phosphorylation but not proliferation in MCL cells. Induction of prolonged early G1-arrest (pG1) by selective inhibition of CDK4/CDK6 with PD 0332991 amplifies and sustains PI3Kδ inhibition, which leads to robust apoptosis. Accordingly, inhibition of PI3Kδ induces apoptosis of primary MCL tumor cells once they have ceased to cycle ex vivo, and this killing is enhanced by PD 0332991 inhibition of CDK4/CDK6. PIK3IP1, a negative PI3K regulator, appears to mediate pG1 sensitization to PI3K inhibition; it is markedly reduced in MCL tumor cells compared with normal peripheral B cells, profoundly induced in pG1 and required for pG1 sensitization to GS-1101. Thus, the magnitude and duration of PI3K inhibition and tumor killing by GS-1101 is pG1-dependent, suggesting induction of pG1 by CDK4/CDK6 inhibition as a strategy to sensitize proliferating lymphoma cells to PI3K inhibition.
Keywords: CDK4, CDK6, GS-1101, PD 0332991, PI3Kα, PI3Kδ, PIK3IP1, mantle cell lymphoma
Introduction
The phosphatidylinositol-3-kinase (PI3K) signaling pathway plays a central role in regulating cell proliferation, survival, metabolism and migration, and it is constitutively activated at a high frequency in human cancers.1,2 Activation of the class I PI3K catalytic subunits (PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ), together with their regulatory subunits, results in the production of phosphatidylinositol-3,4,5-triphosphate (PIP3) from phosphatidylinositol-4,5-triphosphate (PIP2). This leads to phosphorylation of the serine/threonine kinase AKT by mTORC2, which facilitates activation of AKT by 3-phosphoinositide-dependent protein kinase (PDK).1 PI3Kα (p110α) and PI3Kβ (p110β) are widely expressed across cell types, but the expression of PI3Kδ (p110δ) is restricted to cells of the hematopoietic lineage.3 Given that PI3K signaling mediates B cell receptor-dependent survival4 and is constitutive in B-cell malignancies, PI3Kδ represents a rational, lineage-restricted therapeutic target in B-cell lymphomas.
Interest in evaluating the therapeutic utility of selective PI3K inhibition has led to the development of GS-1101 (CAL-101), an orally bioavailable, PI3Kδ-specific inhibitor. GS-1101 has exhibited potent preclinical antitumor activity in chronic lymphocytic leukemia (CLL), Hodgkin lymphoma (HL) and certain subtypes of non-Hodgkin lymphomas (NHL).5-8 Furthermore, it has demonstrated promising single-agent clinical activity in CLL and indolent lymphomas. On this basis, further development of GS-1101 therapy is underway (www.clinicaltrials.gov). GS-1101 has also shown encouraging clinical activity in previously treated mantle cell lymphoma (MCL) in a phase I single-agent study, but tumor control appeared less durable in this disease than in indolent lymphomas or CLL.9 Because cell cycle dysregulation in lymphoma is frequently amplified in relapse/refractory disease, we hypothesize that targeting the cell cycle may sensitize non-indolent lymphomas to selective inhibition of PI3Kδ and tested this hypothesis in MCL.
MCL constitutes ~6% of all NHL, and remains incurable due to the eventual development of drug resistance.10 A hallmark of MCL is aberrant expression of cyclin D1 as a consequence of t(11;14)(q13;q32) chromosomal translocation and elevation of CDK4.11,12 This leads to unrestrained cell cycle progression and proliferation of tumor cells,13 which underlies disease progression.14 Although success in targeting the cell cycle in human cancer with broad-spectrum CDK inhibitors has been limited by a lack of selectivity and high toxicity, PD 0332991, a specific and potent inhibitor for CDK4/CDK6,15 has shown new promise. PD 0332991 rapidly inhibited CDK4 and CDK6 and induced early G1 arrest in primary human myeloma cells ex vivo, providing the first evidence for its bioactivity in human cancer cells.16 It was similarly effective in other Rb-positive (Rb+) cancer cells, such as MCL, acute myeloid leukemia cells (AML), T-cell leukemia, Burkitt lymphoma and breast cancer cells, ex vivo and in animal models.16-23 Reinforcing the specificity of PD 0332991, the therapeutic response of primary breast cancer cells to PD 0332991 ex vivo is strictly dependent on Rb, the substrate for CDK4 and CDK6.23 By inhibition of CDK4/CDK6 and induction of prolonged early G1 arrest (pG1) with PD 0332991, we have further developed a novel strategy that both inhibits tumor cell proliferation and reprograms them for killing by a partner agent.24 Importantly, in a single-agent phase Ib clinical study in MCL, PD 0332991 inhibited CDK4/CDK6 and induced pG1 in vivo, resulting in a durable clinical response with an excellent toxicity profile.25 Furthermore, prolonged inhibition of CDK4/CDK6 with PD 0332991 profoundly enhanced progression-free survival of breast cancer patients treated with letrozole, from 7.5–26.1 mo.26
In this study, we provide the first evidence that induction of pG1 by PD 0332991 both inhibits proliferation of MCL cells and sensitizes them to GS-1101 killing through cooperative inhibition of p-AKT and induction of PIK3IP1, a negative PI3K regulator. Sequential combination of specific CDK4/CDK6 inhibition with selective PI3Kδ inhibition, therefore, may offer a mechanism-based therapy for MCL and potentially other proliferative B-cell malignancies.
Results
PI3Kδ is the predominant PI3K catalytic subunit expressed, and activation of AKT is constitutive in primary MCL cells
To investigate PI3K signaling in MCL, we first determined, by whole-transcriptome sequencing (WTS, RNA-sequencing), the mRNA abundance of PI3K subunits in four primary MCL tumors (MCL1–4), one MCL cell line (JEKO-1) and, as a control, CD19+ peripheral blood B-cells (PBC)s comprising mainly non-cycling B cells from three healthy volunteers (Fig. 1A; Table S1). PIK3CD and PIK3R1 were the predominant class IA PI3K catalytic and regulatory subunits expressed in primary MCL cells and PBCs, whereas PIK3CA and PIK3R2 mRNA were less abundant. PIK3CB and PIK3R3 mRNA were modestly expressed in MCL cells but barely detectable (10 reads) in PBCs. By contrast, despite comparable expression of PIK3CG, expression of the class IB PI3K regulatory subunit was either markedly reduced in MCL cells (PIK3R5) or marginal in both MCL cells and PBCs (PIK3R6). Since PIK3R5 is necessary for PIK3CG activation,27 the class IB PI3K activity is likely impaired in MCL cells.
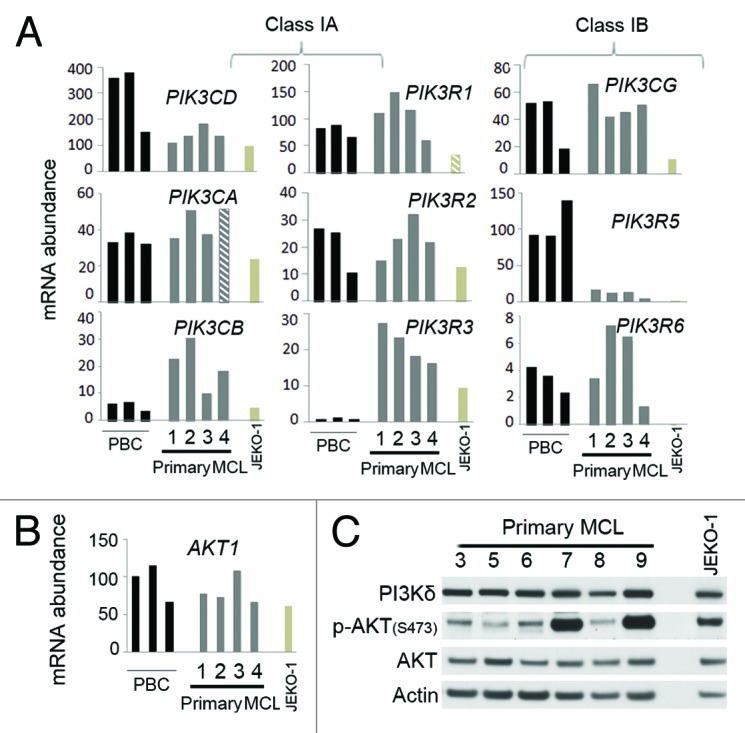
Figure 1. Predominant expression of PI3Kδ and constitutive AKT phosphorylation in primary MCL cells. WTS analysis of mRNA abundance and non-synonymous SNVs in the coding region of PI3K subunits (A) and AKT (B) in primary MCL tumors (MCL1–4), CD19+ peripheral blood B-cells (PBC)s from three healthy volunteers, and JEKO-1. PIK3CA (PI3Kα), PIK3CB (PI3Kβ), PIK3CD (PI3Kδ), PIK3CG (PI3Kγ), PIK3R1 (p85α), PIK3R2 (p85β), PIK3R3 (p55γ), PIK3R5 (p101), PIK3R6 (p84/87), AKT1 (AKT). For mRNA abundance (Table S1), all values were normalized to the expression of Actin (ACTB). Hatched bars indicate the detection of a significant non-synonymous SNV (detailed in Table S2). (C) Immunoblotting of PI3Kδ, p-AKT (S473) and AKT.
Only few non-synonymous single-nucleotide variants (SNVs) were detected in the coding sequences (CDSs) of analyzed PI3K subunits (Fig. 1A). They were predicted to be benign by the Provean and the SIFT programs (Table S2), consistent with reports that lymphoma cells, unlike solid tumors, rarely carry oncogenic mutations in PI3K genes.28-30 Likewise, no SNVs were detected in the CDSs of AKT1, the primary downstream effector of PI3K; PDK1, required for PI3K activation; or PTEN, which inhibits PI3K activation, although they were all expressed in primary MCL cells at the levels of PBCs (Fig. 1B; Fig. S1). Similar results were obtained in WTS analysis of additional 23 primary tumors (Chiron, Di Liberto, Mason and Chen-Kiang, unpublished). Collectively, our WTS data suggest that in MCL, coding sequence mutations are uncommon in the PI3K/AKT signaling pathway; the class IB PI3K activity is likely impaired and PIK3CD is the predominant PI3K catalytic subunit expressed.
Correspondingly, the PI3Kδ protein was highly expressed in primary MCL tumors, as was AKT, consistent with reported high levels of AKT protein expression in leucocytes and malignant B cells (Fig. 1C).3,5,8 Moreover, ATK was phosphorylated on serine 473 (S473), indicating that PI3K is activated in MCL cells (Fig. 1C). PI3Kδ-AKT signaling is thus constitutive in primary MCL cells, reinforcing the rationale for targeting PI3Kδ.
Selective inhibition of PI3Kδ does not inhibit the cell cycle in proliferating MCL cells
As in primary MCL cells, the PI3Kδ protein was highly expressed in multiple MCL cell lines while undetected in the control MM cell lines (Fig. 2A). The AKT protein was also abundant and constitutively phosphorylated on serine 473 (Fig. 2B). GS-1101 has been shown to modestly increase the proportion cells in G1 in two HL cell lines.8 However, it did not induce cell cycle arrest in the MCL cell lines we have tested, as determined by BrdU-pulse labeling (Fig. 2C). With the exception of dose-dependent cytotoxic killing shown by the ToPro-3 assay in SP53 cells, GS-1101 (0.1–10 µM) also did not induce cell death in all other five MCL cell lines characterized (Fig. 2D).
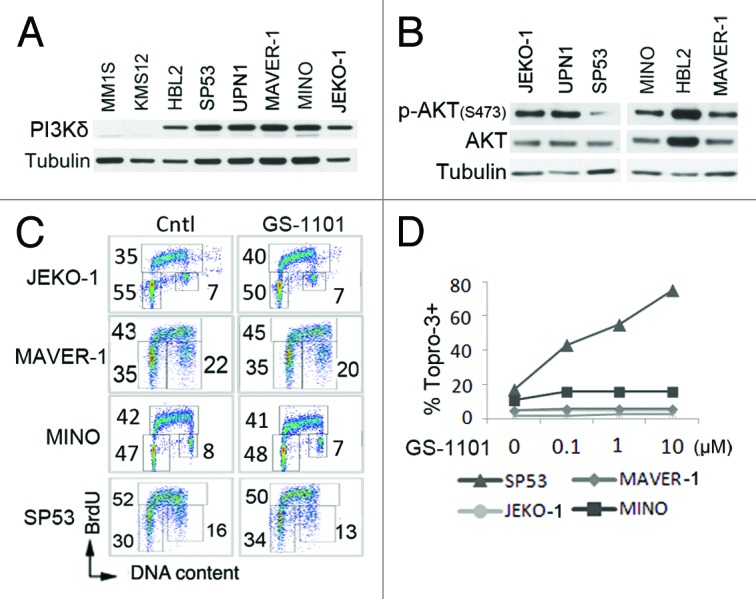
Figure 2. Inhibition of PI3Kδ by GS-1101 does not induce cell cycle arrest or apoptosis in MCL cell lines. (A and B) Immunoblotting of PI3Kδ, p-AKT (S473) and AKT in MCL cell lines. Myeloma cell lines (MM1S, KMS12) were used as a negative control. (C) MCL cells were cultured with GS-1101 for 72 h (5 µM for JEKO-1, MAVER-1 and MINO and 0.1 µM for SP53). BrdU was added 30 min before cell harvest for FACS analysis. Number in the FACS profile indicates the percentage of gated live cells in G1, S and G2/M cell cycle phases. (D) MCL cell lines were cultured with GS-1101 at indicated concentrations for 72 h. DNA fragmentation was determined by ToPro-3 staining and FACS analysis.
The core G1 cell cycle genes are largely intact in MCL cells and controlled by selective inhibition of CDK4/CDK6
GS-1101, however, is highly effective in indolent lymphomas. Since induction of prolonged early G1 arrest (pG1) by selective inhibition of CDK4/CDK6 with PD 0332991 sensitizes primary tumor cells to cytotoxic killing by a partner drug,24 we hypothesize that it will also sensitize proliferating MCL cells to killing by GS-1101. To test this hypothesis, we first determined the transcript abundance and SNVs of core G1 cell cycle genes in primary MCL cells by WTS (Fig. 3A; Table S1). Compared with PBCs, primary MCL cells expressed very high level of Cyclin D1 mRNA, CDK4 but not CDK6 mRNA, comparable levels of RB1 and Cyclin D2 mRNAs and reduced Cyclin D3 mRNA. They also expressed elevated CDK2, E2F1 and the CDK4/CDK6 inhibitor p18INK4c (CDKN2C), which is known to be induced during B cell activation,31 and reduced p19INK4d (CDKN2D), but not p16INK4a (CDKN2A) or p15INK4b (CDKN2B) mRNAs (Fig. 3A; Table S1). No mutations were detected in the CDSs of these cell cycle genes, except for one homozygous non-synonymous SNV in exon 1 of Cyclin D1, which is predicted to be deleterious, in one primary MCL tumor (MCL4) (Fig. 3A; Table S2). This result corroborates the detection of the same mutation by massive parallel genomic sequencing in 19/102 MCL tumors.32 The absence of mutations in CDK4, CDK6 and RB1 further suggest that CDK4/CDK6 are stable molecular targets for therapeutic intervention.

Figure 3. Selective inhibition of CDK4/CDK6 induces early G1 arrest in MCL cells. (A) WTS analysis as in Figure 1A. CCND1 (cyclin D1), CCND2 (cyclin D2), CCND3 (cyclin D3), CDKN2A (p16), CDKN2B (p15), CDKN2C (p18), CDKN2D (p19). (B) Immunoblotting of cyclin D1, cyclin D2, p18, CDK4, CDK6, pSRb (ser 807/811) and Rb. (C) Immunoblotting of pSRb (ser 807/811) and Rb in prolonged early G1 arrest (pG1) induced by PD for 48 and 96 h. (D) MAVER-1 (Rb-positive) and UPN1 (Rb-negative) cells were cultured with PD 0332991 (PD; 0.5 µM) for 24 h and analyzed as in Figure 2C.
Accordingly, primary MCL cells express cyclin D1 and CDK4 proteins and cyclin D2 and p18 proteins at variable levels (Fig. 3B). CDK4/CDK6-specific phosphorylation of Rb on serine 807–811 (pSRb) confirmed that the CDK4/cyclin D1 holoenzyme was active in MCL cells, along with CDK6/cyclin D1 in MCL7 and CDK4/cyclin D2 in MCL6. By contrast, the low level of cyclin D2 and CDK6 expressed in PBCs appears to be insufficient for activation of CDK6 given the absence of pSRB (Fig. 3B). Recapitulating these findings, MCL cell lines express cyclin D1, CDK4 and pSRb, except for the Rb-negative UPN1 cells (Fig. S2A). Inhibition of CDK4/CDK6 with PD 0332991 led to G1 arrest in all Rb+ MCL cell lines tested, as evidenced by the loss of pSRb (Fig. 3C), and cessation of DNA synthesis (Fig. 3D; Fig. S2B), except in UPN1 cells, which do not express Rb (Fig. 3D). Thus, despite a multitude of genomic abnormalities, the core G1 cell cycle genes are largely intact in MCL and controlled by inhibition of CDK4/CDK6.
Inhibition of CDK4/CDK6 sensitizes proliferating MCL cells to apoptosis induced by PI3Kδ inhibition
To determine whether induction of pG1 sensitizes proliferating MCL cells to GS-1101 killing, MCL cell lines were arrested in early G1 with PD 0332991 (0.5 µM for 24 h) before addition of GS-1101 (Fig. S3A). pG1 sensitized refractory, proliferating MCL cells to time- and dose-dependent apoptosis in response to GS-1101, as shown by the dramatic increase in apoptotic (Annexin V-positive) and dead cells (AnnexinV/PI double-positive) at 120 h, (Fig. 4A; Fig. S3B) and caspase-3 and PARP cleavage as early as 48 h (Fig. 4B). Consequently, pG1 prevented the expansion of MCL cells and virtually eradicated them when combined with GS-1101, including the GS-1101-sensitive SP53 cells (Fig. S3C), but did not affect the expansion or survival of Rb- UPN1 cells (Fig. 4C).

Figure 4. CDK4/CDK6 inhibition sensitizes MCL cell lines to GS-1101 killing. (A) Apoptosis was assayed by AnnexinV-FITC (A-V) and PI double staining at 96 and 120 h after GS-1101 addition, with or without prior induction of pG1 by PD 0332991. (B) Immunoblotting of caspase-3 (Casp3) and PARP cleavage (marked by arrow) in JEKO-1 cells treated with sequential combination of PD 0332991 and GS-1101 (5 µM) for time indicated. (C) Total number of live cells at indicated time of GS-1101 treatment, in the presence or absence of pG1 induced by PD. Cell viability was determined by Trypan blue exclusion staining in triplicate. *p < 0.05, **p < 0.005
Reinforcing the importance of prior pG1 sensitization, apoptosis was appreciable by 66 h of GS-1101 addition in JEKO-1 cells pretreated for 24 h with PD 0332991, but not until 90 h and less marked when GS-1101 was added concurrently (Fig. S3D). Furthermore, pG1 similarly enhanced the killing of MCL cells by a pan-PI3K inhibitor GDC-0941 that inhibits PI3Kδ, PI3Kα and PI3Kβ, demonstrating that pG1 sensitizes MCL cells to PI3K inhibition in general (Fig. S4).
Inhibition of CDK4/CDK6 enhances the killing of primary MCL cells by PI3Kδ inhibition
These findings led us to investigate the G1 dependence of GS-1101 killing in primary MCL tumor cells. While long-term proliferation of primary MCL cells ex vivo is not yet attainable, proliferation of freshly isolated CD19+ MCL cells can be extended transiently during the initial hours of isolation in co-culture with HS-5 bone marrow stromal cells (BMSCs) in the presence of IL-4, IL-6, IGF-1, BAFF and CD40L.
The primary MCL cells from five patients characterized (MCL8, 10–13) were all sensitive to GS-1101 (1–5 µM) once they ceased to proliferate ex vivo, as shown by the loss of viability by 48 h of GS-1101 addition, which was saturating at 1 µM except for MCL8 (Fig. 5A and B). Thus, despite stromal support, and that cytokines and growth factors that are known to activate the PI3K/AKT pathway in normal and malignant B cells,33 cell cycle arrest ex vivo rendered primary MCL cells sensitive to GS-1101.

Figure 5. Inhibition of CDK4/CDK6 enhances killing of primary MCL cells by PI3Kδ inhibition ex vivo. (A and B) Primary MCL cells from patients (MCL8, MCL10–13) were co-cultured in the presence or absence of PD 0332991 (0.5 µM) or GS-1101 (1 or 5 µM), or both, for time indicated as described in “Materials and Methods.” Number of live cells are represented as the % of input on day 0; *p < 0.05, **p < 0.005. (C) (left) Number of live cells (MCL 8, 12, 13) was represented as the % of control cells (without treatment); (right) Immunoblotting of pSRb (ser 807/811) and Rb at 24 h after PD 0332991 addition.
Since prolonged inhibition of CDK4/CDK6 with PD 0332991 leads to tumor regression in MCL patients25 and sensitizes MCL cell lines to GS-1101 killing (Fig. 4), it may induce cell death or enhance GS-1101 killing in primary MCL cells. Indeed, PD 0332991 modestly reduced the viability of primary MCL cells and exacerbated and accelerated the loss of viability induced by GS-1101 (Fig. 5B and C). This was a consequence of PD 0332991 inhibition of CDK4/CDK6, since pSRb was markedly reduced by 24 h (Fig. 5C). By contrast, PD 0332991 did not significantly enhance the marginal killing of CD19+ PBCs by GS-1101, in which CDK6 is expressed but inactive due to insufficient cyclin D expression (Fig. S5; Fig. 3B). Together, these results demonstrate that proliferating primary MCL cells are responsive to PI3Kδ inhibition once they have ceased to proliferate ex vivo, and that PI3Kδ inhibition is exacerbated and accelerated by prolonged inhibition of CDK4/6 and induction of pG1 with PD 0332991.
Cooperative inhibition of AKT phosphorylation by CDK4/CDK6 and PI3Kδ inhibition
Further investigations revealed that pG1 markedly reduced p-AKT (S473) in primary MCL cells (MCL8, 13) and JEKO-1 and MAVER-1 MCL cell lines, but not in the Rb- UPN1 cells (Fig. 6A). Moreover, phosphorylation of AKT by both mTORC2 (p-AKTS473) and PDK (p-AKT T308) was maintained in proliferating MCL cells at 72 h of GS1101 addition, but reduced in pG1, and further when GS-1101 was added to MCL cells 24 h after they have been arrested in early G1 by PD 0332991 for 24 h (Fig. 6B). Thus, correlating with induction of apoptosis by GS-1101, pG1 inhibits pAKT phosphorylation and sensitizes MCL cells to PI3Kδ inhibition.

Figure 6. Cooperative inhibition of AKT phosphorylation by CDK4/CDK6 and PI3Kδ inhibition. (A) Immunoblotting of pSRb (ser 807/811), Rb, p-AKT (S473) and AKT in primary MCL cells (MCL8, MCL13), Rb-positive JEKO-1, MAVER-1 cells and Rb-negative UPN1 cell line in the presence of PD 0332991 for 72 h (B) Immunoblotting of p-AKT (S473), p-AKT (T308) and AKT. (C) (left) Immunoblotting of p-AKT (S473) and AKT. Cells were cultured at a starting cell concentration of 1.5 × 105 cells/ml and harvested at time indicated, in the presence of GS-1101 (5 µM) (lanes 5–8) or PD 0332991 (lanes 9–13) or both (lanes 14–17). (right) The ratio of p-AKT/AKT was determined using the ImageJ Program.
However, GS-1101 alone has been shown to rapidly reduce p-AKT in MCL cell lines.7 We therefore hypothesized that inhibition of PI3Kδ by GS-1101 may transiently reduce p-AKT in proliferating MCL cells, and pG1 sustains the loss of p-AKT by PI3Kδ inhibition. Indeed, a time course study in JEKO-1 and MAVER-1 cells demonstrated that p-AKT was markedly reduced by GS-1101 alone within 24 h, but restored by 48 h and maintained thereafter (Fig. 6C). pG1 exacerbated the reduction of p-AKT in response to GS-1101 in 24 h, and sustained the loss of p-AKT in MCL cells (Fig. 6C).
Induction of PIK3IP1 in pG1 mediates pG1 sensitization to GS-1101
To investigate the mechanism for pG1 sensitization to PI3Kδ inhibition in MCL cells, we discovered by WTS analysis that PIK3IP1, a negative regulator of PI3K,34 was markedly downregulated (3–30-fold) in proliferating primary MCL cells compared with resting PBCs (Fig. 7A). Inversely correlating with proliferation, the PIK3IP1 mRNA level was reduced further in MCL cell lines compared with primary MCL tumor cells and elevated prominently (20–40-fold) within 72 h of PD 0332991-induced early G1 arrest (Fig. 7B). GS-1101 moderately induced PI3KIP1 in the sensitive SP53 cells in 24 h (Fig. S6A), but less effectively in JEKO-1 and MAVER-1 cells even after 48 h (Fig. 7B). However, induction of pG1 in combination with GS-1101 led to a 40–80-fold increase in PIK3IP1 mRNA (Fig. 7B), which correlated with marked induction of cell death (Figs. 2D and 4).
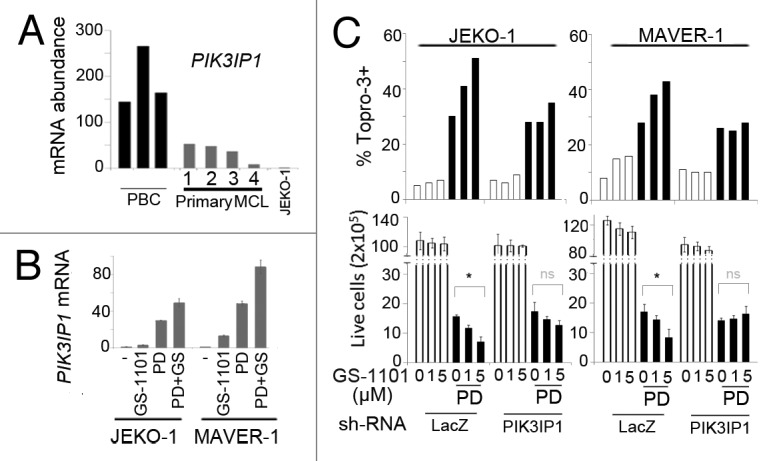
Figure 7. Prolonged early G1 arrest sensitizes MCL cells to GS-1101 through induction of PIK3IP1. (A) WTS analysis of PIK3IP1 mRNA abundance (B) q-RT-PCR analysis of PIK3IP1 mRNA expression in JEKO-1 and MAVER-1 cell cultured with sequential combination of PD 0332991 (72 h) and GS-1101 (48 h). (C) Cells infected with PIK3IP1 or LacZ shRNA lentivirus were treated with PD 0332991 (72 h) and GS-1101 (48 h) at indicated concentration. DNA fragmentation (Topro-3) was determined by FACS analysis (upper panel) and cell viability by Tripan blue exclusion staining in triplicate. *p < 0.05.
Further, partial depletion of PIK3IP1 expression by sh-RNA knockdown significantly blunted the killing of JEKO-1 and MAVER-1 cells by GS-1101 in pG1 and attenuated the reduction of lives cells (Fig. 7C). These data strongly suggest that PIK3IP1 is induced in early G1 arrest to mediate pG1 sensitization to GS-1101 killing in MCL cells.
Discussion
By inducing prolonged early G1 arrest (pG1) through selective inhibition of CDK4/CDK6, we have developed a strategy to both inhibit the expansion of proliferating MCL cells and sensitize them to killing by selective inhibition of PI3Kδ. This study presents a novel sequential combination of selective CDK4/CDK6 inhibition with a selective partner, the PI3Kδ-specific inhibitor GS-1101, in primary human cancer cells and the first therapeutic strategy for reprogramming MCL cells by cell cycle control.
The approach to target CDK4/CDK6 in MCL is rooted in our understanding that (1) proliferation of MCL tumor cells in patients in vivo, as indicated by Ki67 expression, strongly correlates with poor prognosis;14 (2) unrestrained proliferation of MCL cells appears to be driven by aberrant expression of cyclin D1 and CDK4 (Fig. 3A and B); and (3) CDK4 and CDK6 are inhibited by PD 0332991 in MCL patients in vivo25 and in freshly isolated primary MCL cells ex vivo (Fig. 5).
The combination of selective CDK4/CDK6 inhibition with selective PI3Kδ inhibition takes advantage of the predominant expression of PI3Kδ in hematologic lineage cells3 and the essential role of PI3Kδ in B-cell physiology.35 By WTS analysis, we show that PI3Kδ is also the predominant class IA PI3K catalytic subunit expressed in primary MCL cells, and that mutations in the CDSs of genes in the PI3K-AKT signaling pathway are uncommon, except for an apparently non-damaging mutation in PI3Kα in one of the four primary tumors characterized (Fig. 1A; Table S2). These findings are consistent with a recent study of WTS in a larger cohort32 and reinforce the rationale for selective targeting of PI3Kδ in MCL.
Importantly, we demonstrated that inhibition of PI3Kδ does not induce cell cycle arrest or apoptosis in the majority of proliferating MCL cells (Fig. 2). However, induction of pG1 by CDK4/CDK6 inhibition sensitizes refractory MCL cells to apoptosis induced by GS-1101 (Fig. 4) and accelerates and enhances the killing of primary MCL cells by GS-1101 in the presence of bone marrow stromal cells and growth factors (Fig. 5). Inhibition of PI3Kδ by GS-1101 reduces phosphorylation of AKT as previously reported,7 but only transiently in proliferating MCL cells unless they are first arrested in early G1, which sustains and exacerbates the loss of p-AKT by PI3Kδ inhibition (Fig. 6). These data have important implications for the mechanism of resistance and sensitization to selective inhibition of PI3Kδ by GS-1101 in MCL and potentially other non-indolent lymphomas.
Although p-AKT serves as a primary readout for PI3K activation and correlates with cell survival, AKT is not exclusively activated by PI3Kδ. Thus, the restoration of p-AKT in proliferating MCL cells does not necessarily indicate development of resistance to GS-1101. It could also be due to functional compensation by other PI3K isoforms, such as PI3Kα, which has been reported to be expressed at higher levels in MCL progression.36 Such a compensatory mechanism is consistent with the reduction of live cells by the pan-class I PI3K inhibitor GDC-0941 and not by the PI3Kδ-selective inhibitor (Fig. S4). However, we found that induction of pG1 also reduces p-AKT and markedly enhances GDC-0941 killing of MCL cells. Thus, pG1 sensitizes MCL cells to inhibition of PI3K in general and disrupts the potential functional compensation by PI3K isoforms (Fig. S4).
Sustaining PI3K inhibition by induction of pG1 has important implications for mechanism-based targeting of lymphoma. For example, it has been shown that PI3K activation cooperates with c-Myc in a mouse model of Burkitt lymphomagenesis that mimics the human disease,37,38 and that PD 0332991 potently suppresses Burkitt lymphomagenesis.22 Cooperative inhibition of PI3K by selective inhibition of PI3Kδ and CDK4/CDK6 thus represents a promising mechanism-based therapy for Burkitt lymphoma. In addition, inhibition of Bruton’s tyrosine kinase (BTK) by ibrutinib has also emerged as an highly effective single agent therapy for CLL and MCL, in part through inhibition of PI3K downstream.39 However, relapse to both GS-1101 in CLL and Ibrutinib in CLL and MCL is common and accompanied by more aggressive disease, further reinforcing the rational to target PI3K in combination with inhibition of CDK4/CDK6.
We have previously demonstrated that inhibition of CDK4/CDK6 with PD 0332991 halts gene expression in early G1 and prevents the expression of genes programmed for other cell cycle phases, thereby forcing an imbalance in gene expression in pG1 that sensitizes myeloma cells to killing by a partner agent.24 To optimize the schedule for combination, we have further demonstrated the cell cycle targeting specificity of a chosen cytotoxic partner can be determined by cell cycle synchronization through selective and reversible inhibition of CDK4/CDK6 with PD 0332991.24 The finding that inhibition of CDK4/CDK6 with PD 0332991 can antagonize the cytotoxic response to anthracyclin therapy40 poses a note of caution in targeting CDK4/CDK6, and reinforces the critical importance of mechanism-base cytotoxic partner selection and schedule optimization.
Here we show that the expression of PIK3IP1 is markedly reduced in tumor cells in MCL patients compared with normal B cells and is strikingly induced in pG1 in MCL cell lines, greater in cooperation with PI3Kδ inhibition by GS-1101 (Fig. 7). These novel findings, coupled with the requirement of PIK3IP1 for pG1 sensitization to PI3Kδ inhibition (Fig. 7C), suggest that PIK3IP1 is programmed for expression in early G1 and mediates pG1-sensitization. PIK3IP1 was discovered as a novel p110-interacting protein that prevents its activation by p85 family adaptor proteins.34 Its expression is reduced in hepatocellular carcinoma and may contribute to liver carcinogenesis.41 However, PIK3IP1 appears to also have a physiologic role in inhibiting T cell activation42 and in inducing apoptosis of neural progenitor.43 How PIK3IP1 mediates pG1-sensitization to apoptosis induced by PI3Kδ inhibition remains to be elucidated. Nonetheless, it is tempting to speculate that in MCL cells, induction of PIK3IP1 in pG1 disrupts a feedback loop that activates PI3K/AKT, thereby enhancing and prolonging PI3Kδ inhibition by GS-1101.
Inhibition of PI3Kδ by GS-1101 has already shown promising clinical efficacy in indolent lymphomas and CLL (www.clinical trials.gov). Induction of pG1 by CDK4/CDK6 inhibition with PD 0332991 has also led to encouraging durable response in MCL patients with an excellent toxicity profile25 and stable disease in advanced liposarcoma,44 as well as an impressive improvement in progression-free-survival in combination therapy in breast cancer.26 pG1 sensitization of proliferating MCL cells to GS-1101 through cooperative inhibition of p-AKT and induction of PIK3IP1 thus provides a mechanism-based strategy for therapeutic targeting of CDK4/CDK6 in combination with selective targeting of PI3Kδ in MCL and, potentially, other aggressive lymphomas.
Materials and Methods
Whole-transcriptome sequencing (WTS) and analysis
Our RNA-Seq analysis pipeline used BWA and SAMtools. For mRNA abundance, all values were normalized to the expression of actin (ActB) (Table S1). Significant single nucleotide variants (SNVs) present in coding (CDSs, non-synonymous) regions were identified using Genesifter (Geospiza), which uses SAMtools and the Genome Analysis Toolkit (GATK) for base quality score recalibration and local re-alignment for indels. Raw reads were trimmed for the first and last five base pairs before alignment to remove low-quality bases. SNVs that were also detected in the libraries of CD19+ peripheral blood B-cells (PBC)s from three healthy volunteers were removed. Finally, a threshold of 30× coverage as well as a score > 10045 were applied resulting in the list of candidate mutations (Table S2). For each experimental condition, 100 ng of high quality total RNA (RIN > 0.8 on the Agilent BioAnalyzer 2100) was isolated using the RNAEasy kit according to the manufacturer’s instructions (QIAGEN). All RNAs were converted to cDNA, isolated with magnetic beads from the TruSeq mRNA prep kit (v2) and then ligated to Illumina adapters, as per the standard TruSeq Illumina protocol. Using these multi-plexed cDNA libraries, we generated clusters on the Illumina cBot station and paired-end sequenced each sample to 50 × 50 bp on the Illumina HiSeq2000 at the Weill Cornell Medical College (WCMC) Epigenomics Core. Cluster generation, sequencing and processing of the images were done using the real-time analysis (RTA) software on the HiSeq2000 and post-processing with CASAVA (v.1.8.2). To optimize library preparation, we used a TACON high-throughput RNA prep station. Raw data were filtered for high median quality (Q-value > 20) and then sent to Cornell’s high performance computing (HPC) cluster to be run through our RNA-seq analysis pipeline. The RNA-seq data presented in this study, which include libraries from MCL tumor cells of four patients, PBCs from three healthy volunteers and JEKO-1 MCL cell lines have been deposited in the gene expression omnibus (GEO).
Primary cells isolation and culture
Mantle cell lymphoma (MCL) biopsies were obtained from patients at the New York-Presbyterian Hospital after informed consent as part of a study approved by the Institutional Review Board. Primary MCL cells were purified using MACS CD19 MicroBeads (Miltenyi Biotec). The percentage of MCL tumor cells (CD19+, CD5+, CD23−) was determined to be > 90% by flow cytometry. B cells from healthy volunteers were isolated from peripheral blood (PBC)s using the same protocol. Primary MCL cells were cultured in RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated FBS (HyClone), 2 mM L-glutamine and 100 U/ml penicillin/streptomycin (Invitrogen) together with mitomycin-C arrested HS-5 (GFP+) human stromal cells in the presence of recombinant human IL-6 (40 U/ml), soluble IL-6R (40 U/ml), human IGF-1 (100 ng/ml), sCD40L (0.5 µg/mL), human BAFF (100 ng/mL), human IL4 (100 ng/mL) (all from R&D Systems) and β-mercapto-ethanol (50 µM). MCL cells were cultured in the presence of PD 0332991 (Sellek Chemical) or GS-1101 (Gilead), or both, at the concentrations and for the times indicated. For cell lines, PD 0332991 was added daily (0.25 µM) to maintain prolonged G1 arrest (pG1).
Cell lines
Several human MCL cell lines were evaluated: JEKO-1 was obtained from DSMZ; MAVER-1 and MINO from ATCC; UPN1 was provided by Dr Samir Parekh (Albert Einstein College of Medicine), HBL2 by Dr O’Connor (Columbia University) and SP53 by Dr Jiangao Tao (Moffit Cancer Center). The human multiple myeloma (MM) cell lines MM1S and KMS12PE have been described previously.24
Immunoblotting
Proteins were analyzed with the following antibodies: serine phosphorylation on 807/811of Rb (pSRb) (#9308), CDK4 (#2906), cyclinD1 (#2926), cyclinD2 (#3741), AKT (#9272), p-AKT-Ser473 (#9271), PARP (#9542), caspase-3 (#9662) and tubulin (#2144) (all from Cell Signaling Technology); CDK6 (sc-177), actin (sc-1615), p18 (sc-865) and PI3Kδ (sc-7176) from Santa Cruz Biotechnology, and Rb (#554136) from BD Pharmigen.
shRNA knockdown
Cells were infected with PIK3IP1 (TRCN0000135363/TRCN0000135429), or LacZ (TRCN0000072229) control shRNA (The RNAi Consortium at Broad Institute). Knockdown was validated by quantitative RT-PCR at 60–72 h post-transduction. Cell cycle analysis and cell viability assays were performed as previously described.24
Supplementary Material
Acknowledgments
We thank Drs Jihye Paik and Langdon Miller for helpful comments and suggestions, Drs Adriana Rossi and David Jayabalan for help in the studies of normal B cells, Dr Jenny Zhang for RNA-Sequencing and Suzhen Chen for technical assistance.
Glossary
Abbreviations:
- BTK
Bruton’s tyrosine kinase
- CLL
chronic lymphocytic leukemia
- CDK
cyclin-dependent kinase
- CDS
coding sequences
- HL
Hodgkin lymphoma
- MCL
Mantle cell lymphoma
- NHL
non Hodgkin lymphoma
- p-AKT
phospho-AKT
- pG1
prolonged early G1 arrest
- pSRb
CDK4/CDK6-specific phosphorylation of Rb on serine 807-811
- PDK
3-phosphoinositide-dependent protein kinase
- q-RT-PCR
quantitative real-time PCR
- Rb
retinoblastoma protein
- SNV
single nucleotide variants
- WTS
whole-transcriptome sequencing
Disclosure of Potential Conflicts of Interest
B.J.L. is employed by Gilead. The other authors declare that they have no competing interests
Grant Support
This work is supported in part by postdoctoral fellowships from The Cancer Research and Treatment Fund and the Philippe Foundation to DC, NIH/NCI R44HG005297 to C.E.M., and grants from the Lymphoma Research Foundation to J.L. and S.C.-K. and the Leukemia and Lymphoma Society Translational Research Program (6217) and NIH/NCI R01120531 to S.C.-K.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24928
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24928
References
- 1.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 2.Fruman DA, Rommel C. PI3Kδ inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov. 2011;1:562–72. doi: 10.1158/2159-8290.CD-11-0249. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997;94:4330–5. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–86. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–88. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118:3603–12. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–4. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meadows SA, Vega F, Kashishian A, Johnson D, Diehl V, Miller LL, et al. PI3Kδ inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood. 2012;119:1897–900. doi: 10.1182/blood-2011-10-386763. [DOI] [PubMed] [Google Scholar]
- 9.Kahl BBJ, Flinn IW, et al. Clinical Safety and Activity In a Phase1 Study of CAL-101, An Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase P110{delta}, In Patients with Relapsed or Refractory Non-Hodgkin Lymphoma. ASH Annual Meeting Abstracts 2010; 116:1777. [Google Scholar]
- 10.Dreyling M, Weigert O, Hiddemann W, European MCL Network Current treatment standards and future strategies in mantle cell lymphoma. Ann Oncol. 2008;19(Suppl 4):iv41–4. doi: 10.1093/annonc/mdn193. [DOI] [PubMed] [Google Scholar]
- 11.Hernández L, Beà S, Pinyol M, Ott G, Katzenberger T, Rosenwald A, et al. CDK4 and MDM2 gene alterations mainly occur in highly proliferative and aggressive mantle cell lymphomas with wild-type INK4a/ARF locus. Cancer Res. 2005;65:2199–206. doi: 10.1158/0008-5472.CAN-04-1526. [DOI] [PubMed] [Google Scholar]
- 12.Korz C, Pscherer A, Benner A, Mertens D, Schaffner C, Leupolt E, et al. Evidence for distinct pathomechanisms in B-cell chronic lymphocytic leukemia and mantle cell lymphoma by quantitative expression analysis of cell cycle and apoptosis-associated genes. Blood. 2002;99:4554–61. doi: 10.1182/blood.V99.12.4554. [DOI] [PubMed] [Google Scholar]
- 13.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 14.Determann O, Hoster E, Ott G, Wolfram Bernd H, Loddenkemper C, Leo Hansmann M, et al. European Mantle Cell Lymphoma Network and the German Low Grade Lymphoma Study Group Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood. 2008;111:2385–7. doi: 10.1182/blood-2007-10-117010. [DOI] [PubMed] [Google Scholar]
- 15.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–38. [PubMed] [Google Scholar]
- 16.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 17.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108:1744–50. doi: 10.1182/blood-2006-04-016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, et al. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008;68:5519–23. doi: 10.1158/0008-5472.CAN-07-6404. [DOI] [PubMed] [Google Scholar]
- 20.Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell. 2012;22:452–65. doi: 10.1016/j.ccr.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Wang J, Blaser BW, Duchemin AM, Kusewitt DF, Liu T, et al. Pharmacologic inhibition of CDK4/6: mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood. 2007;110:2075–83. doi: 10.1182/blood-2007-02-071266. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–20. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012;11:2756–61. doi: 10.4161/cc.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120:1095–106. doi: 10.1182/blood-2012-03-415984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597–607. doi: 10.1182/blood-2011-10-388298. [DOI] [PubMed] [Google Scholar]
- 26.CDK Inhibitor Triples PFS in Breast Cancer Cancer Discov. 2013;3:4. [Google Scholar]
- 27.Brazzatti JA, Klingler-Hoffmann M, Haylock-Jacobs S, Harata-Lee Y, Niu M, Higgins MD, et al. Differential roles for the p101 and p84 regulatory subunits of PI3Kgamma in tumor growth and metastasis. Oncogene. 2012;31:2350–61. doi: 10.1038/onc.2011.414. [DOI] [PubMed] [Google Scholar]
- 28.Bousquet M, Recher C, Queleen C, Demur C, Payrastre B, Brousset P. Assessment of somatic mutations in phosphatidylinositol 3-kinase gene in human lymphoma and acute leukaemia. Br J Haematol. 2005;131:411–3. doi: 10.1111/j.1365-2141.2005.05784.x. [DOI] [PubMed] [Google Scholar]
- 29.Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108:1668–76. doi: 10.1182/blood-2006-04-015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bretz J, Garcia J, Huang X, Kang L, Zhang Y, Toellner KM, et al. Noxa mediates p18INK4c cell-cycle control of homeostasis in B cells and plasma cell precursors. Blood. 2011;117:2179–88. doi: 10.1182/blood-2010-06-288027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meissner B, Kridel R, Lim RS, Rogic S, Tse K, Scott DW, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121:3161–4. doi: 10.1182/blood-2013-01-478834. [DOI] [PubMed] [Google Scholar]
- 33.Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni M, Pavan A, et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111:5142–51. doi: 10.1182/blood-2007-07-103481. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Z, He X, Johnson C, Stoops J, Eaker AE, Stoffer DS, et al. PI3K is negatively regulated by PIK3IP1, a novel p110 interacting protein. Biochem Biophys Res Commun. 2007;358:66–72. doi: 10.1016/j.bbrc.2007.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–63. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyengar S, Clear A, Bödör C, Maharaj L, Lee A, Calaminici M, et al. P110α-mediated constitutive PI3K signaling limits the efficacy of p110δ-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121:2274–84. doi: 10.1182/blood-2012-10-460832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sander S, Calado DP, Srinivasan L, Köchert K, Zhang B, Rosolowski M, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–79. doi: 10.1016/j.ccr.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sander S, Rajewsky K. Burkitt lymphomagenesis linked to MYC plus PI3K in germinal center B cells. Oncotarget. 2012;3:1066–7. doi: 10.18632/oncotarget.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burger JA, Hoellenriegel J. Phosphoinositide 3′-kinase delta: turning off BCR signaling in Chronic Lymphocytic Leukemia. Oncotarget. 2011;2:737–8. doi: 10.18632/oncotarget.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11:2747–55. doi: 10.4161/cc.21127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He X, Zhu Z, Johnson C, Stoops J, Eaker AE, Bowen W, et al. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 2008;68:5591–8. doi: 10.1158/0008-5472.CAN-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeFrances MC, Debelius DR, Cheng J, Kane LP. Inhibition of T-cell activation by PIK3IP1. Eur J Immunol. 2012;42:2754–9. doi: 10.1002/eji.201141653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt-Strassburger U, Schips TG, Maier HJ, Kloiber K, Mannella F, Braunstein KE, et al. Expression of constitutively active FoxO3 in murine forebrain leads to a loss of neural progenitors. FASEB J. 2012;26:4990–5001. doi: 10.1096/fj.12-208587. [DOI] [PubMed] [Google Scholar]
- 44.Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II Trial of the CDK4 Inhibitor PD0332991 in Patients With Advanced CDK4-Amplified Well-Differentiated or Dedifferentiated Liposarcoma. J Clin Oncol. 2013 doi: 10.1200/JCO.2012.46.5476. Epub Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.