

Abstract
Reversible ubiquitylation of proteins contributes to their integrity, abundance and activity. The RE1-silencing transcription factor (REST) plays key physiological roles and is dysregulated in a spectrum of disease. It is rapidly turned over and is phosphorylated, polyubiquitylated and degraded en masse during neuronal differentiation and cell cycle progression. Through siRNA screening we identified the deubiquitylase USP15 as a key regulator of cellular REST. Both antagonism of REST polyubiquitylation and rescue of endogenous REST levels are dependent on the deubiquitylase activity of USP15. However, USP15 depletion does not destabilize pre-existing REST, but rather specifically impairs de novo REST synthesis. Indeed, we find that a small fraction of endogenous USP15 is associated with polysomes. In accordance with these findings, USP15 does not antagonize the degradation of phosphorylated REST at mitosis. Instead it is required for the rapid accumulation of newly synthesized REST on mitotic exit, thus playing a key role in its cell cycle oscillations. Importantly, this study reveals a novel role for a DUB in specifically promoting new protein synthesis.
Keywords: NRSF, ubiquitin specific peptidase 15, deubiquitination, G1, co-translational, post-translational modification, protein degradation, cell cycle
Introduction
Ubiquitylation is a highly dynamic reversible post-translation modification that is often likened to phosphorylation. However, ubiquitin is a more versatile tag. It may act as a signal for protein degradation, but can also regulate protein activity or localization, ultimately controlling cellular signaling pathways and transcription through diverse mechanisms.1-3 Either monoubiquitin or alternative polyubiquitin chains can be appended to substrates by a cascade of E1, E2 and E3 enzymes. A family of approximately 90 deubiquitylating enzymes (DUBs) mediate the cleavage of ubiquitin and can reverse signals or stabilize proteins.4 As the individual DUBs are gradually assigned to specific substrates and associated with key signaling pathways, we are beginning to understand their physiological significance and relevance to disease.2,5-8 Moreover, complex regulatory networks are emerging, revealing how ubiquitin coordinates progressive processes and highlighting the extent to which alternative ubiquitin ligases or DUBs may regulate the specific temporal or spatial ubiquitylation of a given protein.1-3
Ubiquitylation exhibits multifaceted roles in protein homeostasis, participating not only in proteasomal degradation, but also in lysosomal degradation and autophagy.9 Reversible ubiquitylation also plays a key role in the endoplasmic reticulum-associated degradation (ERAD) pathway that handles misfolded proteins in the ER.10 In addition, accumulating evidence indicates that ubiquitylation of newly synthesized proteins is common. It was reported over a decade ago that around 30% of all newly synthesized proteins are ubiquitylated and degraded by the proteasome.11 More recently, ubiquitin remnant profiling by mass spectrometry found that a substantial fraction of the ubiquitylated proteome failed to accumulate when proteasome inhibition was coupled with cycloheximide treatment.12 These observations suggest that co-translational ubiquitylation may function as an integral component of protein synthesis.
The RE1 silencing transcription factor (REST),13 also known as neuron-restrictive silencer factor (NRSF),14 is a physiologically important transcriptional repressor that is acutely regulated by ubiquitylation.15-17 REST can potentially bind several thousand degenerate RE1 sites in the genome via a multiple zinc finger DNA binding domain.18,19 It recruits a suite of co-factors through bipartite repression domains to nucleate heterochromatin and regulate expression of specific protein-coding genes or microRNAs.20-22 However, it is apparent that REST function is gene-specific, tissue-specific and temporally regulated. REST acts as a master controller during neurogenesis, yet also coordinates vital cellular processes in differentiated neurons and in non-neuronal cells. As such, its expression and function must be tightly regulated. Abnormal REST activity is associated with a spectrum of disorders, such as Huntington’s disease, Down syndrome and epilepsy (for a review, see refs. 20 and 21) and diverse tumor types including medulloblastoma, glioma, neuroblastoma, lung, breast, colon and prostate cancers (for a review, see refs. 23–25). REST exhibits context-dependent roles as an oncogene or a tumor suppressor, as loss of REST in epithelial cancers licenses inappropriate gene expression that can convey a growth advantage,26-29 while REST re-expression in neuronal tumors promotes stem-like characteristics.30-32
REST is part of the embryonic pluripotency network and is downregulated in neural progenitors as they differentiate along a neuronal program.33 This is predominantly achieved through phosphorylation-dependent polyubiquitylation, which targets REST for proteasomal degradation.17 Acute ubiquitin-driven destruction of REST is not restricted to sustained cell lineage decisions, but is similarly employed as a transient mechanism during cell cycle progression; the ubiquitin E3 ligase SCFβTrCP is the common driver in both cases.16,17 Intriguingly, although forced βTrCP expression leads to an oncogenic transformation of human mammary epithelial cells that is dependent on REST degradation,17 βTrCP-dependent degradation of REST also enhances mitotic checkpoint fidelity.16 Thus not only absolute REST levels, but the context and timing of REST degradation, and of its subsequent re-accumulation, influence cellular physiology.
Here we report an unbiased screen through which we identified USP15 as a DUB that regulates REST abundance in lung cancer cells. We show that USP15 does not antagonize degradation of pre-existing REST or protect phosphorylated REST at mitosis. Instead, the physiological role of USP15 is to promote new REST synthesis to restore its cellular level at mitotic exit. Importantly, through examination of REST as a new substrate for USP15, we have uncovered a novel mechanism of action for a DUB in specifically opposing the turnover of the newly synthesized pool of a protein.
Results
Screening for DUBs that modulate REST in lung cancer cells
REST expression varies dramatically among cells derived from different types of lung cancer, such that the protein is undetectable in neuroendocrine small cell lung cancer (SCLC) and present at variable levels in non-small cell lung cancer (NSCLC) cell lines.34 We employed the A549 NSCLC cell line as a model system, as it retains levels of REST comparable to that in normal lung and maintains transcriptional repression of classical REST target genes.26,34 Consistent with its role as a transcriptional repressor, the full-length isoform of REST mainly resides in the nucleus, where it is largely seen as a form with an apparent molecular weight of 180–220 kDa that is suggested to be O-glycosylated.35-37 The 220 kDa form of REST is predominant in A549 cells (Fig. S1A) and is destabilized by siRNA depletion or pharmacological inhibition of O-linked N-acetylglucosamine (GlcNAc) transferase (OGT) (Fig. S1C and 1D).
To investigate the control of REST protein stability in lung cancer cells, we employed an unbiased screen to identify any DUBs that modulated REST levels under normal growth conditions. A549 cells were transfected with a custom library of siRNA pools representing the majority of active human DUBs, and the abundance of 220 kDa REST in the nucleoplasmic protein fraction was ranked (Fig. 1A). Four DUBs whose depletion induced the most marked decreases in REST were subjected to validation. For two of these, USP47 and USP49, we were unable to replicate the effect seen for the siRNA pool in the original screen (Fig. S2). Depletion of a third DUB, USP52 (PAN2), was reproducible but failed to deconvolute, and USP52 lacks residues essential for deubiquitylase activity.38 We therefore focused our attention on the emergent candidate DUB, USP15. We first examined the other subcellular protein fractions to ensure that the USP15-dependent change in nucleoplasmic REST reflected reduced abundance rather than relocalization within the cell. REST was detected at comparable levels in the nucleoplasmic and chromatin fractions of A549 cells, where it was decreased to similar degrees by USP15 siRNA (Fig. 1B). This was attributable to reduced protein abundance, as there was no redistribution of REST to the cytoplasm. Interestingly, endogenous USP15 was mainly restricted to the cytoplasmic fraction (Fig. 1B), which is consistent with the observed localization of GFP-tagged USP1539 and suggests that O-glycosylated REST and USP15 largely occupy different cellular compartments.
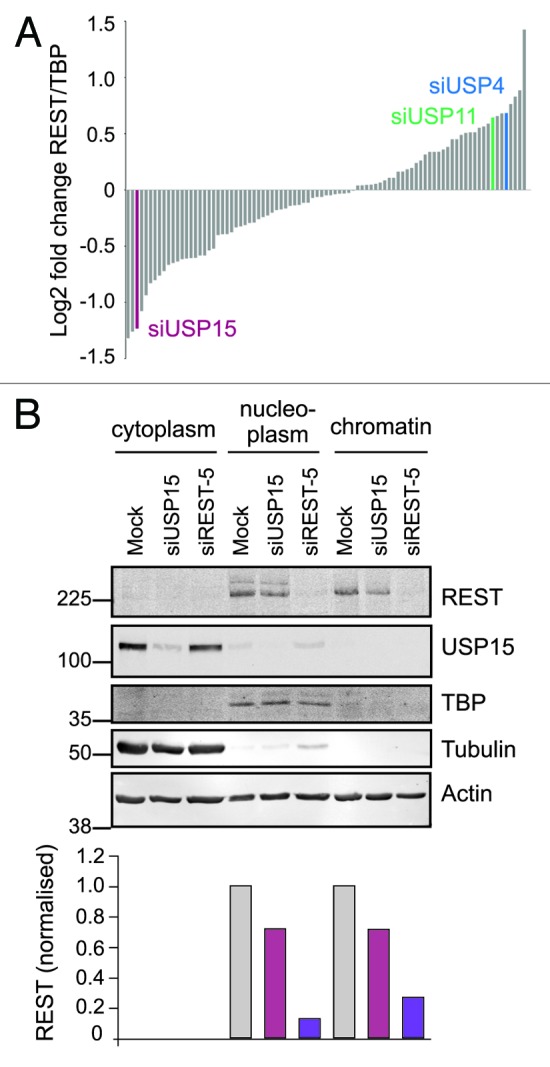
Figure 1. A screen for DUBs that modulate the amount of REST in lung cancer cells identifies USP15. (A) Identification of USP15 from an unbiased DUB siRNA screen. A549 cells were transfected with REST siRNA or a library of pooled siRNA oligos targeting 85 human DUBs. REST levels in the nucleoplasmic fraction were determined 72 h later by immunoblotting and normalized to TATA binding protein (TBP). The candidates for stabilization of REST were USP49, USP47, USP15 and USP52, in contrast to the USP15 paralogs USP4 and USP11. (B). USP15 depletion reduces the amount of REST without influencing its subcellular localization. Following transfection with siRNA targeting REST (siREST-5), pooled siRNAs targeting USP15 or no oligonucleotide (mock), A549 cells were fractionated into cytoplasm, nucleoplasm and a pellet containing chromatin. Extracts were immunoblotted and probed for REST and USP15 (monoclonal); tubulin and TBP were used as cytoplasmic and nucleoplasmic markers, respectively. Quantification of REST normalized to actin, which was detected in each fraction, is shown below.
To negate the possibility that the effect on REST abundance was due to an off-target effect of the USP15 siRNA pool, we examined the consequence of transfection with independent siRNA sequences including three from the original pool (siUSP15-1, -2, -4) and two additional siRNAs (siUSP15-5, -17). Those siRNA oligonucleotides that most efficiently depleted USP15 (Fig. 2B) also decreased REST levels (Fig. 2A). Thus, the USP15 siRNA pool successfully deconvoluted. USP15 is closely related to two other DUBs, USP4 and USP11, with which it shares 71 and 60% similarity, respectively,40 and such close paralogs might exhibit functional redundancy. However, neither USP4 nor USP11 depletion reduced REST in the initial screen; in fact, both these DUBs fell at the opposite end of the ranked data set (Fig. 1A). We further confirmed that REST was specifically targeted by USP15, rather than its paralogs, using independent siRNA pools for USP4 and USP11; neither reduced REST protein levels (Fig. 2A and B). Overall, the amounts of REST and USP15 exhibited a positive correlation across all the samples in these experiments (Fig. 2C) confirming that steady-state REST levels are dependent on the abundance of USP15.
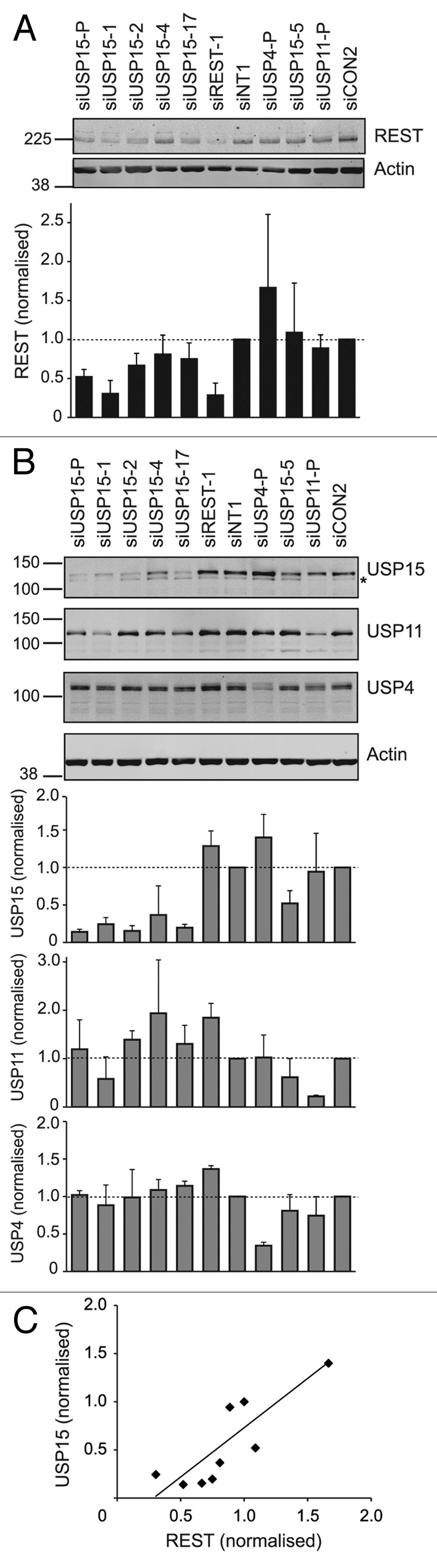
Figure 2. REST abundance is dependent on the amount of cellular USP15. (A) Multiple siRNAs for USP15 but not USP4 or USP11 reduce the amount of REST. siRNA-mediated knockdown was repeated with five individual USP15 oligos and compared with pooled siRNAs targeting USP4 and USP11. After 72 h, proteins were prepared from subcellular fractions and immunoblotted. Representative blots for REST in the nucleoplasmic fraction are shown with mean quantification normalized to actin and the appropriate siRNA control below (n = 3 individual experiments, error bars show standard deviation). (B) Confirmation of specificity for siRNAs targeting the USP15 paralogs. Cytoplasmic protein extracts from the experiments shown in (A) were immunoblotted for USP15 (polyclonal, *indicates cross-reactivity with USP11), USP4 and USP11 to evaluate knockdown efficiency. (C) The amount of REST correlates with USP15 levels. A scatter plot representing the mean quantification of REST (x-axis) plotted against that of USP15 (y-axis) for each of the controls or samples treated with siRNA targeting USP15, USP4 or USP11 in these experiments; linear regression R2 = 0.69.
USP15 can regulate REST protein stability and ubiquitylation
Several DUBs, including USP4 and USP15, interact with mRNA-processing proteins, raising the possibility that USP15 affects REST abundance through altered transcription or splicing.41,42 In fact, we have recently shown that USP15 influences MAP kinase signaling not only through direct deubiquitylation of the KSR E3 ligase BRAP/IMP, but also by affecting CRAF at the mRNA level.43 In the case of REST, the observed decrease in protein abundance was not predicated on a reduction in REST mRNA (Fig. 3A). Although one USP15 targeting sequence did diminish the REST transcript, importantly this was not observed with another individual siRNA that also reduced REST protein abundance, or with the siRNA pool used in the initial screen. We next sought to determine whether ubiquitin dynamics influenced the abundance of REST. Inhibition of the proteasome with epoxomicin could rescue REST levels following USP15 depletion with two independent siRNAs (Fig. 3B, compare lane 5 with 6, and lane 7 with 8), suggesting this was indeed an effect on protein turnover.

Figure 3. USP15 can exert a post-translational effect on REST. (A) The effect of USP15 depletion on REST protein abundance is not predicated on reduction of REST mRNA. A549 cells were transfected with siRNAs as indicated and RNA was prepared 72 h later. REST or USP15 transcripts were quantified relative to β-actin (ACTB) by qRT-PCR; the mean of four independent experiments is shown (bars show standard error). (B) The loss of REST in USP15-depleted cells can be rescued by proteasome inhibition. A549 cells were transfected with siRNA as indicated for 72 h and treated with 50 nM epoxomicin for the final 6 h prior to whole-cell lysis and immunoblotting. Representative blots are shown with the mean fold induction of REST following proteasome inhibition plotted below [siUSP15-1, n = 5, *p = 0.045 (epoxomicin/DMSO); siUSP15-2, n = 3, p = 0.098; bars show standard error]. (C) Catalytically active USP15 can partially rescue levels of nascent REST. HEK-293T cells were transfected with the indicated plasmids 24 h after treatment with USP15 siRNA or control reagents. Whole-cell protein extracts were prepared 48 h later for immunoblotting. The amount of unglycosylated 120 kDa REST or O-glycosylated 220 kDa REST were determined from three independent experiments and normalized to actin; all values are expressed relative to cells transfected with control siRNA and a plasmid expressing GFP alone (lane 1) (n = 3, error bars show standard deviation). (D) Catalytic activity is required for USP15 to reverse ubiquitylation of REST. HEK-293T cells were transfected with the indicated constructs for 48 h and treated with epoxomicin for the last 6 h, before immunoprecipitation with an anti-REST antibody and immunoblotting for myc-tagged ubiquitin. A representative blot from two independent experiments is shown.
To establish whether the deubiquitylase activity of USP15 could rescue REST, we employed HEK-293T cells, which, unlike A549 cells, are amenable to high efficiency plasmid transfection. We detected substantially more of the unmodified 120 kDa form of REST in protein extracts from HEK-293T cells than we did in A549 extracts (Fig. S1), suggesting that they glycosylate REST less fully. We were now able to confirm that USP15 depletion not only altered the abundance of O-glycosylated 220 kDa REST, but also reduced the amount of nascent 120 kDa REST (Fig. 3C, compare lanes 1 and 2). Furthermore, transfection of siRNA-resistant GFP-USP15 into HEK-293T cells that had previously been depleted of endogenous USP15 could rescue 120 kDa REST (Fig. 3C, compare lanes 3 and 4). This was dependent on the deubiquitylase activity of USP15, as REST levels were not affected by GFP-USP15(C269S), which has an inactivating mutation in the catalytic site. However, we noticed that the level of 220 kDa REST was not restored by GFP-USP15, suggesting that the degradation of this mature form was not directly antagonized. Thus, the selective rescue of 120 kDa REST indicated that USP15 might preferentially act on newly synthesized protein.
At this point in our experimental work, it was reported that another DUB, USP7 (also known as HAUSP), could stabilize REST in neural progenitor cells.44 We had not identified USP7 as a candidate DUB for REST in our initial siRNA library screen of lung cancer cells (Fig. S3A). This was born out using a second independent USP7 siRNA pool that neither reduced REST nor potentiated the effect of USP15 knockdown in A549 cells under normal growth conditions (Fig. S3B). We next tested whether USP15 could, like USP7,44 deubiquitylate REST in HEK-293T cells co-transfected with REST, βTrCP and the GFP-tagged DUBs. Immunoprecipitation revealed a smear of polyubiquitylated REST migrating above 120 kDa (Fig. 3D, lane 1), consistent with ubiquitylation of the non-glycosylated species. In agreement with the previous data from Huang and colleagues, REST ubiquitylation was substantially diminished by expression of GFP-USP7 (Fig. 3D, lane 2). Importantly, expression of GFP-USP15 could also abrogate polyubiquitylation of REST (Fig. 3D, lane 3). This effect was contingent on the deubiquitylase activity of USP15, as REST ubiquitylation was unchanged by expression of catalytically inactive GFP-USP15(C269S) (Fig. 3D, lane 4). While we were unable to detect specific ubiquitylation of endogenous REST in our experimental setting, these results suggest that both DUBs are equally capable of rescuing REST from ubiquitylation in a heterologous expression system. Crucially, only USP15, and not USP7, determines endogenous REST levels in A549 lung cancer cells.
USP15 primarily stabilizes newly synthesized REST
To establish whether endogenous USP15 could stabilize cellular REST, we next employed a cycloheximide chase protocol. The half-life of REST in asynchronous A549 cells was approximately 2 h (Fig. 4A), similar to that previously reported in embryonic stem cells.17 Thus, REST is subject to rapid constitutive turnover in lung cancer cells. Depletion of USP15 failed to accelerate the turnover of REST under conditions where translation was inhibited (Fig. 4A). We therefore concluded that USP15 does not oppose the degradation of pre-existing REST. As the earlier experiment in HEK293T cells had revealed selective rescue of 120 kDa REST (Fig. 3C), we wondered whether the primary role of USP15 might be regulating the levels of newly synthesized REST. To test this idea, we inhibited translation with cycloheximide for 4 h to allow the majority of cellular REST to degrade and then monitored the recovery of REST following removal of the translational block (Fig. 4B). Importantly for these experiments, USP15 was markedly more stable than REST in A549 cells (Fig. 4A and B), in keeping with a reported half-life of 47 h in NIH-3T3 cells.45 We found that REST was rapidly resynthesized in control cells, with newly synthesized REST first evident between 30 min and 1 h after reversal of the translational block (Fig. S4). Significantly, the recovery of REST was impaired in cells depleted of USP15, with a marked reduction in the accumulation of newly synthesized 220 kDa REST, which recovered at only 60% of the rate for control cells (Fig. 4B). On resumption of translation, we observed that newly synthesized 220 kDa REST accumulates with similar kinetics to 120 kDa REST (Fig. 4D; Fig. S4). This would be consistent with rapid glycosylation of translated REST. Recovery of both forms of REST was similarly retarded in USP15-depleted cells. Although USP15 activity therefore appears to be primarily directed toward nascent REST, USP15 depletion did not exert a generic effect on translation, as re-synthesis of the transcription factor NRF2 was not inhibited (Fig. 4C).

Figure 4. USP15 primarily stabilizes newly synthesized REST. (A) USP15 depletion does not accelerate REST turnover in the absence of protein synthesis. A549 cells were transfected with siRNA as indicated and 68 h later cycloheximide (CHX) was added. Cells were sampled over 4 h, and whole-cell protein extracts were analyzed by immunoblotting. Quantification of REST normalized to actin is shown below for three independent experiments; the half-life under control conditions is indicated by the dotted line. Mean values for REST are normalized to the level before CHX treatment; error bars show standard deviation (siCON2: R2 = 0.926, gradient -0.208; siUSP15-P: R2 = 0.934, gradient -0.201). (B) USP15 depletion impairs accumulation of newly translated REST. A549 cells were transfected with siRNA, and after 68 h (−4 h) treated with cycloheximide for 4 h, before washing the cells and releasing into fresh medium (0 h). Whole-cell protein extracts were sampled and analyzed by immunoblotting. Quantification of 220 kDa REST normalized to actin is shown below for three independent experiments. Mean values for REST are normalized to the maximum recovery (3 h with siCON2); error bars show standard deviation (siCON2: R2 = 0.996, gradient 0.266; siUSP15-P: R2 = 0.990, gradient 0.159). (C) USP15 depletion does not generically block translational recovery. A549 cells were transfected and treated as in (B); whole-cell extracts were probed for NRF2. (D) USP15 depletion impairs accumulation of both nascent (120 kDa) and glycosylated (220 kDa) REST on cycloheximide washout. A549 cells were transfected with siRNA and treated with cycloheximide and analyzed as described in (B). (E) The effect of USP15 on accumulation of nascent REST is not due to altered REST mRNA. A549 cells were transfected and treated as in (B); RNA was prepared and transcript levels were determined relative to ACTB by QPCR. Data are shown normalized to mock-transfected cells prior to cycloheximide treatment (n = 3 independent experiments, error bars show standard deviation). (F) USP15 exerts a post-translational effect on newly synthesized REST. A549 cells were transfected with siRNA and after 68 h (−4 h) were treated with cycloheximide for 4 h before washing the cells and releasing into fresh medium containing 50 nM epoxomicin. Whole-cell protein extracts were sampled and analyzed by immunoblotting. A non-specific band is indicated by an asterisk (*ns). Quantification of 120 kDa REST normalized to actin is shown below, 3 h into recovery from the translational block in the absence or presence of epoxomicin (n = 3 independent experiments, error bars show standard deviation, *p < 0.05).
REST mRNA did not differ between control and USP15-depleted cells either during the translational block or upon translational recovery, suggesting that the observed requirement for USP15 was post-transcriptional (Fig. 4E). In order to dissect out whether USP15 could stabilize newly synthesized REST by protecting it from degradation, we released cells from the cycloheximide block in the presence of a proteasome inhibitor and optimized lysis conditions to preserve 120 kDa REST. We found that 3 h into the resumption of protein synthesis, epoxomicin treatment significantly rescued the level of 120 kDa REST (Fig. 4F). Taken together, these data suggest that nascent REST may be rapidly turned over by ubiquitylation, and that USP15 might restrict this process.
As our data implicates USP15 in the deubiquitylation of nascent substrates, we set out to establish whether it directly associates with the translational machinery. HEK-293T cell extracts were fractionated by sucrose gradient sedimentation. The ribosome and polysome containing fractions were identified from the characteristic A254nm profile and confirmed by immunoblotting for the ribosome subunit protein RPS6. As expected, the bulk of USP15 fractionated with soluble proteins at the top of the gradient. However, in contrast to the typical monophasic decline of soluble GAPDH along the gradient, we detected a selective enrichment of endogenous USP15 in the region coinciding with the polysomes (Fig. 5A). That the polysome-associated portion represents only a small proportion of the total cellular USP15 pool is consistent with both the pleiotropic cellular functions of USP15, and our observations that not all newly synthesized proteins are dependent on USP15. This experiment was repeated with parallel extracts, one of which was treated with RNase prior to centrifugation to promote polysome disassembly and release free 80S ribosomes (Fig. 5B). This caused a quantitative shift of USP15 away from the denser fractions toward those containing the 80S ribosomes. We conclude that USP15 can associate with the translational machinery and thus may be poised to antagonize co-translational ubiquitylation of some of its substrates.

Figure 5. A small fraction of endogenous USP15 associates with polysomes. (A) USP15 co-fractionates with polysomes. HEK-293T cells were subject to gradient density centrifugation and fractions immunoblotted for the small ribosomal subunit protein RPS6, the soluble protein GAPDH and USP15. The A254nm profile was used to determine the fractions containing polysomes, free ribosomes and ribosomal subunits. (B) Dissociation of polysomes to 80S ribosomes shifts USP15 but not GAPDH within the density gradient. The experiment in (A) was repeated including a replicate sample that was treated with RNase for 1 min prior to gradient density centrifugation.
USP15 depletion opposes REST accumulation during the cell cycle
We next turned our attention to the physiological requirement for the activity of USP15 toward nascent REST. It is known that REST undergoes phosphorylation-dependent polyubiquitylation and degradation during the cell cycle,16 and we wondered whether USP15 played a role in controlling REST periodicity. To establish the dynamics of REST expression in cycling lung cancer cells, A549 were synchronized by double-thymidine block, and protein abundance was followed on release from G1/S into media with or without nocodazole. 220 kDa REST accumulated as cells progressed through G2, then, as previously described,16 became phosphorylated and was subsequently degraded during pro-metaphase arrest with nocodazole (Fig. 6A and B). Again oscillations in 120 kDa REST (data not shown) mapped exactly to those of 220 kDa REST, which was more robustly detected and was followed in subsequent experiments. We noticed that USP15 levels accumulate along with REST during G2; however, prometaphase arrest disengaged their correlation (Fig. 6A) co-incident with the appearance of a phosphorylated form of USP15 that persisted during mitosis (Fig. 6C). To follow the recovery of REST levels after mitosis, we monitored detached prometaphase cells on release from a nocodazole block. The cells exclusively expressed phosphorylated REST that was rapidly degraded as they underwent mitosis. Importantly, non-phosphorylated REST promptly re-accumulated following mitotic exit, corresponding with the dephosphorylation of USP15 (Fig. 6D). Thus, REST is stabilized during G2 and is only transiently degraded at the onset of mitosis before being rapidly replenished in early G1.
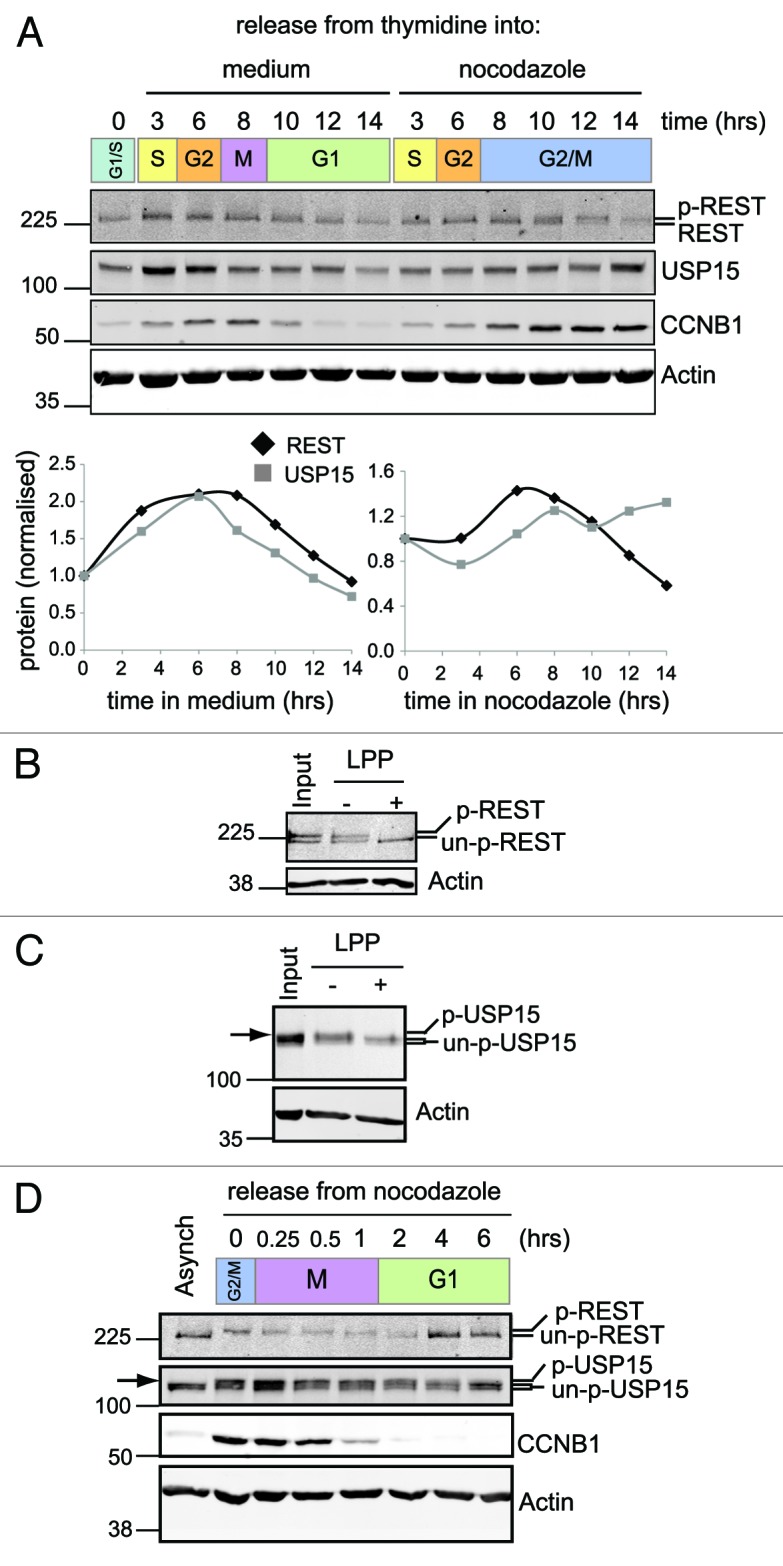
Figure 6. REST is degraded at mitosis and resynthesised as cells enter G1. (A) REST accumulates on release from G1/S arrest and degrades in cells arrested at G2/M. A549 cells were synchronized with thymidine and released from G1/S into media (left) or media containing nocodazole (right). Protein expression was monitored by immunoblotting over 14 h. Total REST and USP15 were quantified from duplicate gels, normalized to actin and plotted relative to the level before release from the thymidine block. (B and C) Phosphorylated forms of both REST (B) and USP15 (C) accumulate during prometaphase arrest. Thymidine synchronized A549 cells were released into nocodazole for 12 h and lysed with E1A buffer. Protein extract was incubated in buffer alone (−) or with lambda protein phosphatase (LPP, +) prior to immunoblotting for REST or USP15; untreated extract (input) is shown for comparison. P-USP15 is indicated with an arrow, above the USP15 doublet seen in asynchronous cells. (D) Phosphorylated REST degrades at mitosis and unphosphorylated REST accumulates as cells enter G1. A549 cells were synchronized with thymidine and then arrested in nocodazole for 14 h before shaking off the mitotic cells and releasing them from G2/M arrest (0 h) into fresh medium. Protein expression was monitored by immunoblotting.
To test whether these correlations signify a role for USP15 in accumulating newly synthesized REST during G2 and in early G1, we transfected cells with USP15 siRNA immediately prior to their synchronization. On release of USP15-depleted cells from G1/S arrest into media containing nocodazole, the accumulation of REST during G2 was attenuated, but its degradation during prometaphase arrest was unaffected (Fig. S5). In detached prometaphase cells depleted of USP15, the degradation of phosphorylated REST during mitosis also proceeded as normal. Therefore, USP15 does not antagonize βTrCP at mitosis. Importantly, however, the reappearance of REST in G1 was substantially impaired in USP15-depleted cells (Fig. 7A). This failure to re-accumulate REST in early G1 was highly reproducible (Fig. 7B) and was replicated by independent USP15 siRNA sequences (Fig. 7C). Thus, in the context of the cell cycle, USP15 does not oppose the rapid degradation of phosphorylated REST that occurs at mitosis, but rather promotes accumulation of REST during G2 ahead of mitosis and again on entry into G1 after mitotic exit (Fig. 7D).

Figure 7. USP15 is required for the recovery of REST levels on mitotic exit. (A) USP15 depletion does not prevent degradation of phosphorylated REST at mitosis but limits its recovery. A549 were transfected with siRNA prior to thymidine synchronization and then arrested in nocodazole for 14 h before shaking off the mitotic cells and releasing from G2/M arrest (0 h). Protein expression was monitored for 4 h, and a representative immunoblot is shown. (B) USP15 depletion impairs recovery of REST at mitotic exit. Cells were treated as described in (A) and extracts collected for immunoblotting before release from G2/M (0 h) or in early G1 (4 h). Mean quantification of REST normalized to actin is plotted, relative to that in asynchronous cells, for four independent experiments (bars show standard error, **p = 0.0005). (C) Individual USP15 siRNAs recapitulate the block on REST accumulation in early G1. Cells were transfected with siRNAs as indicated, then treated and extracts collected as in (B). A representative immunoblot from two independent experiments is shown at 4 h post-release from nocodazole. (D) A model for REST regulation by USP15. USP15 does not oppose the phosphorylation-dependent eradication of REST at mitosis (M). Instead, during G1 and G2, USP15 counteracts ubiquitylation of newly synthesized REST, allowing its levels to accumulate.
Taken together, our data support a model where USP15 primarily counteracts degradation of newly synthesized REST. One important physiological role for USP15 is in ensuring that REST levels are efficiently replenished at mitotic exit to maintain cellular homeostasis of REST in actively cycling cells.
Discussion
REST was initially characterized as a transcriptional silencer that prevented the expression of a handful of neuronal genes in non-neuronal cells.13,14 As our appreciation of the diverse palette of gene expression under the contextual control of REST expands, it has become apparent that the cellular environment dictates REST functionality through complex mechanisms. The precise control of REST availability is one facet of this regulation and is in part dictated by reversible ubiquitylation. During the radical cellular transitions of neural differentiation,17 cell division16 and adenoviral infection,15 phosphorylation triggers the acute polyubiquitylation of REST by SCFβTrCP, leading to its degradation. To date, reversal of this process has been demonstrated only for USP7 acting as an antagonist of neural differentiation.44,46 It remained unclear whether such phosphorylation-dependent ubiquitylation was the only mechanism for cellular turnover of REST, or, indeed, whether all REST ubiquitylation could be counteracted by a single DUB.
We identified USP15 from an unbiased siRNA library screen for DUBs that could regulate the normal cellular levels of REST in the absence of any specific stress or stimuli. USP15 appears to exert its effects on REST through influencing protein stability, as we mechanistically excluded both cellular relocalization and altered transcription, while proteasome inhibition could rescue REST levels. Moreover, expression of USP15, but not a catalytically inactive USP15 mutant, is both capable of deubiquitylating REST in a heterologous setting and restoring the synthesis of endogenous REST in HEK-293T cells. Intriguingly, we observed selective rescue of the 120 kDa nascent form of REST by USP15. Given the predominantly cytosolic localization of GFP-USP15,39 USP15 may have preferential access to a transient pool of freshly synthesized REST, rather than the major 220 kDa glycosylated species that resides in the nucleus. Importantly, we established through cycloheximide chase and washout experiments that USP15 preferentially stabilized newly synthesized over pre-existing REST. We conclude that depletion of USP15 reduced the level of nuclear 220 kDa REST in both HEK-293T and A549 cells, not through accelerating its degradation, but rather through diminishing the supply of newly synthesized REST available to replenish its levels. Interestingly, in support of this model, we also find that a fraction of USP15 associates with polysomes.
To our knowledge, this is the first example of a DUB that preferentially targets a nascent protein, perhaps through opposing its co-translational ubiquitylation. The prevalence of the ubiquitylation of newly synthesized proteins has been known for some time and is regarded as a quality control process that handles mistranslated or misfolded proteins.11 A substantial proportion of the proteome requires continued protein synthesis to accumulate in a ubiquitylated form, including NRF2 and HIF1α, both proteins that are known to be modified by ubiquitin in a regulatory manner.12 This raises the possibility that co-translational ubiquitylation not only deals with defective translation, but may also be integral to normal protein synthesis. Furthermore, it could be exploited to tightly regulate the cellular level of certain proteins, such as these inherently unstable transcription factors. Our data would suggest extensive reversible ubiquitylation of newly synthesized REST, and we speculate that this might be employed as a mechanism to control its cellular availability.
Given the half-life of REST it must be continuously turned over in cells in addition to the periodic destruction at mitosis. Although SCFβTrCP executes phosphorylation-induced ubiquitylation of REST, alternative E3s might regulate its constitutive turnover during interphase, perhaps independently of phosphorylation. Continuous synthesis of transcription factors coupled with their tonic ubiquitylation/deubiquitylation would provide a poised system which can rapidly respond to changing cellular requirements. Indeed, regulation by multiple E3s is emerging as a common mechanism to tightly control transcription factor activity.1,47 One example is NRF1, which, like REST, is ubiquitylated in the nucleus by SCFβTrCP; however, the ER-associated E3 ligase HRD1 together with p97/VCP autonomously controls cytoplasmic NRF1 availability.48 Although HRD1 executes quality control through ERAD, both HRD1 and another predominantly ERAD-associated E3 Doa10/MARCH6, reside in the ER-NE membrane and are known to ubiquitylate transcription factors, including P53 and MATα2.49,50 The p97/VCP ortholog cdc48 is also a component of the recently identified yeast ribosomal associated decay (RAD) complex RQ1.51 Intriguingly, USP15 has been implicated in the processing functions of P97/VCP52 through its association with the COP9 signalosome.53,54
As the DUBs are gradually partnered with their cognate substrates, increasingly complex regulatory networks are emerging. In the same way that many substrates are targeted by alternative E3 ligases, it is becoming evident that ubiquitylation of a particular substrate may also be reversed by alternative DUBs. This is perhaps best exemplified by the complex regulation of p53 stability and activity (for a review see ref. 1) and by the monoubiquitylation of histone H2A, which is reversed by a plethora of different DUBs, allowing its activity around certain gene promoters to be titrated in response to specific stimuli (for a review, see ref. 2). Although USP7 was recently identified as a nuclear DUB that could oppose SCFβTrCP-induced degradation of REST in models of induced neural stem cell differentiation,44 we found that, unlike USP15, depletion of USP7 did not influence the abundance of REST in lung cancer cells under normal growth conditions. As a predominantly nuclear DUB, USP7 may access chromatin-associated REST to antagonize SCFβTrCP activity, which, in the case of E1A-induced REST ubiquitylation at least, is entirely restricted to the nuclear compartment.15 In contrast, our data suggest that the major role for USP15, a largely cytoplasmic DUB, is to oppose co-translational degradation of nascent REST, thus determining its cellular availability.
In synchronized cells, we found that REST accumulated during G2, and its phosphorylation-dependent degradation was tightly restricted to mitosis. However, mitotic REST degradation was followed by its rapid replenishment. We reasoned that, as a DUB that could stabilize newly synthesized REST, USP15 might instead play an important role in promoting its recovery at mitotic exit. Interestingly, we observed that a proportion of USP15 became phosphorylated at mitosis, which could conceivably alter its activity toward REST. Indeed, USP15 depletion impaired the normal periodic accumulation of REST in early G1, suggesting that it plays an important role in cellular REST homeostasis. Rather than directly antagonizing the rapid degradation of phosphorylated REST that occurs en masse at mitosis, USP15 acts sequentially with SCFβTrCP (Fig. 7D). Through reversing the ubiquitylation of newly synthesized REST, USP15 allows nuclear REST to be replenished in early G1 after mitotic exit. Thus USP15 joins a list of DUBs that fulfill specific roles at distinct stages of the cell cycle including BAP1,55 CYLD,56 USP2A,57 USP7,58 USP8,59 USP4460,61 and USP50.62
The action of USP15 is a vital component of the oscillatory mechanism for the transient degradation and recovery of REST during the cell cycle, such that USP15 acts to reset interphase REST levels after each round of cell division. Although bulk destruction of REST is reported to release expression of lineage-specific genes like TUBB3 (TUJ1) during neural differentiation44 and both TUBB3 and MAD2 at mitosis,16,63 more subtle changes in the interphase availability of REST may equally impact on transcriptional programs. Transcription is, broadly speaking, shut down during mitosis, and transcription factors are stripped from chromatin as the chromosomes condense,64 which may be the main driver for the acute degradation of REST at mitosis. However, on mitotic exit, nuclei are already competent for transcription by telophase.65 Although lineage determinant transcription factors can retain some residency or leave epigenetic marks to sustain the phenotype of the progeny,66 REST may need to be rapidly replenished in daughter cells to suppress a neuronal transcription program. Thus the correctly coordinated periodicity of REST that is achieved through reversible ubiquitylation will be important for normal cellular physiology. The RE1 REST-binding motif has proven to be surprisingly common and quite diverse, such that the several thousand potential REST target genes18 bear distinct RE1 variants that exhibit differential in vivo binding affinity for REST.67 Changes in the efficiency with which USP15 deubiquitylates nascent REST, and in the cellular availability of REST during interphase, may influence expression of the broader palette of REST target genes.
USP15 is a widely and relatively highly expressed DUB.45,68 It was recently found to be overexpressed in glioblastoma,69 a tumor type where REST can be upregulated and act as an oncoprotein.70,71 USP15 is emerging as a multifunctional DUB that regulates diverse cancer proteins and signaling pathways, including the HPV E6 oncoprotein,72 APC tumor suppressor,53 NFκB inhibitor IκBα,73 pro-apoptotic caspase-3,74 the TGFβ receptor69 and its R-SMAD effectors,75 CRAF and BRAP/IMP.43 To this list we have now added the context-dependent tumor suppressor and oncoprotein REST. Importantly, our results suggest that USP15 specifically targets the synthesis of a subset of proteins, of which REST is one example. Indeed, USP15 was reported to control NFκB activity by supporting the de novo re-accumulation of IκBα, following its signal-induced degradation, through a post-transcriptional mechanism.73 We have recently also shown that USP15 may regulate the availability and competence of CRAF mRNA.43 While we have uncovered different mechanisms for the regulation of REST and CRAF, both would be compatible with our novel discovery that USP15 can associate with polysomes.
In a broader sense, this study has unveiled an as yet unique preference of a DUB for targeting a nascent form of its substrate. There is currently increasing interest in co-translational ubiquitylation, and our observation prompts speculation that this may be a reversible modification that can be antagonized by DUBs. Two elegant studies recently described the extent and role of co-translational ubiquitylation in yeast76 and in human cells, where 12–15% of nascent polypeptides are ubiquitylated.77 Ribosome-associated ubiquitylation may not only promote degradation of misfolded proteins, but could be envisaged to act as a more sophisticated quality control mechanism. Elsewhere, the ubiquitylation of ER-associated proteins has been hypothesized to chaperone their folding on the cytosolic side of the ER.78 In that model, the periodic intervention of DUBs was proposed to enable evaluation of whether the protein is sent for ERAD, exits the ER or enters another round of folding. Similarly, repetitive rounds of co-translational ubiquitylation and deubiquitylation could also act to supervise the correct cytosolic folding of other proteins with complex secondary structures, including transcription factors like REST.
Materials and Methods
Cell culture
A549 and HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% non-essential amino acids at 37°C and 5% CO2. A549 cells were authenticated by 16 loci STR profiling (LGC Standards cell authentication service) on 11/08/2011, and all cells were cultured for limited passage numbers.
Antibodies
Rabbit polyclonal or mouse monoclonal USP15 antibodies were purchased from Bethyl (A300-923A) or Abnova (H00009958-M01), respectively; the monoclonal was specific for USP15 by immunoblotting, whereas the polyclonal also weakly cross-reacted with USP11 (* in Fig. 2B). Other antibodies used were rabbit: anti-REST (07-579, Millipore), anti-TBP (sc-204, Santa Cruz), anti-USP4 (A300-830A, Bethyl), anti-USP11 (A301-613A, Bethyl), anti-USP7 (ab4080, Abcam), anti-NRF2 (ab62352, Abcam), anti-GAPDH (14C10, Cell Signaling), anti-OGT (5368, Cell Signaling) and anti-actin (A2066, Sigma); or mouse: anti-cyclinB1 (05-373, Millipore), anti-β-actin (ab6276, Abcam), anti-α-tubulin (clone B512, Sigma), anti-myc (clone 4A6, Millipore), anti-RPS6 (Santa Cruz) and anti-ubiquitin P4G7 (MMS-258R, Covance). Polyclonal affinity-purified sheep anti-GFP was a gift from Ian Prior.
RNA interference
A custom DUB siRNA library consisting of pools of four siGenome oligos for each of 85 human DUBs was purchased from Dharmacon. Individual siGenome siRNAs were used for follow-up studies: siUSP15-1 (D-006066-01), siUSP15-2 (D-006066-02), siUSP15-4 (D-006066-04), siUSP15-17 (D-006066-17), siREST-1 (CAACGAAUCUACCCAUAUUUU), siREST-5 (CAUCCUACUUGUCCUAAUAUU), together with the controls siCON1 (D-001210-01) and siCON2 (D-001210-02). On-target plus siRNA (Dharmacon) were used as oligo pools against USP4 (L-004974-00), USP11 (L-006063-00) and USP7 (L-006097-00) or as a single oligo siUSP15-5 (J-006066-05) and the control siNT1 (D-001810-01). OGT siRNA (SI00053508) and the All-Stars negative control siRNA (1027281, siC) were purchased from Qiagen. For the siRNA library screen and subsequent experiments, A549 cells were seeded at 6 × 104 cells per well in 6-well plates and transfected the following day with 50 nM siRNA using Oligofectamine (Invitrogen); cells were analyzed 72 h later.
Plasmid DNA constructs
USP15 (NM_006313) and USP7 (NM_003470.2) were cloned using the Gateway system into pDONR233 entry constructs, sequence verified and shuttled into expression vectors as previously described.39 Catalytically inactive, siRNA-resistant or catalytically inactive/siRNA-resistant forms of USP15 were generated by Quickchange site-directed mutagenesis in pDONR233 using complementary primer pairs, forward primers: USP15(C296S) 788-GTAACTTGGGAAATACGAGTTTCATGAACTCAGC, or USP15-siRes1 1858-GCATACATGAAGAAGGGAGCCCAAGTGAAATGG. The RE-EX1 plasmid expressing full-length untagged REST was a gift from Gail Mandel (Howard Hughes Medical Institute, USA). FLAG-tagged βTrCP1 was a gift from Ger Strous (Utrecht). The Myc-Ubiquitin plasmid was a gift from John O’Bryan (University of Illinois).
Cell lysis and immunoblotting
Sequential buffers were used to prepare fractionated protein extracts. The cytoplasmic fraction was obtained by lysis in ice-cold NP-40 buffer (0.5% NP-40, 25 mM Tris pH7.9, 100 mM NaCl). A nucleoplasm enriched fraction was obtained from the NP40-insoluble pellet by incubation at 4°C in Dignam buffer C (20 mM HEPES pH7.9, 25% glycerol, 0.42 mM NaCl, 1.5 mM MgCl, 0.2 mM EDTA) for 40 min with vortexing at 2,000 rpm. Following centrifugation, the insoluble pellet containing chromatin was extracted in Laemmli buffer (50 mM Tris pH6.8, 2% SDS, 10% glycerol) at 110°C for 20 min with intermittent vortexing. Alternatively, whole-cell extracts were prepared by direct addition to cells of hot Laemmli buffer or hot 2% SDS buffer (2% SDS, 1 mM EDTA, 50 mM NaF), and incubation at 110°C for 10 min with intermittent vortexing. The protein concentration of each sample was determined after suitable dilution using a Bicinchoninic Acid (BCA) assay (Thermo Scientific). Equal amounts of lysates for comparator samples were subject to immunoblotting. For fractionated extracts, we loaded 10 μg of cytoplasmic extract and 3 μg of the nuclear or chromatin fractions; this typically represented a 1:5:5 ratio based on cell number. Following resolution by SDS-PAGE, proteins were transferred to BiotraceNT membrane (VWR) and incubated with primary antibodies. Proteins were visualized using donkey anti-mouse, anti-rabbit or anti-sheep secondary antibodies conjugated to the IRDyes IR680, IR680-LT or IR800 (LI-COR) and the LI-COR Odyssey 2.1 system; 16-bit images were analyzed and quantified using the Odyssey analysis software.
RNA extraction and real-time PCR
Total RNA was extracted using RNeasy columns (Qiagen), and cDNA was reverse transcribed from 1 µg RNA with RevertAid H-minus M-MuLV reverse transcriptase (Fermentas) using an oligo-dT primer (Promega). Quantitative real-time RT-PCR (qRT-PCR) was performed in triplicate using SYBR Green supermix and an IQ5 real-time PCR detection system (Bio-Rad). Primer sequences were: ACTB (For: 5′-CACCTTCTACAATGAGCTGCGTGTG-3′, Rev: 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′), REST (For: 5′-GCATGTTAGAACTCATACAGG-3′, Rev: 5′-TTCTCACCTGAATGAGTACG-3′) to detect full-length REST, and USP15 (For: 5′-CAGACAGCACCATTCAGGATGC-3′, Rev: 5′-GAGTTTTTCACATTAGGAGTAG-3′) to detect all USP15 variants. Samples underwent two-step amplification at 94°C (30 sec) and 60°C (60 sec); melt curves were analyzed after 40 cycles. The Ct values for test genes were normalized to ACTB and relative expression represented as 2−[ΔΔCt].
DUB siRNA library screen
A549 cells in 6-well plates were transfected with the custom siRNA library representing 85 human DUBs (Dharmacon). Cellular protein fractions were prepared 72 h later, and 3 μg of the nucleoplasmic extract was subject to SDS-PAGE. Samples were split between four 8% gels that were run and processed in parallel, each included extracts from mock (no oligo) and siREST-5-transfected cells. Following immunoblotting for REST, actin and TBP, immunoreactive bands were quantified using the Odyssey system. The amount of REST was expressed relative to the amount of TBP to standardize between samples. The value for each sample was then normalized to the mean value for the corresponding immunoblot, before collation and ranking of data across the four gels.
Rescue experiments
HEK-293T were seeded at 9 × 104 per well in 6-well plates and transfected the following day with 50 nM siRNA using Oligofectamine (Invitrogen). After 24 h, the cells were transfected with 1 µg of plasmid using GeneJuice (Novagen). The cells were lysed 48 h later in pre-heated 2% SDS buffer.
Immunoprecipitation
HEK-293T cells were seeded at a density of 3 × 105 cells in 6 cm dishes and transfected with 5 μg of plasmid mixtures using GeneJuice (Novagen) for 48 h. Fifty nM epoxomicin (Calbiochem) was added to the cells for the last 6 h before cell lysis in pre-heated 2% SDS buffer. One mg of lysate was diluted in 4 volumes of dilution buffer (2.5% Triton X-100, 12.5 mM Tris pH 7.5, 187.5 mM NaCl with protease and phosphatase inhibitors). Samples were precleared by centrifugation at 4°C, and the supernatants subject to overnight immunoprecipitation at 4°C with 1 µg REST antibody (Millipore 07-579) and protein A agarose beads (Sigma). Immunoprecipitates were washed three times with wash buffer (2% Triton X-100, 0.4% SDS, 10 mM Tris pH7.5, 150 mM NaCl) and then once with 10 mM Tris pH7.5 before preparation for SDS-PAGE.
Inhibition of protein synthesis, proteasomal degradation and O-glycosylation
The OGT inhibitor 6-diazo-5-oxo-L-norleucine (DON, 100 μM) or the OGT agonist (PuGNaC, 100 μM) (both from Sigma) were added to cells in complete media for 15 h to assay the effects on steady-state REST levels. For protein turnover and synthesis experiments, A549 or HEK293T cells were treated with control reagents or siRNA for 68 h before incubation with 10 μg/ml cycloheximide (Sigma). Protein turnover was monitored over a 4-h cycloheximide chase. Alternatively, after 4 h of cycloheximide treatment, cells were released from the translational block by washing three times in PBS and the addition of full media; protein synthesis was then monitored over 3 h. For analysis of newly synthesized protein while the proteasome was inhibited, 50 nM epoxomicin was added at the time of cycloheximide removal. Cells were lysed in Laemmli buffer and processed for immunoblotting.
Polysome fractionation
HEK-293T cells were treated with cycloheximide for 30 min prior to harvesting in polysome lysis buffer containing 200 μg/ml heparin to inhibit RNase, as described by Durfee et al.79 Briefly, lysates were centrifuged at 16,300 × g and 4°C for 10 min, before the supernatants were loaded onto 7–47% w/v sucrose gradients containing 200 μg/ml cycloheximide. For RNase treatment, the heparin was omitted from the lysis buffer and RNase was added to a final concentration of 1 μg/ml for 1 min prior to application onto the sucrose gradient. Gradients were centrifuged at 222,000 × g for 90 min at 4°C in a Beckman SW41Ti rotor. Fractions were collected from each gradient and the A254nm profile determined. TCA (10% final concentration) was added to each 1 ml fraction, and the precipitated proteins were analyzed by immunoblotting.
Cell synchronization
Three protocols were employed for synchronization of A549 cells, and, where required, cells were transfected with siRNA 6 h earlier. To follow cells progressing from G1/S through mitosis, cells were subject to a double thymidine block (2 mM thymidine (Sigma) for 18 h, release into fresh media for 8 h, arrest with thymidine for 17 h) then released into full medium and monitored over 14 h (49–63 h post-siRNA transfection). To analyze cells arrested in prometaphase, a double thymidine block was performed before cells were released into full medium containing 100 ng/ml nocodazole (Sigma) and then monitored over 14 h (49–63 h post-siRNA transfection). To analyze cells exiting G2/M arrest and progressing through mitosis, cells were synchronized with double thymidine and released into nocodazole for 14 h; mitotic cells were collected by knocking off the dish and replating in full medium; cells were then monitored for up to 6 h (63–69 h post-siRNA transfection). For analysis of each timepoint, adherent cells were trypsinised, pooled with non-adherent cells collected by centrifugation from the medium and then lysed in pre-heated Laemmli buffer.
Phospho-protein analysis
A549 cells were synchronized in thymidine and held in nocodazole for 12 h, prior to lysis in E1A buffer (50 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES) pH 7.5, 250 mM NaCl, 0.1% Triton X-100) at 4°C for 10 min with intermittent vortexing. Forty-five µg of lysate was incubated for 30 min at 30°C, either with 400 U lambda protein phosphatase (New England Biolabs) or without the enzyme, before inactivation with 5 mM of EDTA and incubation for 1 h at 65°C. Equivalent proportions of each reaction were analyzed by 7% SDS-PAGE and immunoblotting.
Supplementary Material
Acknowledgments
This project was funded by a North West Cancer Research Fund grant (ref: CR850), L.D. was funded by a Wellcome Trust Vacation Scholarship, and S.D. is funded by a Wellcome Trust Prize studentship. S.U. is the recipient of a Cancer Research UK Senior Research Fellowship. We thank Larissa Durfee and Jon Huibregste for advice on polysome fractionation protocols, and Gail Mandel, Ger Strous and John O’Bryan for constructs.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
L.D. and J.M.C. performed the siRNA library screen. S.D. performed comparison of USP7 with USP15 and glycosylation analysis. V.P. performed polysome association and translational recovery experiments. M.F. performed all other experiments and linked USP15 to stabilization of new protein during the cell cycle. J.M.C. wrote the paper and directed the work with input from S.U. and M.C. All authors discussed the work, analyzed the data and commented on the manuscript.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25035
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25035
References
- 1.Hammond-Martel I, Yu H, Affar B. Roles of ubiquitin signaling in transcription regulation. Cell Signal. 2012;24:410–21. doi: 10.1016/j.cellsig.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Clague MJ, Coulson JM, Urbé S. Cellular functions of the DUBs. J Cell Sci. 2012;125:277–86. doi: 10.1242/jcs.090985. [DOI] [PubMed] [Google Scholar]
- 3.Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. 2011;12:295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 5.Sacco JJ, Coulson JM, Clague MJ, Urbé S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life. 2010;62:140–57. doi: 10.1002/iub.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, et al. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One. 2011;6:e15891. doi: 10.1371/journal.pone.0015891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fulda S, Rajalingam K, Dikic I. Ubiquitylation in immune disorders and cancer: from molecular mechanisms to therapeutic implications. EMBO Mol Med. 2012;4:545–56. doi: 10.1002/emmm.201100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussain S, Zhang Y, Galardy PJ. DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle. 2009;8:1688–97. doi: 10.4161/cc.8.11.8739. [DOI] [PubMed] [Google Scholar]
- 9.Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–5. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 10.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–90. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–4. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 12.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–40. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chong JA, Tapia-Ramírez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–57. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 14.Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–3. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 15.Guan H, Ricciardi RP. Transformation by E1A oncoprotein involves ubiquitin-mediated proteolysis of the neuronal and tumor repressor REST in the nucleus. J Virol. 2012;86:5594–602. doi: 10.1128/JVI.06811-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, et al. Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature. 2008;452:365–9. doi: 10.1038/nature06641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–4. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto SJ, McCorkle SR, Hover J, Conaco C, Han JJ, Impey S, et al. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J Neurosci. 2007;27:6729–39. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 20.Gopalakrishnan V. REST and the RESTless: in stem cells and beyond. Future Neurol. 2009;4:317–29. doi: 10.2217/fnl.09.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bithell A. REST: transcriptional and epigenetic regulator. Epigenomics. 2011;3:47–58. doi: 10.2217/epi.10.76. [DOI] [PubMed] [Google Scholar]
- 22.Gao Z, Ding P, Hsieh J. Profiling of REST-Dependent microRNAs Reveals Dynamic Modes of Expression. Front Neurosci. 2012;6:67. doi: 10.3389/fnins.2012.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coulson JM. Transcriptional regulation: cancer, neurons and the REST. Curr Biol. 2005;15:R665–8. doi: 10.1016/j.cub.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 24.Faronato M, Coulson JM. REST (RE1 silencing transcription factor) Atlas Genet Cytogenet Oncol Haematol. 2011;15:208–13. [Google Scholar]
- 25.Majumder S. REST in good times and bad: roles in tumor suppressor and oncogenic activities. Cell Cycle. 2006;5:1929–35. doi: 10.4161/cc.5.17.2982. [DOI] [PubMed] [Google Scholar]
- 26.Coulson JM, Edgson JL, Woll PJ, Quinn JP. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: a potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000;60:1840–4. [PubMed] [Google Scholar]
- 27.Gurrola-Diaz C, Lacroix J, Dihlmann S, Becker CM, von Knebel Doeberitz M. Reduced expression of the neuron restrictive silencer factor permits transcription of glycine receptor alpha1 subunit in small-cell lung cancer cells. Oncogene. 2003;22:5636–45. doi: 10.1038/sj.onc.1206790. [DOI] [PubMed] [Google Scholar]
- 28.Coulson JM, Fiskerstrand CE, Woll PJ, Quinn JP. Arginine vasopressin promoter regulation is mediated by a neuron-restrictive silencer element in small cell lung cancer. Cancer Res. 1999;59:5123–7. [PubMed] [Google Scholar]
- 29.Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–48. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 30.Lawinger P, Venugopal R, Guo ZS, Immaneni A, Sengupta D, Lu W, et al. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat Med. 2000;6:826–31. doi: 10.1038/77565. [DOI] [PubMed] [Google Scholar]
- 31.Su X, Kameoka S, Lentz S, Majumder S. Activation of REST/NRSF target genes in neural stem cells is sufficient to cause neuronal differentiation. Mol Cell Biol. 2004;24:8018–25. doi: 10.1128/MCB.24.18.8018-8025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuller GN, Su X, Price RE, Cohen ZR, Lang FF, Sawaya R, et al. Many human medulloblastoma tumors overexpress repressor element-1 silencing transcription (REST)/neuron-restrictive silencer factor, which can be functionally countered by REST-VP16. Mol Cancer Ther. 2005;4:343–9. doi: 10.1158/1535-7163.MCT-04-0228. [DOI] [PubMed] [Google Scholar]
- 33.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–57. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Moss AC, Jacobson GM, Walker LE, Blake NW, Marshall E, Coulson JM. SCG3 transcript in peripheral blood is a prognostic biomarker for REST-deficient small cell lung cancer. Clin Cancer Res. 2009;15:274–83. doi: 10.1158/1078-0432.CCR-08-1163. [DOI] [PubMed] [Google Scholar]
- 35.Kreisler A, Strissel PL, Strick R, Neumann SB, Schumacher U, Becker CM. Regulation of the NRSF/REST gene by methylation and CREB affects the cellular phenotype of small-cell lung cancer. Oncogene. 2010;29:5828–38. doi: 10.1038/onc.2010.321. [DOI] [PubMed] [Google Scholar]
- 36.Pance A, Livesey FJ, Jackson AP. A role for the transcriptional repressor REST in maintaining the phenotype of neurosecretory-deficient PC12 cells. J Neurochem. 2006;99:1435–44. doi: 10.1111/j.1471-4159.2006.04190.x. [DOI] [PubMed] [Google Scholar]
- 37.Lee JH, Shimojo M, Chai YG, Hersh LB. Studies on the interaction of REST4 with the cholinergic repressor element-1/neuron restrictive silencer element. Brain Res Mol Brain Res. 2000;80:88–98. doi: 10.1016/S0169-328X(00)00129-7. [DOI] [PubMed] [Google Scholar]
- 38.Ye Y, Scheel H, Hofmann K, Komander D. Dissection of USP catalytic domains reveals five common insertion points. Mol Biosyst. 2009;5:1797–808. doi: 10.1039/b907669g. [DOI] [PubMed] [Google Scholar]
- 39.Urbé S, Liu H, Hayes SD, Heride C, Rigden DJ, Clague MJ. Systematic survey of deubiquitinase localization identifies USP21 as a regulator of centrosome- and microtubule-associated functions. Mol Biol Cell. 2012;23:1095–103. doi: 10.1091/mbc.E11-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faronato M, Urbé S, Coulson JM. USP15 (ubiquitin specific peptidase 15) Atlas Genet Cytogenet Oncol Haematol. 2011;15:645–51. [Google Scholar]
- 41.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song EJ, Werner SL, Neubauer J, Stegmeier F, Aspden J, Rio D, et al. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010;24:1434–47. doi: 10.1101/gad.1925010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes SD, Liu H, MacDonald E, Sanderson CM, Coulson JM, Clague MJ, et al. Direct and indirect control of mitogen-activated protein kinase pathway-associated components, BRAP/IMP E3 ubiquitin ligase and CRAF/RAF1 kinase, by the deubiquitylating enzyme USP15. J Biol Chem. 2012;287:43007–18. doi: 10.1074/jbc.M112.386938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Z, Wu Q, Guryanova OA, Cheng L, Shou W, Rich JN, et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol. 2011;13:142–52. doi: 10.1038/ncb2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 46.Huang Z, Zhou W, Bao S. Role of deubiquitylase HAUSP in stem cell maintenance. Cell Cycle. 2011;10:1182–3. doi: 10.4161/cc.10.8.15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubenstein EM, Hochstrasser M. Redundancy and variation in the ubiquitin-mediated proteolytic targeting of a transcription factor. Cell Cycle. 2010;9:4282–5. doi: 10.4161/cc.9.21.13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsuchiya Y, Morita T, Kim M, Iemura S, Natsume T, Yamamoto M, et al. Dual regulation of the transcriptional activity of Nrf1 by β-TrCP- and Hrd1-dependent degradation mechanisms. Mol Cell Biol. 2011;31:4500–12. doi: 10.1128/MCB.05663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamasaki S, Yagishita N, Sasaki T, Nakazawa M, Kato Y, Yamadera T, et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007;26:113–22. doi: 10.1038/sj.emboj.7601490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–43. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–54. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cayli S, Klug J, Chapiro J, Fröhlich S, Krasteva G, Orel L, et al. COP9 signalosome interacts ATP-dependently with p97/valosin-containing protein (VCP) and controls the ubiquitination status of proteins bound to p97/VCP. J Biol Chem. 2009;284:34944–53. doi: 10.1074/jbc.M109.037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang X, Langelotz C, Hetfeld-Pechoc BK, Schwenk W, Dubiel W. The COP9 signalosome mediates beta-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J Mol Biol. 2009;391:691–702. doi: 10.1016/j.jmb.2009.06.066. [DOI] [PubMed] [Google Scholar]
- 54.Hetfeld BK, Helfrich A, Kapelari B, Scheel H, Hofmann K, Guterman A, et al. The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr Biol. 2005;15:1217–21. doi: 10.1016/j.cub.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 55.Eletr ZM, Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys. 2011;60:3–11. doi: 10.1007/s12013-011-9184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez-Ferreria MA, Bashkurov M, Mullin M, Gingras AC, Pelletier L. CEP192 interacts physically and functionally with the K63-deubiquitinase CYLD to promote mitotic spindle assembly. Cell Cycle. 2012;11:3555–8. doi: 10.4161/cc.21574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim J, Kim WJ, Liu Z, Loda M, Freeman MR. The ubiquitin-specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle. 2012;11:1123–30. doi: 10.4161/cc.11.6.19550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giovinazzi S, Morozov VM, Summers MK, Reinhold WC, Ishov AM. USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death Differ. 2013;20:721–31. doi: 10.1038/cdd.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meijer IM, van Leeuwen JE. ERBB2 is a target for USP8-mediated deubiquitination. Cell Signal. 2011;23:458–67. doi: 10.1016/j.cellsig.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, et al. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest. 2012;122:4362–74. doi: 10.1172/JCI63084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–81. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- 62.Aressy B, Jullien D, Cazales M, Marcellin M, Bugler B, Burlet-Schiltz O, et al. A screen for deubiquitinating enzymes involved in the G₂/M checkpoint identifies USP50 as a regulator of HSP90-dependent Wee1 stability. Cell Cycle. 2010;9:3815–22. doi: 10.4161/cc.9.18.13133. [DOI] [PubMed] [Google Scholar]
- 63.Shibazaki M, Maesawa C, Akasaka K, Kasai S, Yasuhira S, Kanno K, et al. Transcriptional and post-transcriptional regulation of βIII-tubulin protein expression in relation with cell cycle-dependent regulation of tumor cells. Int J Oncol. 2012;40:695–702. doi: 10.3892/ijo.2011.1291. [DOI] [PubMed] [Google Scholar]
- 64.Martínez-Balbás MA, Dey A, Rabindran SK, Ozato K, Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 65.Prasanth KV, Sacco-Bubulya PA, Prasanth SG, Spector DL. Sequential entry of components of the gene expression machinery into daughter nuclei. Mol Biol Cell. 2003;14:1043–57. doi: 10.1091/mbc.E02-10-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zaidi SK, Young DW, Montecino M, Lian JB, Stein JL, van Wijnen AJ, et al. Architectural epigenetics: mitotic retention of mammalian transcriptional regulatory information. Mol Cell Biol. 2010;30:4758–66. doi: 10.1128/MCB.00646-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bruce AW, López-Contreras AJ, Flicek P, Down TA, Dhami P, Dillon SC, et al. Functional diversity for REST (NRSF) is defined by in vivo binding affinity hierarchies at the DNA sequence level. Genome Res. 2009;19:994–1005. doi: 10.1101/gr.089086.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baker RT, Wang XW, Woollatt E, White JA, Sutherland GR. Identification, functional characterization, and chromosomal localization of USP15, a novel human ubiquitin-specific protease related to the UNP oncoprotein, and a systematic nomenclature for human ubiquitin-specific proteases. Genomics. 1999;59:264–74. doi: 10.1006/geno.1999.5879. [DOI] [PubMed] [Google Scholar]
- 69.Eichhorn PJ, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med. 2012;18:429–35. doi: 10.1038/nm.2619. [DOI] [PubMed] [Google Scholar]
- 70.Kamal MM, Sathyan P, Singh SK, Zinn PO, Marisetty AL, Liang S, et al. REST regulates oncogenic properties of glioblastoma stem cells. Stem Cells. 2012;30:405–14. doi: 10.1002/stem.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blom T, Tynninen O, Puputti M, Halonen M, Paetau A, Haapasalo H, et al. Molecular genetic analysis of the REST/NRSF gene in nervous system tumors. Acta Neuropathol. 2006;112:483–90. doi: 10.1007/s00401-006-0102-8. [DOI] [PubMed] [Google Scholar]
- 72.Vos RM, Altreuter J, White EA, Howley PM. The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. J Virol. 2009;83:8885–92. doi: 10.1128/JVI.00605-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NFkappaB by deubiquitinylation of IkappaBalpha. EMBO J. 2007;26:1532–41. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu M, Takanashi M, Oikawa K, Tanaka M, Nishi H, Isaka K, et al. USP15 plays an essential role for caspase-3 activation during Paclitaxel-induced apoptosis. Biochem Biophys Res Commun. 2009;388:366–71. doi: 10.1016/j.bbrc.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 75.Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. 2011;13:1368–75. doi: 10.1038/ncb2346. [DOI] [PubMed] [Google Scholar]
- 76.Duttler S, Pechmann S, Frydman J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol Cell. 2013;50:379–93. doi: 10.1016/j.molcel.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang F, Durfee LA, Huibregtse JM. A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol Cell. 2013;50:368–78. doi: 10.1016/j.molcel.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feldman M, van der Goot FG. Novel ubiquitin-dependent quality control in the endoplasmic reticulum. Trends Cell Biol. 2009;19:357–63. doi: 10.1016/j.tcb.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 79.Durfee LA, Huibregtse JM. The ISG15 conjugation system. Methods Mol Biol. 2012;832:141–9. doi: 10.1007/978-1-61779-474-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.