

Abstract
Microvascular barrier dysfunction is a serious problem that occurs in many inflammatory conditions, including sepsis, trauma, ischemia–reperfusion injury, cardiovascular disease, and diabetes. Barrier dysfunction permits extravasation of serum components into the surrounding tissue, leading to edema formation and organ failure. The basis for microvascular barrier dysfunction is hyperpermeability at endothelial cell–cell junctions. Endothelial hyperpermeability is increased by actomyosin contractile activity in response to phosphorylation of myosin light chain by myosin light chain kinase (MLCK). MLCK-dependent endothelial hyperpermeability occurs in response to inflammatory mediators (e.g., activated neutrophils, thrombin, histamine, tumor necrosis factor alpha, etc.), through multiple cell signaling pathways and signaling molecules (e.g., Ca++, protein kinase C, Src kinase, nitric oxide synthase, etc.). Other signaling molecules protect against MLCK-dependent hyperpermeability (e.g., sphingosine-1-phosphate or cAMP). In addition, individual MLCK isoforms play specific roles in endothelial barrier dysfunction, suggesting that isoform-specific inhibitors could be useful for treating inflammatory disorders and preventing multiple organ failure. Because endothelial barrier dysfunction depends upon signaling through MLCK in many instances, MLCK-dependent signaling comprises multiple potential therapeutic targets for preventing edema formation and multiple organ failure. The following review is a discussion of MLCK-dependent mechanisms and cell signaling events that mediate endothelial hyperpermeability.
Keywords: myosin light chain kinase (MLCK), protein kinases, edema, endothelial barrier dysfunction, microvascular permeability
1. INTRODUCTION
The microvascular endothelium is a protective barrier that controls the exchange of fluid and plasma components between blood and biological tissues (for review see1, 2). Under inflammatory conditions, or upon exposure to inflammatory agents (e.g., histamine, thrombin, etc.), microvascular endothelial (micro-VE) barriers are compromised, leading to tissue edema, decreased perfusion, and multiple organ failure. Micro-VE barrier dysfunction permits plasma components and blood cells (e.g., neutrophils) to infiltrate surrounding tissues. This occurs in many adverse conditions, including acute lung injury (ALI), cardiovascular disease/atherosclerosis, dermal burn injury, ischemia–reperfusion (I/R), and traumatic brain injury (TBI), affecting the lungs, heart, gut, brain, or other tissues.
Endothelial barrier dysfunction is triggered by misregulation of intracellular signaling events that normally maintain barrier integrity, or by inappropriate activation of cell signaling pathways that induce endothelial hyperpermeability.1, 3–5 In many cases, cell signaling events that induce barrier dysfunction act by increasing the activity of myosin light chain kinase (MLCK). A normal amount of basal MLCK activity is required to maintain physiological microvascular permeability. In contrast, pathologically elevated MLCK activity induces microvascular barrier dysfunction (reviewed in3). Appropriate control of MLCK activity in endothelium is necessary for preventing barrier dysfunction in inflammatory conditions.
The main biological function of MLCK is to phosphorylate the regulatory myosin light chain (MLC-2).3, 6 Phosphorylation of MLC-2 at Ser-19, and subsequently at Thr-18, induces ATP-dependent actomyosin contraction (Fig. 1)3, 7, 8; MLCK-dependent actomyosin contractility regulates arteriolar vasoconstriction, and capillary/venule permeability. MLCK-dependent actin-myosin mediated cytoskeletal contraction followed by endothelial cell membrane retraction and intercellular gap formation is the classical mechanism of endothelial barrier dysfunction. In some instances, MLCK activity causes barrier dysfunction through phosphorylation and decreased expression of endothelial cell–cell junction proteins, via mechanisms that are poorly understood. The following review is an examination of cell signaling events related to MLCK-dependent endothelial barrier dysfunction, and is directed at understanding the role of MLCK in microvasculature under inflammatory conditions.
Figure 1.

Myosin light chain kinase (MLCK)-dependent control of actin-myosin contraction in endothelium. Increased myosin light chain (MLC) phosphorylation at Ser-18 and Tyr-19 in response to MLCK activation or myosin light chain phosphatase (MLCP) inhibition increases MLC ATPase-driven force generation relative to actin. MLCK activity is increased by Ca2+-calmodulin binding, and/or by kinase-dependent phosphorylation including phosphorylation by protein kinase C (PKC) or tyrosine kinase phosphorylation at Tyr-464 and Tyr-471. MLCK activity is decreased in response to protein kinase A (PKA) activity. Inhibition of MLCP is achieved by ROCK activation downstream of RhoA activation, resulting in phosphorylation-dependent inhibition of MLCP.
2. PHYSIOLOGY, STRUCTURE, AND REGULATION OF MLCK AT ENDOTHELIAL BARRIERS
A. Endothelial Cell–Cell Junctions and Hyperpermeability
The bases for microvascular barrier function are endothelial intercellular junctions that restrict the passage of plasma components into the tissue interstitial space.1, 9, 10 While plasma components can be transported across endothelial barriers in a transcellular fashion, facilitated by vesicles or transporter proteins at the endothelial cell membrane, most plasma extravasation is paracellular, through cell–cell junctions. Endothelial cell–cell junctions consist of integral membrane proteins expressed on the lateral surfaces of adjacent endothelial cells that interact via extracellular domains to form a barrier to the passage of solutes between cells. On the intracellular side, junction proteins are linked to adaptor proteins that are in turn connected to the actin cytoskeleton. These linkages are a physical basis for control of junction integrity, translating cytoskeletal tension into retraction at cell–cell interfaces.
Microvascular cell–cell junctions consist of adherens junctions (AJs) and tight junctions (TJs). AJs are present in the vascular endothelium of most organs and tissues,10–12 whereas TJs are found in the microvasculature of specialized tissues (e.g., the blood–brain barrier, or blood–retinal barrier).13–15 AJs are composed of VE-cadherin and junction adhesion molecules (JAMs), connected intracellularly to the actin cytoskeleton via catenins (α, β, γ, and p120).12, 16–18 TJs have additional specialized junction proteins: occludin and claudins,13, 19, 20 anchored to the actin cytoskeleton via zona occludens (ZO-1 or ZO-2), and α-catenin.12, 16–18
Control of cell–cell junction permeability is dependent upon stability and organization of the actin cytoskeleton. Cytoskeletal actin filaments are stabilized by cross-linking proteins (e.g., spectrin or filamen), and are regulated by molecules that control actin polymerization (e.g., cofilin, gelsolin, or heat shock protein (HSP27)). In addition to their connections at cell–cell junctions, the opposing ends of actin filaments are connected via actin-linking proteins (e.g., talin, paxillin, and vinculin) to transmembrane integrins at focal adhesions.16, 18, 21 Focal adhesions anchor endothelium to the basement membrane extracellular matrix, and provide a fulcrum for actin cytoskeletal tension to control cell–cell junction permeability.17, 22 Therefore, endothelial hyperpermeability can be mediated in three ways, through (i) loss of cell–cell or cell–matrix adhesion due to decreased expression or impaired function of junction proteins, cross-linker proteins, or focal adhesion proteins, (ii) cytoskeletal instability and reorganization, or (iii) actomyosin contractile forces and tension propagated by the actin cytoskeleton.
B. MLCK-Dependent Hyperpermeability
Cytoskeletal tension is controlled dynamically by the phosphorylation status of MLC, which is the result of a balance between phosphorylation by MLCK and dephosphorylation by MLC phosphatase (MLCP) (Fig. 1).3, 18 MLC phosphorylation permits ATP-dependent mechanochemical interaction between actin and myosin, which increases cytoskeletal tension and causes endothelial hyperpermeability.23–25 Conversely, MLC dephosphorylation by MLCP decreases actomyosin contractility, which relaxes the actin cytoskeleton and decreases paracellular permeability.26 In addition, MLCK and MLCP are controlled reciprocally by phosphorylation: MLCK activity is increased while MLCP activity is decreased. MLCP is phosphorylated and inactivated by Rho kinase, downstream of the Rho GTPase, RhoA (Fig. 1).12, 27, 28 MLCP inhibition increases MLC phosphorylation, actomyosin contractility, and endothelial hyperpermeability in a manner that is MLCK-dependent, but does not require increased MLCK activity.
C. MLCK Isoforms in Endothelium
MLCK is a member of the immunoglobin superfamily of proteins (see3, 8). All MLCKs are derived from the genes mylk1–3, on human chromosomes 3 (3q21), 20 (20q13.31), or 16 (16q11.2), respectively,29–31 including muscle MLCK variants: cardiac (cMLCK), skeletal (skMLCK), and smooth muscle (smMLCK), products of genes mylk3, mylk2, and mylk1, respectively,6, 29, 32, 33 and nonmuscle (nmMLCK) isoforms, products of mylk1.8 The structural components required for activity (catalytic kinase domain) and binding to MLC (C2-type immunoglobin-like domain) are highly conserved in all MLCK variants across species. Expression of cMLCK and skMLCK is restricted to cardiac and skeletal muscle, respectively. In contrast, smMLCK and nmMLCK are highly expressed in endothelium, and several other tissues.
smMLCK and nmMLCK, splice variants derived from mylk1, differ in that nmMLCK isoforms have a 922-amino acid insertion near the N-terminus.34 Hence, these variants have markedly different molecular weights: 108 kD for smMLCK and approximately 210 kD for nmMLCK, referred to as MLCK108 and MLCK210, respectively.35, 36 The nmMLCK iso-forms (MLCK1–4), originally known as endothelial MLCK (eMLCK),34 are variations based on the 1914 amino acid sequence of the prototypic isoform, MLCK1.8 Compared to MLCK1, MLCK2 lacks a 69-amino acid regulatory region containing two sites for tyrosine phosphorylation by p60Src.8,37 MLCK 3a and 3b lack 51 amino acids corresponding to exon 30, and are otherwise identical to MLCK1 and MLCK2, respectively. Only partial sequence information is available for MLCK4, and little is known about the function of this isoform. MLCK1–4 have broad tissue distribution, however, MLCK1 and MLCK2 are the most abundant nmMLCK isoforms in endothelial tissue.
D. MLCK Activity and Regulation
The principle function of MLCK activity is to phosphorylate MLC. Control of MLCK activity is a basis for cellular regulation of actomyosin contractile activity and permeability at endothelial intercellular junctions. Ca2+/calmodulin are central to control of MLCK activity6,38 in that calmodulin binding to MLCK causes release of MLCK autoinhibition.32,36,39 In smooth muscle, Ca2+ binding to calmodulin activates smMLCK and induces actin-myosin contraction. However, in endothelium and other nonmuscle cell types, Ca2+/calmodulin binding alone is not sufficient to elicit actomyosin contraction, and requires additional signaling.34 For example, MLCK1 can be activated by tyrosine kinase-mediated phosphorylation at Tyr-464 and Tyr-471.7,40,41 The 922 amino acid insertion to the N-terminal region of nmMLCK contains multiple consensus phosphorylation recognition sequences for protein kinases including protein kinase A (PKA), protein kinase C (PKC), and calmodulin kinase II (CaMKII).7,10,37,42 Signaling to MLCK through these pathways is crucial for barrier function in vascular endothelium (summarized in Fig. 1).
E. MLCK-Dependent Maintenance of Basal Microvascular Permeability
Elevated MLCK activity causes endothelial hyperpermeability and endothelial barrier dysfunction, however, tonic MLCK activity is important for maintenance of basal microvascular permeability. In isolated porcine coronary venules as well as microvessels from rodent skeletal muscle and mesentery, basal microvessel permeability to fluorescein isothiocyanate (FITC)-albumin is significantly attenuated by the MLCK inhibitor ML-7.43,44 In addition, treatment with the protein phosphatase (PP1/PP2A) inhibitor calyculin-A increases both MLC phosphorylation and vascular permeability, supporting the notion that residual MLCK activity is present and maintains normal endothelial permeability in intact microvasculature.
In cultured endothelial cell monolayers, MLCK inhibition often has no effect on basal transendothelial albumin flux as seen in rat heart or lung micro-VE cells, or on transendothelial electrical resistance (TER) as seen in bovine brain micro-VE cells.45–47 This lack of responsiveness may be due to inherent differences between intact microvessels and cultured endothelial monolayers, or to phenotypic heterogeneity between endothelial cells.9 For example, Curry and Adamson have noted that FITC-albumin leakage occurs adjacent to a very small percentage of endothelial cells in microvessels in situ.9,48 Hence homogeneous populations of cultured endothelial cells may display barrier properties that differ from those of heterogeneous populations of endothelial cells in intact microvessels. Barrier properties may also vary between endothelial cells originated from different tissues. In particular, endothelial cells derived from nonexchange vessels, for example, human umbilical vein endothelial cells (HUVEC) or aortic endothelial cells, may display permeability responses that differ from that of exchange microvessels. Endothelial cells in vivo also receive signals from cells in the surrounding tissue. This is well-established for brain endothelial cells where the presence of astrocytes enhances endothelial barrier function, in part by increasing endothelial expression of junction proteins,49,50 or for skin endothelium, where contraction of surrounding fibroblasts modulates microvascular permeability by regulating compaction of the extracellular matrix.51 These dynamic interactions with surrounding nonendothelial cells in situ further explain why some treatments can increase or decrease endothelial permeability differently in cultured micro-VE cells versus microvessels in vivo.
3. MLCK-DEPENDENT SIGNALING AND ENDOTHELIAL HYPERPERMEABILITY
A. Ca++ Channels
In many cases, the molecular basis of MLCK-induced hyperpermeability is not well-understood, and involves mechanisms other than actomyosin contractility, including activation of Ca++ channels.11 For example, in pulmonary artery or brain capillary endothelial cells, hyperpermeability is mediated by activation of store-operated membrane transient receptor potential (TRPC) Ca++ channels (Fig. 2).52,53 Activation of TRPC is prevented by treatment with the MLCK inhibitor ML-9, or by agents that either disrupt or stabilize actin filaments. Therefore, activation of TRPC requires both MLCK activity and the intact actin cytoskeleton. MLCK-dependent endothelial hyperpermeability may also depend upon activation of store-operated Ca++ (SOC) channels, and subsequent decreased expression, or modification, of junction proteins or cytoskeletal linker proteins (Fig. 2). Activation of SOC channels is triggered by depletion of Ca++ from intracellular endoplasmic reticulum (ER) stores, and MLCK activity prior to SOC channel activation depends on Ca++ released from the ER in response to inositol 1,4,5-trisphosphate receptor (IP3R) activation. IP3R activation leads to phosphorylation of MLC, and MLC phosphorylation-dependent phosphorylation of TJ proteins occludin and claudin-5.54,55 MLCK-dependent phosphorylation of occludin and claudin-5 occurs at the blood-brain barrier (BBB) during the hyperpermeability responses to controlled cortical impact TBI, or exposure to C-reactive protein (CRP) or methamphetamine, suggesting a general role for this mechanism in BBB endothelial hyperpermeability.56–58 However, a causal relationship between MLCK activation and SOC channel activation is challenged by the recent observation in immortalized mouse brain endothelial (bend.3) cells that treatment with the TRPC channel inhibitor SKF 96365 prevents both MLC phosphorylation and junction hyperpermeability following hypoxia.53 Ca++-dependent events independent of MLCK activation also contribute to endothelial hyperpermeability. For example, in lung endothelium, TRPC Ca++ channel activation by thrombin induces actin stress fiber formation and hyperpermeability, possibly through activation of RhoA kinase.10,59
Figure 2.
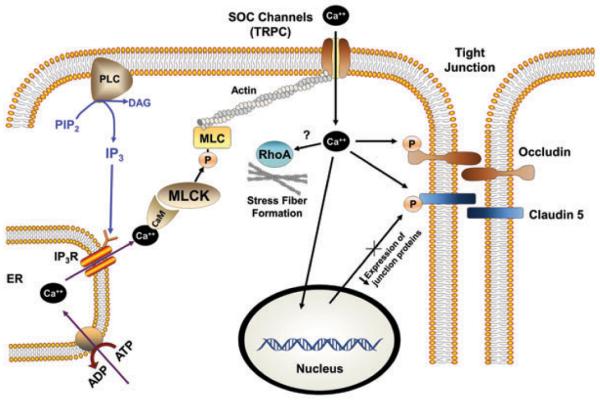
Myosin light chain kinase (MLCK)-dependent endothelial tight junction (TJ) hyperpermeability signaled through store-operated Ca++ (SOC) channel activation. Intracellular stores (endoplasmic reticulum (ER)) Ca++ released in response to inositol triphosphate (IP3) receptor (IP3R) activation by inositol triphosphate (IP3), downstream of phospholipase C (PLC)-mediated hydrolysis of phosphotidylinositol bisphosphate (PIP2), increases MLCK activity through Ca++ binding to calmodulin (CaM). MLCK activity in turn activates SOC channels, further increasing cytosolic Ca++ concentration, resulting in phosphorylation of occludin and claudin-5, transcriptional downregulation of junction proteins and increased actin stress fiber formation.
B. Protein Kinase C
Signaling for endothelial hyperpermeability in response to a variety of inflammatory agents is dependent upon PKC (Fig. 3).1,5,10–12 However, signaling through PKC is complex involving effects on cytoskeletal organization and contractility, Ca++ channels, and multiple cell signaling pathways, including opposing roles for specific PKC isoforms in controlling endothelial hyperpermeability. Classical PKC isoforms α and β are well-known inducers of hyperpermeability. For example, PKC-induced endothelial dysfunction in the progression of diabetes requires PKCβII.60–62 In isolated coronary venules, KDR receptor-mediated vascular endothelial growth factor (VEGF)-induced hyperpermeability is prevented by the phospholipase C (PLC) inhibitor U-73122, or by the classical PKC isoform-selective inhibitor bis-indolylmaleimide (BIM). These treatments also prevent VEGF-induced tyrosine phosphorylation of endothelial nitric oxide synthase (eNOS), suggesting that PKC-mediated hyperpermeability is signaled through increased nitric oxide (NO) synthesis (Fig. 3).63 VEGF binding to KDR also initiated PLC-mediated cytosolic Ca2+ elevation, which may be necessary for activation of classical (Ca++-dependent) PKC isoforms (α/βI/II/γ) and subsequent hyperpermeability. In coronary venules, PKC-induced hyperpermeability is also significantly attenuated by ML-7.43 Therefore, PKC-induced microvascular permeability in isolated coronary venules may occur partially via activation of MLCK. In support of these observations, we observe synergy between elevated intracellular Ca2+ and PMA treatment to increase MLC phosphorylation and hyperpermeability in cultured endothelial (HUVEC) cell monolayers.40 Therefore, activation of PKC isoforms and elevated cytosolic Ca++ may act in concert to signal MLCK-dependent microvascular hyperpermeability.
Figure 3.

MLCK signaling in endothelial hyperpermeability. MLCK activity is increased by multiple signaling pathways in response to inflammatory mediators, downstream of G-protein coupled receptor (GPCR) activation. Activation of phospholipase C (PLC) leads to hydrolysis of phosphatidylinositol bisphosphate (PIP2) into diacylglycerol (DAG) and inositol triphosphate (IP3). IP3 triggers intracellular Ca++ release from intracellular stores (endoplasmic reticulum (ER)) via activation of the IP3 receptor. Stores Ca++ release activates MLCK directly through Ca++ binding to calmodulin (CaM). DAG and Ca++ together activate PKC-α and other classical PKC isoforms. PKC-α activates endothelial nitric oxide synthase (eNOS) (inhibited by PKC-ζ mediated phosphorylation at T497) by phosphorylation at S1179, to increase nitric oxide (NO) production. NO activates many signaling processes, including NO combines with reactive oxygen species (ROS) to form peroxynitrite (ONOO−); NO causes S-nitrosylation of numerous proteins; NO impairs function of complex IV of the mitochondrial oxidative phosphorylation system causing release of sequestered Ca++; and NO activates guanylate cyclase (GC) to increase conversion of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). cGMP can increase or decrease endothelial permeability through various mechanisms depending upon the cell type examined. cGMP activates MLCK through activation of PKG, which may be mediated through activation of the Raf-MEK-ERK pathway. cGMP may also modulate phosphodiesterase activity to increase or decrease abundance of cAMP and correspondingly affect PKA activity.
Because of the existence of at least 11 PKC isoforms and heterogeneous responses of endothelial cells from different vascular segments, PKC signaling for hyperpermeability is complex. Studies of hypoxia in rat brain micro-VE cells show that increased PKC activity and expression of multiple PKC isoforms (βII/γ /η/μ/θ/λ) may contribute to endothelial hyperpermeability during hypoxia/I/R injury.64,65 However, in mouse brain endothelium, TNF-α induced hyperpermeability is mediated through PKC-α.66 Similarly, blood–retinal barrier dysfunction may be mediated through PKC-βII,67,68 PKC-δ,69 or PKC-ζ.70 In contrast, coronary endothelial hyperpermeability is achieved either by overexpression of PKCβII or by inhibition of PKCδ,61 and inflammatory cytokine-dependent hyperpermeability requires both classical PKC and MLCK activity.47 Other investigators have shown in bovine aortic endothelial cells that VEGF-induced hyperpermeability is prevented by PKCζ-dependent phosphorylation of eNOS (at Thr497) and inhibition of eNOS activity,71 or that thrombin-induced eNOS phosphorylation (at Ser1179) and activation is inhibited by expression of dominant negative PKCα or PKCδ.72 Therefore, individual PKC isoforms may induce or prevent endothelial hyperpermeability by phosphorylation of eNOS at sites that either stimulate (Ser1179) or inhibit (Thr497) eNOS activity (Fig. 3).
Clearly, the notion that the effects of Ca++ and PKC are mediated through the effects of eNOS on MLCK is an oversimplification. VEGF induced PLC-mediated hydrolysis of phosphatidyl inositol bisphosphate (PIP2) leads to diacylglygerol (DAG)-mediated effects on classical and novel PKC isoforms, and IP3-induced release of Ca++ from intracellular stores. Release of ER Ca++ activates MLCK (through calmodulin), eNOS, classical PKCs, and many other signaling molecules that affect MLCK activity or cytoskeletal organization, leading to endothelial hyperpermeability (reviewed extensively elsewhere).10,11
C. NO and Cyclic GMP
There is significant debate about the effects of NO and guanosine 3′, 5′-cyclic monophosphate (cGMP) which induce hyperpermeability in intact microvessels, yet are protective against hyperpermeability in cultured endothelial cells.73–75 In porcine coronary venules, NO production and cGMP mediate endothelial shear stress- and agonist-induced hyperpermeability responses.76′78 For example, histamine increases permeability through a PLC′NOS′cGMP signaling cascade (Fig. 3), where permeability is inhibited by the PLC inhibitor 2-nitro-4-carboxyphenyl N,N-diphenylcarbamate (NCDC), the NOS inhibitor NG-monomethyl-L-arginine (L-NMMA), or the guanylate cyclase inhibitor LY83583, respectively.76 The NO donor sodium nitroprusside (SNP) or cGMP analog 8Br-cGMP also induce hyperpermeability to albumin, indicating dependence on NO and cGMP-dependent protein kinase (PKG).78,79 Hyperpermeability in response to these agonists is significantly attenuated by treatment with 10 μM ML-7. Although nmMLCK has consensus sites for phosphorylation by PKG,7 there is no conclusive evidence that MLCK is directly phosphorylated by PKG. Rather, evidence suggests that PKG-dependent activation of MLCK is mediated by mitogen-activated protein kinases (MAPK), and extracellular-regulated kinase (ERK1/2) (Fig. 3). For example, in HUVEC cells, the Raf-1/MAPK/ERK1/2 signaling cascade is activated by PKG downstream of NO, in response to VEGF treatment.80,81 Furthermore, in isolated coronary venules, VEGF-, histamine-, SNP- or 8Br-cGMP-induced hyperpermeability responses are decreased by inhibition of MEK1 and ERK1/2 with U0126 or PD98059.82
In endothelial cell culture systems, NO and cGMP decrease MLC phosphorylation, and decrease endothelial permeability.83,84 Several laboratories have suggested that control of endothelial permeability by cGMP is due to cross-talk between cGMP and cAMP.75,85 The cAMP-dependent protein kinase, PKA suppresses nmMLCK activity and hyperpermeability in most endothelial systems.34,86 In HUVEC cells, differential effects of PKG on phosphodiesterase isoforms can increase or decrease the amount of cellular cAMP in a biphasic fashion dependent upon the concentration of cGMP.87 Thus, the hyperpermeability-inducing versus protective effects of NO-cGMP could be explained by tissue/cell-specific differences in expression or regulation of phosphodiesterase isoforms, resulting in increases or decreases in cellular cAMP in response to cGMP (Fig. 3). This is supported by studies of ventilator-induced lung injury in mice showing that pathological expression of the phosphodiesterase isoform PDE2A in the pulmonary endothelium, coupled with cGMP production, leads to increased hydrolysis of cAMP.88 Therefore, PKG-dependent activation of MLCK and hyperpermeability in some endothelial systems may be signaled through decreased cAMP and decreased PKA activity. A recent study in HUVEC cells, using a fluorescence resonance energy transfer (FRET)-based indicator of cAMP, demonstrated that increased cytosolic Ca++ in response to thrombin treatment also decreases cAMP levels through modulation of adenylate cyclase isoforms (AC5/6).89 Therefore, decreased cAMP may contribute to endothelial hyperpermeability in trauma or prothrombotic conditions.
Direct protective effects of cGMP on endothelial junction integrity are also reported. In rat coronary endothelial cells, treatment with the NO donor DEA-nonoate or the soluble guanylate cyclase activator HMR1766 causes Ca++ sequestration into the ER through phosphorylation of phospholamban and subsequent activation of the ER Ca++ ATPase, leading to decreased intercellular gap formation and increased MLC phosphorylation during I/R.83 Conversely, inhibition of PKG with KT5823 prevents Ca++ sequestration and increases gap formation during I/R injury in these cells. Therefore, PKG activity may affect MLC phosphorylation (and intercellular gap formation) by limiting cytosolic Ca++ available for MLCK activation.
NO affects multiple signaling pathways, and therefore the effects of NO are not confined to signaling through cGMP. For example, NO modifies the activity of many proteins by S-nitrosylation at cysteine residues.90 NO combines with reactive oxygen species (ROS) to form the highly reactive species, peroxynitrate (ONOO−) (Fig. 3).91 NO also impairs mitochondrial function by competing with O2 for binding to complex IV of the electron transport chain, and can thereby interfere with mitochondrial Ca++ sequestration, contributing to a rise in cytosolic Ca++.92 NO affects many other aspects of cell Ca++ homeostasis, including ER Ca++ loading or membrane Ca++ channel activity, and NOS activity is Ca++-dependent.93 Because of the brief and unstable existence of NO, the effects of NO are often localized to specific subcellular or membrane regions where NOS isoforms are expressed.90 Therefore, while NO-induced cGMP production is central to control of endothelial hyperpermeability, the effects of NO signaling on MLCK-dependent actomyosin contraction and endothelial hyperpermeability may be exceptionally complex.
4. PATHOPHYSIOLOGICAL RELEVANCE OF MLCK-DEPENDENT ENDOTHELIAL HYPERPERMEABILITY
A. Leukocytes
In several inflammatory conditions, activated neutrophils induce endothelial hyperpermeability.22,94,95 Some investigators have argued that neutrophil-induced endothelial hyperpermeability is a prerequisite for neutrophil extravasation.95,96 Yet, in some instances there is a lack of temporal correlation between leukocyte adhesion and vascular leakage.97 In general, leukocyte transendothelial migration occurs via two routes: transcellular or paracellular.98 Fluorescence microscopy images of leukocytes migrating across endothelium in a transcellular fashion show podosomes in close apposition with the endothelial vesicular membrane, suggesting that transcellular migration is not accompanied by gap formation or leakage.99,100 Classic histological evidence by Lewis and Granger has shown that neutrophil transmigration across microvessel endothelial wall in a paracellular fashion occurs in the absence of serum protein extravasation.101 In addition, in a rat aseptic dermal injury model with fluorescent-labeled neutrophils and albumin, endothelial hyperpermeability and neutrophil transendothelial migration were temporally separated events, where neutrophil transmigration did not cause nor was accompanied by hyperpermeability.102 It has been suggested that cell junction leakage of chemoattractants is necessary to attract leukocytes to sites of inflammation.103 However, this is refuted by the observation that the chemokine interleukin-8 (IL-8) must be presented at the endothelial luminal surface, by vesicle-mediated transcytosis from the basolateral to the apical cell membrane, for IL-8 to attract leukocytes. Together, these lines of evidence indicate that neutrophil transmigration and neutrophil-induced endothelial hyperpermeability are separate and not necessarily related events.
In isolated coronary venules, activated neutrophil-induced hyperpermeability to albumin is significantly attenuated by treatment with ML-7, indicating the involvement of MLCK.104 The requirement for MLCK in this process was demonstrated in isolated, perfused coronary venules transfected with either an MLCK-inhibiting peptide or dominant negative, inactivated MLCK.104,105 Following transfection with either agent, activated neutrophils were incapable of inducing microvessel hyperpermeability. Although these transfections were directed at the microvessel endothelium, it is possible that decreased neutrophil-induced hyperpermeability is due to inhibition of MLCK in neutrophils. This is supported by the observation that nmMLCK−/− neutrophils fail to attach to wild-type lung endothelium during lipopolysaccharide (LPS)-induced lung injury in mice.106 Related to these phenomena, we have observed that endothelial hyperpermeability is attenuated in nmMLCK−/− aortic endothelial cell monolayers in response to thrombin, but that monocyte transendothelial migration across these monolayers is unaffected by endothelial MLCK gene knockout.107 Thus, monocyte transmigration across aortic endothelium is not dependent upon endothelial MLCK or hyperpermeability. The relative importance of MLCK-dependent signaling in endothelium versus neutrophils with regard to neutrophil-induced endothelial hyperpermeability and neutrophil transmigration remains to be determined.
Neutrophil-induced endothelial hyperpermeability is dependent upon neutrophil granule secretion, including the release of many hyperpermeability-inducing compounds, for example, proteases, elastase, arachadonic acid, serum proteins, and ROS.95 ROS are additionally produced within endothelial cells by NAPH oxidase or xanthine oxidoreductase, therefore, the combination of ROS from intracellular and extracellular sources can be potent mediators of neutrophil-induced endothelial hyperpermeability.91 In brain endothelial cells, ROS generated in response to interleukin-17, CRP, or hypoxia activate MLCK.45,57,108 ROS generation is dependent upon p38MAPK activity in response to CRP, and upon cytosolic Ca++ in response to hypoxia. In porcine brain capillary endothelial cells, hydrogen peroxide (H2O2) increases junctional permeability via a pathway dependent upon ERK1/2 activity downstream of intracellular Ca++ release.109 In pulmonary endothelial cell monolayers, neutrophil-induced endothelial hyperpermeability is dependent upon ROS signaling through PKCα-dependent activation of the transient receptor potential melastatin cation channel (TRPM2).110,111 Therefore, neutrophil-induced ROS generation may cause endothelial hyperpermeability through PKCα-dependent activation of Ca++ channels, and activation of MAP/ERK kinases. ROS activates many other signaling molecules that could play a role in endothelial hyperpermeability. For example, CaMKII is rapidly autophosphorylated and activated by exposure to H2O2,112,113 and is capable of phosphorylating MLCK. Oxidative stress also affects RhoA (known to inhibit MLCP and to induce actin stress fiber formation), though the specific mechanism for RhoA activation by ROS is not known.91 In brain micro-VE cells, H2O2 decreases actin stress fiber formation and decreases expression of TJ proteins, occludin, and claudin-5.114 These effects are prevented by inhibition of RhoA, PI3 kinase, or Protein Kinase B (PKB) (Akt), suggesting that multiple control mechanisms mediate ROS-induced endothelial hyperpermeability.
B. Severe Burn Injury
Severe burns cause systemic inflammation, leading to systemic inflammatory response syndrome (SIRS) characterized by microvascular leak in tissues far removed from the site of injury.115–118 Leakage of fluid across microvascular barriers decreases blood volume, leading to hypovolemic shock and poor tissue perfusion, and increases fluid accumulation in tissues, leading to pulmonary edema, abdominal compartment syndrome, and multiple organ failure.119–122 In clinical patients, these pathologies can be further exacerbated by accompanying smoke inhalation, or infection and sepsis, precipitating further ALI, inflammation, and edema. In addition, the splanchnic microvessels are highly susceptible to vascular leakage during severe dermal burn injury, and are an excellent model for examining microvascular permeability to albumin in rodents.123 Microvascular permeability in the gut microvasculature can be examined either in isolated perfused venules, or in vivo in the intact splanchnic mesentery using fluorescence intravital microscopy.124,125 The time-dependent distribution of fluorescent FITC-albumin in the intravascular versus extravascular space is monitored and used as an indicator of permeability.126 In rat splanchnic mesenteric microvasculature, FITC-albumin leakage is significantly increased over 6 hr following a 25% total body surface area (TBSA) full-thickness scald burn.127 In these experiments, the splanchnic microvascular is far-removed from the injury site, suggesting that hyperpermeability-inducing factors produced by the dermal burn injury travel in the blood circulation and affect microvascular permeability remotely in the gut. To test this hypothesis, isolated, perfused mesenteric venules were treated with plasma collected from rats following burn injury. In this study, burn plasma significantly increased venule permeability to albumin, compared to venules treated with plasma obtained from unburned animals. Therefore, endothelial hyperpermeability is induced by circulating factors present in the plasma during burn injury. In humans and rodents, circulating factors released into the plasma during burn injury include multiple inflammatory cytokines that are known to induce endothelial hyperpermeability.128 However, blocking specific individual inflammatory cytokine pathways has not yielded effective clinical treatments for burn-induced microvascular dysfunction.129 Consistent with this observation, treatment of isolated perfused venules with inhibitors of intermediate inflammatory signaling molecules, for example, Src or PKC, fails to prevent burn plasma-induced microvascular leakage.127 Based on this evidence, we suspect that multiple inflammatory signaling pathways operate in parallel during burn injury to induce endothelial hyperpermeability. A better therapeutic strategy would be to identify and target common terminal effectors of multiple inflammatory pathways that mediate the hyperpermeability response. Because multiple extracellular inflammatory mediators and intracellular signaling intermediaries can cause endothelial hyperpermeability by increasing MLCK-dependent MLC phosphorylation, MLCK is a strong candidate target for preventing burn-induced microvascular dysfunction.130–135 This notion is supported by the observation that treatment of isolated venules with ML-7 blocks MLC phosphorylation and significantly attenuates burn-induced venular hyperpermeability.127 In addition, albumin leakage is fully attenuated in the splanchnic mesentary of MLCK210 knockout mice following severe dermal burn injury, compared to that of wild-type mice,136 further indicating that endothelial MLCK is required for burn-induced endothelial hyperpermeability.
C. Trauma and Sepsis
The lungs are especially vulnerable to trauma- or sepsis-induced inflammation, and are frequently involved in multiple-organ failure.18,137 Increased susceptibility to ALI or acute respiratory distress syndrome is associated with single nucleotide polymorphisms (SNPs) in the human mylk1 gene.138,139 Moreover, SNPs associated with ALI in this study, occurred at loci corresponding to regions that are unique to nmMLCK, whereas SNPs in regions also encoding smMLCK were unrelated to ALI. This suggests that lung microvascular hyperpermeability during ALI is mediated by nmMLCK isoforms. In support of this suggestion, MLCK210 knockout mice have lowered susceptibility to sepsis-induced lung injury.140 In a subsequent study, endothelial-specific transgenic overexpression of MLCK2 in mice greatly enhanced serum protein extravasation into lung tissue during LPS-induced sepsis, or mechanical ventilator injury.141 Although artifacts are associated with protein overexpression models, this suggests that MLCK2 contributes to lung vascular hyperpermeability in response to sepsis or trauma. Thus, a likely future direction for studies of microvascular dysfunction is to distinguish the respective contributions of MLCK isoforms to endothelial hyperpermeability.
D. Chronic Inflammatory Diseases
Endothelial barrier dysfunction also occurs in chronic inflammatory conditions including atherosclerosis, BBB dysfunction in multiple sclerosis, and diabetic microvascular complications in several organs including the peripheral circulation and the blood–retinal barrier. Interestingly, in an apolipoprotein E deficient mouse model of atherosclerosis, macrophage deposition and albumin (Evans Blue) leakage across the aortic wall were attenuated by nmMLCK (−/−) gene knockout.107 Further mechanistic studies revealed that MLCK-dependent barrier dysfunction in aortic endothelium is only partially dependent upon MLCK kinase activity, and that MLCK participates in endothelial barrier dysfunction through both kinase activity-dependent and kinase activity-independent molecular mechanisms.
5. PROTECTIVE STRATEGIES AND THERAPEUTIC TARGETS IN MLCK-DEPENDENT ENDOTHELIAL HYPERPERMEABILITY
A. Sphingosine-1-Phosphate (S1P)
S1P, a derivative of sphingomyelin, enhances the barrier properties of lung endothelium, and protects against hyperpermeability.142,143 S1P in the circulation is supplied by blood cells, including erythrocytes and activated platelets, helping to maintain VE integrity. In mice, selective knockout of erythrocyte SIP eliminates S1P from the blood circulation, causing increased basal endothelial leakage and decreased resistance to hyperpermeablility in response to PAF or histamine.144 S1P binds to G-protein coupled S1P receptor isoforms (S1PR1–5).143 In a study of LPS-induced toxicity with an S1P analog FTY720, and SIPR knock-out mice, it was determined that the protective effects of SIP against vascular leakage in the lungs are mediated by Gi-coupled SIPR1.145 In human pulmonary artery endothelial cells, SIP binding to S1PR1 recruits nmMLCK and multiple tyrosine phosphorylated proteins to caveolin-1 containing lipid rafts in the plasma membrane, and increases TER.146 Recruitment to lipid rafts brings nmMLCK in close proximity with the actin binding proteins cortactin and β-catenin. S1P treatment also triggers tyrosine phosphorylation of both nmMLCK (Y464) and cortactin (Y486) by the actin-binding tyrosine kinase c-Abl, and promotes nmMLCK binding to cortactin coinciding with localization to the peripheral actin ring.147,148 Because MLCK1 and MLCK2 are the most abundant nmMLCK isoforms in endothelium, phosphorylation at Y464 indicates that the protective effects of S1P are mediated through MLCK1. However, in human pulmonary endothelial cells, it was shown that S1P treatment induces tyrosine kinase-dependent mobilization of cortactin and MLCK2 to the cell periphery.149 In this study, it is hypothesized that the tyrosine kinase-dependent association of MLCK with cortactin strengthens the cortical actin band, and improves endothelial barrier function. However, recruitment to the cell membrane also removes MLCK from its usual location within the cell, thereby decreasing the amount of nmMLCK available to induce actomyosin contraction in response to inflammatory agents.
B. MLCK as a Therapeutic Drug Target
Microvascular barrier dysfunction is a consequence of highly complex molecular events and signaling cascades affecting intercellular junctions and cell-matrix adhesion. Many of these events are signaled through MLCK. Therefore, the ability to selectively and appropriately inhibit MLCK activity in vascular endothelium could be useful for treatment of inflammatory conditions. The pharmacological inhibitors, ML-7 and ML-9, target the ATP binding site of MLCK, which is highly homologous with other kinases, including PKA and PKC.150 In addition, these compounds broadly affect all MLCK isoforms in many tissue types, and cause unintended physiological effects in vivo. Hence, these compounds are not suitable for clinical use. Because of this, Watterson and co-workers synthesized a 6-phenyl-3-aminopyridazine compound that inhibits MLCK more specifically at low micromolar concentrations, and prevents LPS-induced Evans blue extravasation and edema in the lungs of mice.151,152 This compound is an improvement over previous MLCK inhibitors because of increased specificity for MLCK.
It is also possible to generate highly specific peptide inhibitors of MLCK. For example, the peptide “membrane permeant inhibitor of MLCK” (PIK) is a fragment of the MLCK inhibitory domain that specifically inhibits MLCK without affecting other kinases such as PKA.150 Inhibition of MLCK with PIK prevents albumin flux through epithelial TJs in the gut during T-cell activation-induced diarrhea in mice.153 Therefore, with improved methods of delivery, inhibition of MLCK with engineered peptides could be a useful therapeutic approach. Optimally, engineered peptides would be delivered specifically to endothelial tissue, and designed to target specific MLCK isoforms. In support of this claim, MLCK210 knock-out mice are resistant to endothelial hyperpermeability, and show no significant decline in cardiovascular function.154 This suggests that targeting specific nmMLCK isoforms would have fewer adverse physiological consequences than does nonselective inhibition of MLCK activity.
C. Targeting Signaling Upstream of MLCK
An alternative strategy for therapeutic intervention in vascular barrier dysfunction is to target cell signaling events that modulate MLCK-dependent endothelial hyperpermeability.155 Many therapeutic compounds currently in clinical use are kinase inhibitors.112 Therefore, selective kinase inhibition may be a useful approach for improving vascular barrier function. For example, p60Src phosphorylates and activates nmMLCK isoforms 1 and 3a in response to thrombin exposure. Src inhibition or src gene knockout also protect mice from vascular leakage at the BBB in experimental cerebral ischemia.156 This suggests that Src kinase is a worthwhile target for inhibition of MLCK activity and attenuation of vascular hyperpermeability in some inflammatory conditions. Other cell signaling pathways decrease MLCK activity and preserve endothelial barrier function. For example, treatments that increase cellular cAMP, such as inhibition of PDE2,157 may improve vascular function, as it has long been known that cAMP decreases MLCK activity and attenuates endothelial hyperpermeability. Further evidence indicates that S1P signaling through the S1P receptor (SIPR1) improves endothelial barrier function by recruiting MLCK to the cortical actin cytoskeleton.146,147,149 Thus, S1P signaling may contain additional molecular targets for inhibition of MLCK-dependent vascular barrier dysfunction.
6. CONCLUSION
Endothelial barrier dysfunction is an integral component of many inflammatory conditions and disease states, including severe burn injury, trauma, and sepsis. Microvascular leakage and tissue edema, due to compromised barrier integrity, are serious pathophysiologies associated with inflammation that are difficult to correct clinically. This is in part due to the lack of effective molecular targets for therapeutic treatment. The data presented here support the conclusion that MLCK activity by way of MLC phosphorylation is a central mediator of endothelial barrier dysfunction. MLCK is an endpoint mediator of microvascular barrier dysfunction in response to many cell signaling pathways triggered by inflammatory mediators, activated neutrophils, and severe trauma or sepsis. Because MLCK is a downstream effector of multiple signaling pathways leading to endothelial hyperpermeability, selective inhibition of MLCK is a reasonable strategy for correcting microvascular barrier dysfunction. More specifically, nmMLCK isoforms mediate endothelial hyperpermeability in response to many inflammatory stimuli, and should be further investigated as drug targets for preventing or treating vascular dysfunction and edema. In addition, signaling pathways that protect endothelial barriers by suppressing MLCK activity, such as the S1P pathway, should be investigated to reveal the downstream molecular mechanisms that prevent MLCK activation. Finally, because endothelial hyperpermeability can be induced by MLCK-independent mechanisms, hyperpermeability-associated events such as actin stress fiber formation, and phosphorylation or decreased expression of cell junction or cytoskeletal adapter proteins need to be further investigated to determine if these events are physiologically important for microvascular barrier dysfunction independent of MLCK activity.
ACKNOWLEDGMENTS
The authors thank Olesya Litovka for assistance with figure preparation.
Contract grant sponsor: NIH; Contract grant numbers: HL61507, HL70752, HL73324, HL84542, and HL96640.
Biographies
Robert R. Rigor is a postdoctoral research fellow with Drs. Sarah Yuan and Mack Wu in the Department of Surgery at the University of California Davis, School of Medicine. Dr. Rigor earned his Ph.D. in 2007 with Dr. Peter M. Cala in the Department of Physiology and Membrane Biology at UC Davis School of Medicine, and was formerly a postdoctoral fellow at the NIH National Institute of Environmental Health Science with Dr. David S. Miller investigating control of P-glycoprotein drug transport activity in brain capillaries. Dr. Rigor's research interest is kinase-dependent cell signaling and control of microvascular endothelial barrier function. His work in the Department of Surgery is focused on blood–brain barrier dysfunction during traumatic brain injury and inflammation, and he recently co-authored a textbook entitled Regulation of Endothelial Barrier Function.
Qiang Shen is a postdoctoral researcher with Drs. Sarah Yuan and Mack Wu in the Department of Surgery at the University of California Davis, School of Medicine. Dr. Qiang Shen earned his M.D. in 1997 at Harbin Medical University, China. He earned his Ph.D. in 2007 with Dr. Judith Hugh in the Department of Laboratory Medicine and Pathology at University of Alberta. Dr. Shen's research focuses on the molecular mechanisms underlying endothelial barrier dysfunction and microvascular leakage in disease conditions including cancer and inflammation.
Christopher D. Pivetti is a research associate in the Department of Surgery at the University of California Davis Medical Center. Chris earned his M.S. in Biology at the University of California San Diego. He was a research associate at Jackson Laboratories in Sacramento, California, as well as a researcher and surgical instructor at UCSD. His work emphasizes in vivo models of microvascular dysfunction and permeability.
Mack H. Wu is a professor in the Department of Surgery at the University of South Florida Morsani College of Medicine. Dr. Wu earned his M.D. at the Second Military Medical College in Shanghai, China. He was at Texas A&M University Health Science Center from 1996 to 2004 where he served as a research faculty in the department of Medical Physiology. Dr. Wu was an associate professor in the Department of Surgery at the University of California Davis, School of Medicine from 2004 to 2011. His research interest focuses on the signal transduction of growth factors in regulating microvascular permeability, molecular control of fibrinogen degradation products-induced microvascular dysfunction, and the mechanism of leaky guts during trauma. Dr. Wu is an elected member of several national societies. He has authored more than 40 scientific articles and book chapters. His work has been supported by NIH and VA.
Sarah Y. Yuan is professor and Chair of the Department of Molecular Pharmacology and Physiology at the University of South Florida Morsani College of Medicine. Dr. Yuan received a M.D. degree and a Ph.D. in pathophysiology from the First and Second Military Medical Universities in China. She completed a surgical residency at the Trauma and Burn Center in Shanghai, a surgical research fellowship at the University of Kentucky, and postdoctoral research training in medical physiology at Texas A&M University College of Medicine. Dr. Yuan was the Raleigh White Chair in Surgery and Director of Surgical Research at Texas A&M until 2004, and was the Pearl Stewart Endowed Professor and Director of Research in the Department of Surgery at the University of California Davis School of Medicine from 2004 to 2011. Dr. Yuan's research interest is cellular and molecular regulation of cardiovascular function in disease and health. Her recent work is focused on cell signaling and microvascular endothelial barrier dysfunction in inflammatory diseases and trauma. She has published extensively in these areas in high-impact journals. Dr. Yuan has served on multiple research review boards and NIH study sections, and has been an associate editor or editorial board member for several peer-reviewed scientific journals including the American Journal of Physiology, Microcirculation, and Microvascular Research. She has authored many research articles, book chapters, and textbooks on the subjects of vascular physiology and endothelial barrier regulation.
REFERENCES
- 1.Yuan SY, Rigor RR. Regulation of endothelial barrier function. In: Granger DN, Granger JP, editors. Colloquium series on integrated systems physiology: From molecule to function to disease. Morgan & Claypool Life Sciences; San Rafael (CA): 2011. pp. 1–143. [Google Scholar]
- 2.Adamson RH, Sarai RK, Altangerel A, Thirkill TL, Clark JF, Curry FR. Sphingosine-1-phosphate modulation of basal permeability and acute inflammatory responses in rat venular microvessels. Cardiovasc Res. 2010;88(2):344–351. doi: 10.1093/cvr/cvq184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87(2):272–280. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7(6 Pt 1):395–403. [PubMed] [Google Scholar]
- 5.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39(4–5):213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 6.Takashima S. Phosphorylation of myosin regulatory light chain by myosin light chain kinase, and muscle contraction. Circ J. 2009;73(2):208–213. doi: 10.1253/circj.cj-08-1041. [DOI] [PubMed] [Google Scholar]
- 7.Verin AD, Gilbert-McClain LI, Patterson CE, Garcia JG. Biochemical regulation of the non-muscle myosin light chain kinase isoform in bovine endothelium. Am J Respir Cell Mol Biol. 1998;19(5):767–776. doi: 10.1165/ajrcmb.19.5.3126. [DOI] [PubMed] [Google Scholar]
- 8.Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK) Genomics. 1999;57(2):256–267. doi: 10.1006/geno.1999.5774. [DOI] [PubMed] [Google Scholar]
- 9.Curry FR, Adamson RH. Vascular permeability modulation at the cell, microvessel, or whole organ level: Towards closing gaps in our knowledge. Cardiovasc Res. 2010;87(2):218–229. doi: 10.1093/cvr/cvq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 11.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 12.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann NY Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 13.Coisne C, Engelhardt B. Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal. 2011;15(5):1285–1303. doi: 10.1089/ars.2011.3929. [DOI] [PubMed] [Google Scholar]
- 14.Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin Immunopathol. 2009;31(4):497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- 15.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 16.Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res. 2009;77(1):53–63. doi: 10.1016/j.mvr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 18.Shen Q, Wu MH, Yuan SY. Endothelial contractile cytoskeleton and microvascular permeability. Cell Health and Cytoskeleton. 2009;1:43–50. doi: 10.2147/chc.s5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol. 1999;5(6):556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- 20.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24(12):719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 21.Wu MH. Endothelial focal adhesions and barrier function. J Physiol. 2005;569(Pt 2):359–366. doi: 10.1113/jphysiol.2005.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan SY, Shen Q, Rigor RR, Wu MH. Neutrophil transmigration, focal adhesion kinase and endothelial barrier function. Microvasc Res. 2012;83(1):82–8. doi: 10.1016/j.mvr.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnittler HJ, Wilke A, Gress T, Suttorp N, Drenckhahn D. Role of actin and myosin in the control of paracellular permeability in pig, rat and human vascular endothelium. J Physiol. 1990;431:379–401. doi: 10.1113/jphysiol.1990.sp018335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai S, Pestic-Dragovich L, O'Donnell ME, Wang N, Ingber D, Elson E, De Lanerolle P. Regulation of cytoskeletal mechanics and cell growth by myosin light chain phosphorylation. Am J Physiol. 1998;275(5 Pt 1):C1349–C1356. doi: 10.1152/ajpcell.1998.275.5.C1349. [DOI] [PubMed] [Google Scholar]
- 25.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133(6):1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verin AD, Patterson CE, Day MA, Garcia JG. Regulation of endothelial cell gap formation and barrier function by myosin-associated phosphatase activities. Am J Physiol. 1995;269(1 Pt 1):L99–L108. doi: 10.1152/ajplung.1995.269.1.L99. [DOI] [PubMed] [Google Scholar]
- 27.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273(34):21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 28.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150(4):797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watterson DM, Schavocky JP, Guo L, Weiss C, Chlenski A, Shirinsky VP, Van Eldik LJ, Haiech J. Analysis of the kinase-related protein gene found at human chromosome 3q21 in a multi-gene cluster: Organization, expression, alternative splicing, and polymorphic marker. J Cell Biochem. 1999;75(3):481–491. [PubMed] [Google Scholar]
- 30.Herring BP, El-Mounayri O, Gallagher PJ, Yin F, Zhou J. Regulation of myosin light chain kinase and telokin expression in smooth muscle tissues. Am J Physiol Cell Physiol. 2006;291(5):C817–C827. doi: 10.1152/ajpcell.00198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potier MC, Chelot E, Pekarsky Y, Gardiner K, Rossier J, Turnell WG. The human myosin light chain kinase (MLCK) from hippocampus: Cloning, sequencing, expression, and localization to 3qcen-q21. Genomics. 1995;29(3):562–570. doi: 10.1006/geno.1995.9965. [DOI] [PubMed] [Google Scholar]
- 32.Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys. 2011;510(2):135–146. doi: 10.1016/j.abb.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ Res. 2008;102(5):571–580. doi: 10.1161/CIRCRESAHA.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia JG, Lazar V, Gilbert-McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: Molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997;16(5):489–494. doi: 10.1165/ajrcmb.16.5.9160829. [DOI] [PubMed] [Google Scholar]
- 35.Gallagher PJ, Herring BP. The carboxyl terminus of the smooth muscle myosin light chain kinase is expressed as an independent protein, telokin. J Biol Chem. 1991;266(35):23945–23952. [PMC free article] [PubMed] [Google Scholar]
- 36.Blue EK, Goeckeler ZM, Jin Y, Hou L, Dixon SA, Herring BP, Wysolmerski RB, Gallagher PJ. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am J Physiol Cell Physiol. 2002;282(3):C451–C460. doi: 10.1152/ajpcell.00333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src) J Biol Chem. 2001;276(11):8567–8573. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- 38.Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276(7):4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 39.Ito M, Guerriero V, Jr., Hartshorne DJ. Structure-function relationships in smooth muscle myosin light chain kinase. Adv Exp Med Biol. 1991;304:3–10. doi: 10.1007/978-1-4684-6003-2_2. [DOI] [PubMed] [Google Scholar]
- 40.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: Role of myosin light chain phosphorylation. J Cell Physiol. 1995;163(3):510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 41.Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: The relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. 1995;130(3):613–627. doi: 10.1083/jcb.130.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW, 2nd, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol. 1986;128(1):96–104. doi: 10.1002/jcp.1041280115. [DOI] [PubMed] [Google Scholar]
- 43.Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: Modulation of basal and agonist-stimulated venular permeability. Am J Physiol. 1997;272(3 Pt 2):H1437–H1443. doi: 10.1152/ajpheart.1997.272.3.H1437. [DOI] [PubMed] [Google Scholar]
- 44.Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am J Physiol Cell Physiol. 2000;279(4):C1285–1289. doi: 10.1152/ajpcell.2000.279.4.C1285. [DOI] [PubMed] [Google Scholar]
- 45.Kuhlmann CR, Tamaki R, Gamerdinger M, Lessmann V, Behl C, Kempski OS, Luhmann HJ. Inhibition of the myosin light chain kinase prevents hypoxia-induced blood-brain barrier disruption. J Neurochem. 2007;102(2):501–507. doi: 10.1111/j.1471-4159.2007.04506.x. [DOI] [PubMed] [Google Scholar]
- 46.Tinsley JH, Teasdale NR, Yuan SY. Myosin light chain phosphorylation and pulmonary endothelial cell hyperpermeability in burns. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L841–L847. doi: 10.1152/ajplung.00341.2003. [DOI] [PubMed] [Google Scholar]
- 47.Tinsley JH, Hunter FA, Childs EW. PKC and MLCK-dependent, cytokine-induced rat coronary endothelial dysfunction. J Surg Res. 2009;152(1):76–83. doi: 10.1016/j.jss.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 48.Adamson RH, Zeng M, Adamson GN, Lenz JF, Curry FE. PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol. 2003;285(1):H406–H417. doi: 10.1152/ajpheart.00021.2003. [DOI] [PubMed] [Google Scholar]
- 49.Cohen-Kashi Malina K, Cooper I, Teichberg VI. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. 2009;1284:12–21. doi: 10.1016/j.brainres.2009.05.072. [DOI] [PubMed] [Google Scholar]
- 50.Roux F, Couraud PO. Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions. Cell Mol Neurobiol. 2005;25(1):41–58. doi: 10.1007/s10571-004-1376-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reed RK, Rubin K. Transcapillary exchange: Role and importance of the interstitial fluid pressure and the extracellular matrix. Cardiovasc Res. 2010;87(2):211–217. doi: 10.1093/cvr/cvq143. [DOI] [PubMed] [Google Scholar]
- 52.Norwood N, Moore TM, Dean DA, Bhattacharjee R, Li M, Stevens T. Store-operated calcium entry and increased endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. 2000;279(5):L815–L824. doi: 10.1152/ajplung.2000.279.5.L815. [DOI] [PubMed] [Google Scholar]
- 53.Hicks K, O'Neil RG, Dubinsky WS, Brown RC. TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am J Physiol Cell Physiol. 2010;298(6):C1583–C1593. doi: 10.1152/ajpcell.00458.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005;29(6):999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- 55.Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. 2007;100(2):324–336. doi: 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- 56.Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29(12):1933–1945. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuhlmann CR, Librizzi L, Closhen D, Pflanzner T, Lessmann V, Pietrzik CU, de Curtis M, Luhmann HJ. Mechanisms of C-reactive protein-induced blood-brain barrier disruption. Stroke. 2009;40(4):1458–1466. doi: 10.1161/STROKEAHA.108.535930. [DOI] [PubMed] [Google Scholar]
- 58.Luh C, Kuhlmann CR, Ackermann B, Timaru-Kast R, Luhmann HJ, Behl C, Werner C, Engelhard K, Thal SC. Inhibition of myosin light chain kinase reduces brain edema formation after traumatic brain injury. J Neurochem. 2010;112(4):1015–1025. doi: 10.1111/j.1471-4159.2009.06514.x. [DOI] [PubMed] [Google Scholar]
- 59.Cioffi DL, Barry C, Stevens T. Store-operated calcium entry channels in pulmonary endothelium: The emerging story of TRPCS and Orai1. Adv Exp Med Biol. 2010;661:137–154. doi: 10.1007/978-1-60761-500-2_9. [DOI] [PubMed] [Google Scholar]
- 60.Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55(6):498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 61.Gaudreault N, Perrin RM, Guo M, Clanton CP, Wu MH, Yuan SY. Counter regulatory effects of PKCbetaII and PKCdelta on coronary endothelial permeability. Arterioscler Thromb Vasc Biol. 2008;28(8):1527–1533. doi: 10.1161/ATVBAHA.108.166975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan SY, Ustinova EE, Wu MH, Tinsley JH, Xu W, Korompai FL, Taulman AC. Protein kinase C activation contributes to microvascular barrier dysfunction in the heart at early stages of diabetes. Circ Res. 2000;87(5):412–417. doi: 10.1161/01.res.87.5.412. [DOI] [PubMed] [Google Scholar]
- 63.Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ. Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. Am J Physiol. 1999;276(2 Pt 2):H535–H542. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]
- 64.Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30(11):1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005;289(5):H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 66.Peng J, He F, Zhang C, Deng X, Yin F. Protein kinase C-alpha signals P115RhoGEF phosphorylation and RhoA activation in TNF-alpha-induced mouse brain microvascular endothelial cell barrier dysfunction. J Neuroinflamm. 2011;8(1):28. doi: 10.1186/1742-2094-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, Smith LE, King GL. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46(9):1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- 68.Aiello LP, Clermont A, Arora V, Davis MD, Sheetz MJ, Bursell SE. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Invest Ophthalmol Vis Sci. 2006;47(1):86–92. doi: 10.1167/iovs.05-0757. [DOI] [PubMed] [Google Scholar]
- 69.Kim JH, Jun HO, Yu YS, Kim KW. Inhibition of protein kinase C delta attenuates blood-retinal barrier breakdown in diabetic retinopathy. Am J Pathol. 2010;176(3):1517–1524. doi: 10.2353/ajpath.2010.090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59(11):2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oubaha M, Gratton JP. Phosphorylation of endothelial nitric oxide synthase by atypical PKC zeta contributes to angiopoietin-1-dependent inhibition of VEGF-induced endothelial permeability in vitro. Blood. 2009;114(15):3343–3351. doi: 10.1182/blood-2008-12-196584. [DOI] [PubMed] [Google Scholar]
- 72.Motley ED, Eguchi K, Patterson MM, Palmer PD, Suzuki H, Eguchi S. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by thrombin. Hypertension. 2007;49(3):577–583. doi: 10.1161/01.HYP.0000255954.80025.34. [DOI] [PubMed] [Google Scholar]
- 73.Duran WN, Breslin JW, Sanchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res. 2010;87(2):254–261. doi: 10.1093/cvr/cvq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Granger DN, Kubes P. Nitric oxide as anti-inflammatory agent. Methods Enzymol. 1996;269:434–442. doi: 10.1016/s0076-6879(96)69044-2. [DOI] [PubMed] [Google Scholar]
- 75.Kemp-Harper B, Schmidt HH. cGMP in the vasculature. In: Schmidt HH, Hofmann F, Stasch J-P, editors. cGMP: Generators, Effectors and Therapeutic Implications. 2008/12/18 edition. Volume 191. Springer-Verlag; New York: 2009. pp. 447–468. Handb Exp Pharmacol. [Google Scholar]
- 76.Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM. Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am J Physiol. 1993;264(5 Pt 2):H1734–H1739. doi: 10.1152/ajpheart.1993.264.5.H1734. [DOI] [PubMed] [Google Scholar]
- 77.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am J Physiol. 1992;263(2 Pt 2):H641–H646. doi: 10.1152/ajpheart.1992.263.2.H641. [DOI] [PubMed] [Google Scholar]
- 78.Wu HM, Huang Q, Yuan Y, Granger HJ. VEGF induces NO-dependent hyperpermeability in coronary venules. Am J Physiol. 1996;271(6 Pt 2):H2735–H2739. doi: 10.1152/ajpheart.1996.271.6.H2735. [DOI] [PubMed] [Google Scholar]
- 79.Huang Q, Yuan Y. Interaction of PKC and NOS in signal transduction of microvascular hyperpermeability. Am J Physiol. 1997;273(5 Pt 2):H2442–H2451. doi: 10.1152/ajpheart.1997.273.5.H2442. [DOI] [PubMed] [Google Scholar]
- 80.Hood J, Granger HJ. Protein kinase G mediates vascular endothelial growth factor-induced Raf-1 activation and proliferation in human endothelial cells. J Biol Chem. 1998;273(36):23504–23508. doi: 10.1074/jbc.273.36.23504. [DOI] [PubMed] [Google Scholar]
- 81.Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, Ledda F, Ziche M. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. J Biol Chem. 1998;273(7):4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- 82.Wu MH, Yuan SY, Granger HJ. The protein kinase MEK1/2 mediate vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules. J Physiol. 2005;563(Pt 1):95–104. doi: 10.1113/jphysiol.2004.076075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kasseckert SA, Schafer C, Kluger A, Gligorievski D, Tillmann J, Schluter KD, Noll T, Sauer H, Piper HM, Abdallah Y. Stimulation of cGMP signalling protects coronary endothelium against reperfusion-induced intercellular gap formation. Cardiovasc Res. 2009;83(2):381–387. doi: 10.1093/cvr/cvp065. [DOI] [PubMed] [Google Scholar]
- 84.Moldobaeva A, Welsh-Servinsky LE, Shimoda LA, Stephens RS, Verin AD, Tuder RM, Pearse DB. Role of protein kinase G in barrier-protective effects of cGMP in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L919–L930. doi: 10.1152/ajplung.00434.2005. [DOI] [PubMed] [Google Scholar]
- 85.Chong TJ, Victorino GP. Cyclic nucleotide second messengers (cAMP and cGMP) play a central role in signal transduction and regulation of mesenteric postcapillary fluid leak. J Trauma. 2005;59(2):302–306. doi: 10.1097/01.ta.0000180385.23675.98. discussion 306–307. [DOI] [PubMed] [Google Scholar]
- 86.Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium. 2000;7(4):287–308. doi: 10.3109/10623320009072215. [DOI] [PubMed] [Google Scholar]
- 87.Surapisitchat J, Jeon KI, Yan C, Beavo JA. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ Res. 2007;101(8):811–818. doi: 10.1161/CIRCRESAHA.107.154229. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DB. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295(6):L1056–L1065. doi: 10.1152/ajplung.90329.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Werthmann RC, von Hayn K, Nikolaev VO, Lohse MJ, Bunemann M. Real-time monitoring of cAMP levels in living endothelial cells: Thrombin transiently inhibits adenylyl cyclase 6. J Physiol. 2009;587(Pt 16):4091–4104. doi: 10.1113/jphysiol.2009.172957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qian J, Zhang Q, Church JE, Stepp DW, Rudic RD, Fulton DJ. Role of local production of endothelium-derived nitric oxide on cGMP signaling and S-nitrosylation. Am J Physiol Heart Circ Physiol. 2010;298(1):H112–H118. doi: 10.1152/ajpheart.00614.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res. 2009;77(1):26–34. doi: 10.1016/j.mvr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 92.Davidson SM, Duchen MR. Endothelial mitochondria: Contributing to vascular function and disease. Circ Res. 2007;100(8):1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 93.Tran QK, Watanabe H. Calcium signalling in the endothelium. In: Moncada S, Higgs A, editors. The Vascular Endothelium I. 2006/09/27 edition. Volume 176. Springer-Verlag; New York: 2006. pp. 145–187. Handb Exp Pharmacol. [Google Scholar]
- 94.Rodrigues SF, Granger DN. Role of blood cells in ischaemia-reperfusion induced endothelial barrier failure. Cardiovasc Res. 2010;87(2):291–299. doi: 10.1093/cvr/cvq090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DiStasi MR, Ley K. Opening the flood-gates: How neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carman CV, Springer TA. Trans-cellular migration: Cell-cell contacts get intimate. Curr Opin Cell Biol. 2008;20(5):533–540. doi: 10.1016/j.ceb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.He P. Leucocyte/endothelium interactions and microvessel permeability: Coupled or uncoupled? Cardiovasc Res. 2010;87(2):281–290. doi: 10.1093/cvr/cvq140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carman CV. Mechanisms for transcellular diapedesis: Probing and pathfinding by `invadosome-like protrusions'. J Cell Sci. 2009;122(Pt 17):3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- 99.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167(2):377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26(6):784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lewis RE, Granger HJ. Diapedesis and the permeability of venous microvessels to protein macromolecules: The impact of leukotriene B4 (LTB4) Microvasc Res. 1988;35(1):27–47. doi: 10.1016/0026-2862(88)90048-9. [DOI] [PubMed] [Google Scholar]
- 102.Kim MH, Curry FR, Simon SI. Dynamics of neutrophil extravasation and vascular permeability are uncoupled during aseptic cutaneous wounding. Am J Physiol Cell Physiol. 2009;296(4):C848–C856. doi: 10.1152/ajpcell.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91(3):385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 104.Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ Res. 2002;90(11):1214–1221. doi: 10.1161/01.res.0000020402.73609.f1. [DOI] [PubMed] [Google Scholar]
- 105.Tinsley JH, Zawieja DC, Wu MH, Ustinova EE, Xu W, Yuan SY. Protein transfection of intact microvessels specifically modulates vasoreactivity and permeability. J Vasc Res. 2001;38(5):444–452. doi: 10.1159/000051077. [DOI] [PubMed] [Google Scholar]
- 106.Xu J, Gao XP, Ramchandran R, Zhao YY, Vogel SM, Malik AB. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat Immunol. 2008;9(8):880–886. doi: 10.1038/ni.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun C, Wu MH, Yuan SY. Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation. 2011;124(1):48–57. doi: 10.1161/CIRCULATIONAHA.110.988915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2009;24(4):1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- 109.Fischer S, Wiesnet M, Renz D, Schaper W. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur J Cell Biol. 2005;84(7):687–697. doi: 10.1016/j.ejcb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 110.Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102(3):347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 111.Hecquet CM, Ahmmed GU, Malik AB. TRPM2 channel regulates endothelial barrier function. Adv Exp Med Biol. 2010;661:155–167. doi: 10.1007/978-1-60761-500-2_10. [DOI] [PubMed] [Google Scholar]
- 112.Cai H, Davis ME, Drummond GR, Harrison DG. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler Thromb Vasc Biol. 2001;21(10):1571–1576. doi: 10.1161/hq1001.097028. [DOI] [PubMed] [Google Scholar]
- 113.Cai H, Liu D, Garcia JG. CaM Kinase II-dependent pathophysiological signalling in endothelial cells. Cardiovasc Res. 2008;77(1):30–34. doi: 10.1093/cvr/cvm010. [DOI] [PubMed] [Google Scholar]
- 114.Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, Blasig IE, Dijkstra CD, Ronken E, de Vries HE. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21(13):3666–3676. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 115.Arturson G. Forty years in burns research-the postburn inflammatory response. Burns. 2000;26:599–604. doi: 10.1016/s0305-4179(00)00069-3. [DOI] [PubMed] [Google Scholar]
- 116.Cioffi WG. What's new in burns and metabolism. J Am Coll Surg. 2001;192(2):241–254. doi: 10.1016/s1072-7515(00)00795-x. [DOI] [PubMed] [Google Scholar]
- 117.Gibran NS, Heimbach DM. Current status of burn wound pathophysiology. Clin Plast Surg. 2000;27(1):11–22. [PubMed] [Google Scholar]
- 118.Lund T, Onarheim H, Reed RK. Pathogenesis of edema formation in burn injuries. World J Surg. 1992;16(1):2–9. doi: 10.1007/BF02067107. [DOI] [PubMed] [Google Scholar]
- 119.Evers LH, Bhavsar D, Mailander P. The biology of burn injury. Exp Dermatol. 2010;19(9):777–783. doi: 10.1111/j.1600-0625.2010.01105.x. [DOI] [PubMed] [Google Scholar]
- 120.Spanholtz TA, Theodorou P, Amini P, Spilker G. Severe burn injuries: Acute and long-term treatment. Dtsch Arztebl Int. 2009;106(38):607–613. doi: 10.3238/arztebl.2009.0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deitch EA. Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg. 1992;216(2):117–134. doi: 10.1097/00000658-199208000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Demling RH. The burn edema process: Current concepts. J Burn Care Rehabil. 2005;26(3):207–227. [PubMed] [Google Scholar]
- 123.Breslin JW, Wu MH, Guo M, Reynoso R, Yuan SY. Toll-like receptor 4 contributes to microvascular inflammation and barrier dysfunction in thermal injury. Shock. 2008;29(3):349–355. doi: 10.1097/shk.0b013e3181454975. [DOI] [PubMed] [Google Scholar]
- 124.Huxley VH, Curry FE, Adamson RH. Quantitative fluorescence microscopy on single capillaries: Alpha-lactalbumin transport. Am J Physiol. 1987;252(1 Pt 2):H188–H197. doi: 10.1152/ajpheart.1987.252.1.H188. [DOI] [PubMed] [Google Scholar]
- 125.Witte S. Microphotometric techniques in intravital microcirculatory studies. J Microsc. 1979;116(3):373–384. doi: 10.1111/j.1365-2818.1979.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 126.Bekker AY, Ritter AB, Duran WN. Analysis of microvascular permeability to macromolecules by video-image digital processing. Microvasc Res. 1989;38(2):200–216. doi: 10.1016/0026-2862(89)90028-9. [DOI] [PubMed] [Google Scholar]
- 127.Huang Q, Xu W, Ustinova E, Wu M, Childs E, Hunter F, Yuan S. Myosin light chain kinase-dependent microvascular hyperpermeability in thermal injury. Shock. 2003;20(4):363–368. doi: 10.1097/01.shk.0000079425.0000.db. [DOI] [PubMed] [Google Scholar]
- 128.Finnerty CC, Przkora R, Herndon DN, Jeschke MG. Cytokine expression profile over time in burned mice. Cytokine. 2009;45(1):20–25. doi: 10.1016/j.cyto.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: Implications in inflammation. Expert Rev Mol Med. 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mileski WJ, Burkhart D, Hunt JL, Kagan RJ, Saffle JR, Herndon DN, Heimbach DM, Luterman A, Yurt RW, Goodwin CW, Hansborough J. Clinical effects of inhibiting leukocyte adhesion with monoclonal antibody to intercellular adhesion molecule-1 (enlimomab) in the treatment of partial-thickness burn injury. J Trauma. 2003;54(5):950–958. doi: 10.1097/01.TA.0000030626.84680.11. [DOI] [PubMed] [Google Scholar]
- 131.Piccolo MT, Wang Y, Verbrugge S, Warner RL, Sannomiya P, Piccolo NS, Piccolo MS, Hugli TE, Ward PA, Till GO. Role of chemotactic factors in neutrophil activation after thermal injury in rats. Inflammation. 1999;23(4):371–385. doi: 10.1023/a:1020213717336. [DOI] [PubMed] [Google Scholar]
- 132.Shimizu S, Tanaka H, Sakaki S, Yukioka T, Matsuda H, Shimazaki S. Burn depth affects dermal interstitial fluid pressure, free radical production, and serum histamine levels in rats. J Trauma. 2002;52(4):683–687. doi: 10.1097/00005373-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 133.Gibran NS, Heimbach DM. Mediators in thermal injury. Semin Nephrol. 1993;13(4):344–358. [PubMed] [Google Scholar]
- 134.Kowal-Vern A, Walenga JM, Sharp-Pucci M, Hoppensteadt D, Gamelli RL. Postburn edema and related changes in interleukin-2, leukocytes, platelet activation, endothelin-1, and C1 esterase inhibitor. J Burn Care Rehabil. 1997;18(2):99–103. doi: 10.1097/00004630-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 135.Kurose I, Wolf R, Miyasaka M, Anderson DC, Granger DN. Microvascular dysfunction induced by nonsteroidal anti-inflammatory drugs: Role of leukocytes. Am J Physiol. 1996;270(2 Pt 1):G363–369. doi: 10.1152/ajpgi.1996.270.2.G363. [DOI] [PubMed] [Google Scholar]
- 136.Reynoso R, Perrin RM, Breslin JW, Daines DA, Watson KD, Watterson DM, Wu MH, Yuan S. A role for long chain myosin light chain kinase (MLCK-210) in microvascular hyperpermeability during severe burns. Shock. 2007;28(5):589–595. doi: 10.1097/SHK.0b013e31804d415f. [DOI] [PubMed] [Google Scholar]
- 137.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 138.Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, Shanholtz C, Yates CR, Meduri GU, Shriver MD, Ingersoll R, Scott AF, Beaty TH, Moitra J, Ma SF, Ye SQ, Barnes KC, Garcia JG. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol. 2006;34(4):487–495. doi: 10.1165/rcmb.2005-0404OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Christie JD, Ma SF, Aplenc R, Li M, Lanken PN, Shah CV, Fuchs B, Albelda SM, Flores C, Garcia JG. Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med. 2008;36(10):2794–2800. doi: 10.1097/ccm.0b013e318186b843. [DOI] [PubMed] [Google Scholar]
- 140.Wainwright MS, Rossi J, Schavocky J, Crawford S, Steinhorn D, Velentza AV, Zasadzki M, Shirinsky V, Jia Y, Haiech J, Van Eldik LJ, Watterson DM. Protein kinase involved in lung injury susceptibility: Evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci USA. 2003;100(10):6233–6238. doi: 10.1073/pnas.1031595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Moitra J, Evenoski C, Sammani S, Wadgaonkar R, Turner JR, Ma SF, Garcia JG. A transgenic mouse with vascular endothelial over-expression of the non-muscle myosin light chain kinase-2 isoform is susceptible to inflammatory lung injury: Role of sexual dimorphism and age. Transl Res. 2008;151(3):141–153. doi: 10.1016/j.trsl.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Komarova YA, Mehta D, Malik AB. Dual regulation of endothelial junctional permeability. Sci STKE. 2007;2007(412):re8. doi: 10.1126/stke.4122007re8. [DOI] [PubMed] [Google Scholar]
- 143.Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77(1):39–45. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119(7):1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, Garcia JG. Differential effects of S1P receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43(4):394–402. doi: 10.1165/rcmb.2009-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao J, Singleton PA, Brown ME, Dudek SM, Garcia JG. Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: Role in barrier enhancement. Cell Signal. 2009;21(12):1945–1960. doi: 10.1016/j.cellsig.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, Lal R, Van Eyk JE, Imam SZ, Garcia JG. Abl tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21(22):4042–4056. doi: 10.1091/mbc.E09-10-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dudek SM, Birukov KG, Zhan X, Garcia JG. Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun. 2002;298(4):511–519. doi: 10.1016/s0006-291x(02)02492-0. [DOI] [PubMed] [Google Scholar]
- 149.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: Roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279(23):24692–24700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 150.Owens SE, Graham WV, Siccardi D, Turner JR, Mrsny RJ. A strategy to identify stable membrane-permeant peptide inhibitors of myosin light chain kinase. Pharm Res. 2005;22(5):703–709. doi: 10.1007/s11095-005-2584-9. [DOI] [PubMed] [Google Scholar]
- 151.Rossi JL, Velentza AV, Steinhorn DM, Watterson DM, Wainwright MS. MLCK210 gene knockout or kinase inhibition preserves lung function following endotoxin-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1327–L1334. doi: 10.1152/ajplung.00380.2006. [DOI] [PubMed] [Google Scholar]
- 152.Behanna HA, Watterson DM, Ranaivo HR. Development of a novel bioavailable inhibitor of the calmodulin-regulated protein kinase MLCK: A lead compound that attenuates vascular leak. Biochim Biophys Acta. 2006;1763(11):1266–1274. doi: 10.1016/j.bbamcr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 153.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115(10):2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ohlmann P, Tesse A, Loichot C, Ralay Ranaivo H, Roul G, Philippe C, Watterson DM, Haiech J, Andriantsitohaina R. Deletion of MLCK210 induces subtle changes in vascular reactivity but does not affect cardiac function. Am J Physiol Heart Circ Physiol. 2005;289(6):H2342–2349. doi: 10.1152/ajpheart.00511.2004. [DOI] [PubMed] [Google Scholar]
- 155.van Nieuw Amerongen GP, van Hinsbergh VW. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul Pharmacol. 2002;39(4–5):257–272. doi: 10.1016/s1537-1891(03)00014-4. [DOI] [PubMed] [Google Scholar]
- 156.Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7(2):222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- 157.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109(3):366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]