

Abstract
Pharmacoperone drugs correct the folding of misfolded protein mutants and restore function (i.e. “rescue”) by correcting the routing of (otherwise) misrouted mutants. Assays for pharmacoperones have not been applied to screen large libraries previously. Currently most pharmacoperones possess intrinsic agonist or antagonist activities since these were identified using high throughput screens aimed at discovering direct agonists or antagonists. Here we describe an ultra-high throughput compatible no-wash assay system designed to specifically identify pharmacoperones of the vasopressin type 2 (V2) receptor (V2R). Development of such assays is important and novel since useful chemical structures with the ability to control cellular trafficking, but lacking intrinsic agonist or antagonist properties have not likely been identified using existing screens. In the described assay, the level of functional hV2R (mutant) present in each test well is quantitated by stimulation with saturating levels of agonist followed by use of a luminescent-based cyclic adenosine monophosphate (cAMP) assay. This allows the assay to identify compounds which increase the trafficking of mutant hV2R[L83Q] in our model system.
Keywords: vasopressin type 2 receptor (V2R), pharmacoperone, chemical library, GPCRs, protein
Introduction
G protein coupled receptors (GPCRs), which include the vasopressin type 2 receptor (V2R), comprise the largest family of validated drug targets — 35-50% of approved drugs derive their benefits by selective targeting of GPCRs. Mutations in GPCRs are frequently misrouted proteins and are known to be responsible for over 30 disorders, including cancers, heritable obesity and endocrine disease. Included in this group is nephrogenic diabetes insipidus, caused by mutations in the hV2R 5. Although this class has been broadly targeted in HTS campaigns in the past, the assays used have typically been optimized for identification of direct agonists and antagonists. Valuable drugs which affect the trafficking of GPCRs may have been overlooked because of this limitation 3; 4.
Normally, GPCRs are subjected to a stringent quality control system (QCS) in the endoplasmic reticulum (ER); this system consists of both protein chaperones that retain misfolded proteins and enzyme-like proteins that catalyze the folding process. The QCS (consisting of endogenous chaperones), which assesses structure but not function, insures that only correctly folded proteins enter the pathway leading to the plasma membrane (PM) 5. Because of this, point mutations may result in the production of misfolded and disease-causing proteins that are unable to reach their functional destinations in the cell because they are retained by the QCS even though they may retain (or regain) function.
The functional rescue of misfolded mutant receptors by small non-peptide molecules, originally screened from libraries to serve as receptor antagonists, has now been demonstrated. A pharmacoperone is a small molecule that enters cells and serves as a “molecular scaffold” to promote correct folding of otherwise misfolded mutant proteins within the cell 9. Misfolded proteins are frequently retained by the cellular quality control system (QCS) of the endoplasmic reticulum (ER), and do not reach their normal site 2. Pharmacoperones can rescue misfolded receptor mutants and restore them to function, which is a potentially useful therapeutic approach when the target is a misfolded/misrouted protein (Fig. 1a). We have summarized the literature for the gonadotropin releasing hormone (GnRH) and vasopressin type 2 (V2) receptor’s pharmacoperones with a view toward moving these compounds in vivo5.
Fig. 1. The canonical pathway of protein translation from mRNA.

(a) emphasizing that misfolded proteins are retained in the cell (ER and elsewhere) and can be rescued by target specific pharmacoperone drugs (gray arrow) that correct misfolding and restore function. Redrawn from 5. (b) The previously reported HTS protocol involving multiple wash steps (gray box) and lengthy incubations. (c) The new optimized ultra-HTS compatible no-wash protocol. Changes from the wash protocol are denoted in bold.
The pharmacoperone approach is a viable alternative to gene therapy since it serves as a means of correcting the function of defective proteins. This view is supported by the consideration that correction of defective protein folding appears significantly less challenging than replacement of a defective gene (or gene product) by a perfect one. The QCS is not protein-specific; it recognizes general aspects of misfolding (e.g. exposure of hydrophobic plates in aqueous environments), frequently with relatively low affinity. Accordingly, GPCRs that retain ligand binding and effector coupling but are recognized as misfolded by such general criteria, are often retained in the ER. Their rescue with pharmacoperones leads to proper folding, passage through the QCS, restoration to the proper site and return of function.
The V2 receptor (V2R, also known as the arginine vasopressin receptor) is expressed in the distal convoluted tubule and the collecting ducts of the kidney. V2R responds to vasopressin by stimulating mechanisms that concentrate the urine and maintain water homeostasis in the organism. When the function of V2R is lost due to mutation, the disease nephrogenic diabetes insipidus (NDI) results. The current goal of treatment is to control the body’s fluid levels and problems with electrolyte imbalances. Patients must drink large quantities of water to offset the loss. If the affected person does not drink enough fluids, high urine output may cause dehydration and high levels of sodium in the blood. NDI is caused by an inborn error of metabolism that is present at birth; it is a chronic condition requiring lifelong treatment. Complications can include dilation of the ureters and bladder, hypernatremia, severe dehydration and shock. Presently, there are no drugs known which reverse this condition, which frequently results in death.
We have previously reported a microtiter plate-based screening assay system designed to detect compounds able to act as pharmacoperones in the V2R system 14. This system relied on several long incubations coupled with multiple washing steps to remove the variable test compounds prior to stimulation of active V2R with vasopressin. While this assay format resulted in a robust signal, the multi-step wash assay protocol was not amenable to ultra-high throughput screening. We now report the further optimization of this assay and the development of a no-wash mix and read protocol well suited to large scale screening for pharmacoperones active in the V2R system.
Materials and Methods
Materials
SR121463 V2R antagonist, used in the current study as a pharmacoperone drug, was generously provided by Dr. Claudine Serradeil at Sanofi-Aventis and used as received. The scaling library (LOPAC) was obtained from Sigma Aldrich (St. Louis, MO) and stored as 10 mM DMSO stock solutions at −20 °C in sealed polypropylene plates. 3-Isobutyl-1-methylxanthine (IBMX, Sigma Aldrich, St. Louis, MO), vasopressin (Tocris Biosciences, Bristol, England UK) and fetal bovine serum (FBS, Hyclone, Logan, UT) were obtained as indicated.
uHTS Optimized Primary V2R Pharmacoperone Assay
HeLa cells stably expressing L83Q hV2R under the control of a tetracycline-controlled transactivator were cultured in growth media (1× DMEM + 10% FBS and 1 mg/L gentamicin10. All reagents were added by a BioRaptor (Beckman Coulter) unless specified otherwise below. On the day of screening, cells were trypsinized and added to plates (4 μl/well, 3000 cells/well). This followed immediately by pin-tool addition (Kalypsys) of test compounds and controls (positive control is 100 nM SR121463, negative control is carrier only) in 10 nl of DMSO. The drugged plates were incubated for 17 hours at 37 °C, 5% CO2 prior to addition of 1 μM vasopressin (final) in 1 μl stimulation buffer (growth media plus 2.5 mM IBMX). After stimulation, plates were incubated for 2 hours at 37 °C, 5% CO2 then allowed to equilibrate at room temperature for 10 minutes followed by addition of 1 μl of 6X cAMP-Glo detection reagent (Promega). After incubation at room temperature for 20 minutes, 6 μl of 1X Kinase-Glo (Promega) was added to each well to quantitate residual ATP levels and produce the final luminescent endpoint in the cAMP-Glo detection protocol Kinase-Glo is a part of the cAMP-Glo detection reagent system. Plates were incubated a further 10 minutes at room temperature and luminescence was measured using a PerkinElmer Viewlux plate imager. The optimized counter screen was identical to the primary screen described except that cells were cultured in the presence of 1 μg/ml doxycycline for 36 hours prior to plating and during all phases of the experiment.
Data Processing
Raw luminescence values were normalized to positive and negative controls to give percent response scores. Hits in screening runs were identified as compounds showing activity more than three standard deviations away from the negative control population6. Dose response curves were fit using a four-parameter variable slope sigmoidal curve in Graphpad Prism 5.02 (GraphPad Software, Inc). Competitive binding curves were fit using a one site – fit Ki model in Prism 5.02.
Results and Discussion
Selection of Positive Control Compound
In the present study a competitive antagonist of the V2R (SR121463) known to act as a pharmacoperone and able to rescue mutants of the V2R and restore them to function 12 served as the positive control compound. As in the case of other systems with other pharmacoperones 8, this pharmacoperone rescued many mutants of the V2R and was used as a positive control in our assay development, although the antagonism it shows to the V2R makes it a sub-optimum drug 1. This is because the antagonistic side makes it necessary to first rescue, then wash out the drug to allow agonism by the endogenous ligand. When applied in vivo, a short-term treatment with a V1a receptor antagonist showed that patients given this molecule decreased both 24-h urine volume and water intake. Maximum increase in urine osmolality was observed on day 3 and sodium, potassium, creatinine excretions and plasma sodium were constant throughout the study 1. Unfortunately, the trade-off between antagonism and pharmacoperone activity resulted in complex pharmacology that did not allow full exploitation of pharmacoperone activity. In the present study SR121463 served as a positive control for establishment of the assay.
The Primary Assay
The primary assay system for this project uses a HeLa cell line constitutively expressing hV2R[L83Q] coupled to a tetracycline-regulated transactivator 10; 14. In the absence of doxycycline (stable analog of tetracycline), the mutant is expressed, then misrouted and retained in the endoplasmic reticulum (ER). Following pretreatment with pharmacoperone, the mutant is rescued and trafficked to the plasma membrane. The rescued mutant is then responsive to native vasopressin and coupled to the production of cAMP, which is quantified using the commercially available cAMP-Glo enzyme-linked reagent system 11.
The basis of the assay is that the mutant hV2R[L83Q] is retained in the ER in untreated cells. Addition of pharmacoperone rescues hV2R[L83Q] and the receptor is trafficked to the plasma membrane. hV2R[L83Q] on the plasma membrane is now responsive to agonism by vasopressin. The level of functional hV2R (mutant) is proportional to the magnitude of cAMP.
In our previous wash assay protocol (Fig. 1b), cells were plated and allowed to grow for 24 hours prior to addition of pharmacoperone compounds. Following a 17 hour incubation with test compounds, the plates were washed three times by removing growth media and then incubating with fresh media at 37 °C. After washing, the cells were challenged with an EC100 of vasopressin, followed by lysing and quantitation of cAMP levels. The total time for this protocol from cell plating to data acquisition was 44 hours, with seven bulk liquid additions, four liquid removal steps, and eight timed incubations. In our newly optimized no-wash system (Fig. 1c), cells are plated immediately following trypsinization and drugged immediately. This is followed by a 17 hour incubation after which they are challenged with an EC50 of vasopressin, lysed and cAMP levels quantified. The no-wash protocol has a total time of 19.5 hours, four bulk liquid additions and four timed incubations. With no removal of SR121463, the apparent EC50 of vasopressin shifts by approximately two-hundred fifty fold (Fig. 2a). When the raw data from the vasopressin response using the no-wash protocol is fitted to a competitive binding model, a Kd value of 1.2 to 2.7 nM results (Fig. 2b), consistent with the literature reported Kd (1.6 nM) for vasopressin and V2R 15.
Fig. 2. Responses of controls in the 384-well formatted hV2R[L83Q] Pharmacoperone Assay.

(a)Comparison of vasopressin titrations in the absence of doxycycline using the heterogenous and no-wash protocols. Note, the shifted EC50 for the no-wash protocol (in the presence of 100 nM SR121463) indicative of the competitive antagonistic properties of this pharmacoperone. (b)Raw luminescence values from a vasopressin titration performed using the no-wash protocol in the presence of 100 nM SR121463 fit to a one site competitive binding model. Literature values (0.94 nM) 13 for the Ki of SR121463 were used to perform the fit. The resulting Kd for vasopressin is consistent with the literature value of 1.6 nM 15. (c) Vasopressin titrations. In the absence of doxycycline (“Dox”), hV2R[L83Q] is synthesized and retained in the ER; a vasopressin challenge yields no cAMP response (triangles). Following pre-treatment with 100 nM of the pharmacoperone SR121463 (“SR”), hV2R[L83Q] is rescued and trafficked to the plasma membrane, and a robust cAMP response to vasopressin challenge is observed (circles). Also included are the results of challenging the cells with vasopressin after preincubation with SR and 1 μg/ml doxycycline (squares). (d) Pharmacoperone titrations. In the absence of Dox, increasing concentrations of pharmacoperone SR121463 increased the amount of hV2R[L83Q] on the surface of the cells, with the result of increasing cAMP responses after challenge with an EC100 of vasopressin (circles). An EC100 challenge of vasopressin to cells in the absence of doxycycline (squares) or pretreated with 1 μg/ml doxycycline (triangles) results in no appreciable cAMP response. Each dataset is normalized to either the 100% response of either VP (left) or SR (right) dose-response curve.
The Counter Screen Assay
In high-throughput screening (HTS) efforts, a counter screen assay assists in the identification and triage of compounds that have off-target activity or optically interfere with measurement of signal. The counter screen protocol selected for HTS is identical to the primary HTS assay protocol with the exception that the hV2R[L83Q] cells are incubated in the presence of doxycycline (“Dox”) for 36 hours prior to assay. During this time, the gene for the mutant is off and no measurable mutant remains in the cell. Accordingly the hV2R[L83Q] primary assay and counter screen protocols confirm the expected pharmacology of positive and negative controls (Fig. 2c-d).
The SR121463 pharmacoperone yielded an EC50 of 1.97 ± 0.25 nM in the presence of 1 μM vasopressin using the no-wash protocol in the absence of doxycycline, a Z’ value of 0.7, and a signal-to-background ratio (S/B) of ~7 (n=4). In light of these promising results, we proceeded with a small-scale high throughput screen to confirm the performance of our optimized assay conditions.
LOPAC pilot screen
To determine the performance of our optimized hV2R[L83Q] pharmacoperone assay under HTS conditions, we used the assay to screen the Sigma LOPAC (Library of Pharmacologically Active Compounds). Briefly, compounds were analyzed at a single concentration of 6 μM (0.4% DMSO) using SR121463 as a positive control. Although this screen was not performed in 1536 well plates, low volume 384 well plates were used. These plates have similar surface area to volume ratios as those found in 1536 well format. Therefor we expect the described system will perform similarly in 1536 well format. Each plate contained high (100 nM SR121463) and low (DMSO only) signal control wells, which were used in Z’ factor calculations. An activity scatterplot of all compounds tested, as well as positive and negative controls, is shown in Fig. 3.
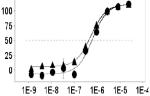
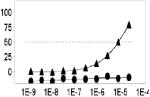
Fig. 3. Scatter plot analysis of the high-throughput LOPAC pilot screen.

(a) All data from all assay plates (n=17, triplicate results), including controls, are displayed. Large separation in activity (Z’ =0.85 ± 0.05, S/B=7.3 ± 0.6) between wells dosed with SR121463 (“high control”) and DMSO (“low control”), as well as a broad distribution of hits indicate an excellent HTS assay. (b) Representative graph of compound activity correlating plate replicas. The best fit line has an r2=0.86, indicative of the high fidelity of the no-wash assay hit identification.
As indicated from the positive and negative control scatterplots (Fig. 3a), the assay demonstrated a high Z’ factor (0.85 ± 0.05) for the entire LOPAC screen, indicative of an excellent assay window. Reproducibility of single compound activities was also excellent. A scatterplot of replicate measurements yields an r2= 0.86 (Fig. 3b) indicating highly reliable hit identification. It is important to note that a high number of hits are expected from LOPAC screens due to the high proportion of pharmacologically active compounds present in the LOPAC itself.
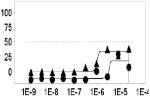
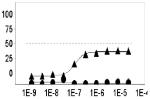
Another important parameter to assess for HTS readiness is the susceptibility of an assay to artifact. For example, in the case of the hV2R[L83Q] pharmacoperone assay compounds that non-specifically activate cyclase activity, agonists of endogenous GPCRs, cytotoxic compounds, and compounds that interfere with the luciferase-based detection reagents (via biochemical or optical means) may be falsely identified as hits. To assess the pharmacology of hits, as well as the contribution of artifact to the primary assay readout, we cherry-picked and titrated all hits from the LOPAC screen. Compounds were tested starting from 25 μM with 3-fold series dilution steps for 10 points (n=3) in both the primary hV2R[L83Q] pharmacoperone assay as well as its “ Dox” counter screen (i.e. pretreatment of the same cells with doxycycline 36 hours prior to assay). All hits confirmed activity in the titration assay, albeit with varying degrees of potency; a subset of the selected hits are shown in Table 1.
Table 1.
Primary assay and counterscreen results for three hits from the LOPAC screen. For EC50 determination compounds were titrated starting from 25 μM with 3 fold series dilution steps for 10 points (n=3). The x-axis of the dose response curve is the concentration of the tested compound in M and the y-axis is the percent activity determined by normalization of the endpoint to positive (100 nM SR121463) and negative (DMSO alone) control values.
| Structure | Vendor Information |
LOPAC Screen Average %Response +/−Std Dev |
EC50(μM) −Dox |
EC50(μM) +Dox |
Name | Selectivity | Dose Response Curves Triangles = −Dox Circles = +Dox |
|---|---|---|---|---|---|---|---|
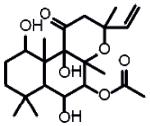
|
Sigma Aldrich F6886 |
104+/−0 | 0.43 | 0.59 | Forskolin | Activates adenylate cyclase |
|
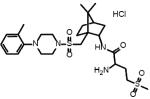
|
Sigma Aldrich L2540 |
74+/−2 | 8.3** | I.A. | L-368,899 | Non-peptide oxytocin receptor antagonist |
|
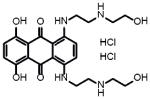
|
Sigma Aldrich M6545 |
51+/−1 | 0.90* | ~2* | Mitoxantrone | DNA synthesis inhibitor (cytotoxic) |
|
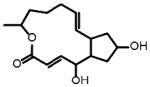
|
Sigma Aldrich B7651 |
43+/−1 | 0.27* | I.A. | Brefeldin A | Disrupts structure and function of the Golgi |
|
50% activity not reached with certainty for these compounds.
EC50 value reported is the concentration of compound needed to produce a response equal to one half that of 100 nM SRS121463. Saturating signal was not observed for these compounds.
As expected, the counter screen was effective at identifying false positives such as forskolin, as well as cytotoxic compounds (such as Mitoxantrone). Remarkably, some compounds showed activity only in the primary screen, such as L-368,899, an oxytocin receptor agonist. Given that the oxytocin receptor shares homology with the vasopressin receptor, this compound may be useful for further characterization. Another compound showing doxycycline dependent activity was brefeldin A, a fungal toxin known to disrupt the structure of the Golgi. While unlikely to be a specific pharmacoperone for V2R, it is interesting that a compound normally expected to decrease protein trafficking is, in this case, apparently increasing it. The fact that our assay system was able to detect this phenomenon underscores its overall utility in identifying small molecules with previously unknown and unexpected activities. Combined, these results demonstrated the ability of the hV2R[L83Q] pharmacoperone assay to identify potent hits, and the usefulness of the counter screen to triage off-target hits.
While this assay system is well behaved and is likely to detect compounds which allosterically increase trafficking of V2R to the cell surface without acting as direct agonists or antagonists, there are some limitations in the overall approach in identifying all compounds able to function as pharmacoperones in a given test library. Namely, highly potent antagonists or vasopressin competitive compounds with very slow off rates able to increase V2R trafficking may not be detected. However, for this target, this is likely advantageous, since direct antagonists or agonists often result in complex pharmacology complicating clinical use of pharmacoperones acting in this manner. Furthermore, an antagonistic compound would need to have a Ki value in the pico-molar range to be undetectable using this approach. In this system the positive control, SR121463, is an antagonist with a Ki of ~1 nM and is detectable at levels down to 1 nM, well below typical primary screening concentrations.
To our knowledge, this is the first report of a truly ultra-high throughput amenable assay system specifically designed to detect pharmacoperones. All pharmacoperone drugs for this target identified to date came from hits in screens for receptor antagonists and were repurposed as pharmacoperones. Unfortunately, these must be removed after rescue to preclude competition with agonists.
The importance of such non-antagonistic pharmacoperones was shown in a study of patients with X-linked nephrogenic diabetes insipidus (NDI). Mutant vasopressin 2 receptors in NDI resulted in misrouted proteins that were trapped in the ER, degraded and did not reach the plasma membrane in the collecting ducts of the kidney where they would normally reabsorb water. In vitro studies indicated that a non-peptide V1a receptor antagonist rescued cell surface expression and function of mutant V2 receptors. This would, in fact, be an unexpected requirement since one could imagine pharmacoperones that might stabilize the correctly routed form of the receptor and not show any antagonism (or agonism). Accordingly, identification of non-antagonistic pharmacoperones is a reasonable and therapeutically important goal.
Two additional observations are important, since these extend the therapeutic potential of these drugs.
First, pharmacoperone drugs need not be present at the time of protein synthesis, but can rescue ER-retained proteins that have already accumulated 7. This observation increases the therapeutic reach, since misfolded mutants need not be (first) degraded and then replaced by newly synthesized protein (i.e. the portion synthesized in the presence of pharmacoperone).
Second, while pharmacoperones are specific for individual proteins, those that rescue one mutant of an individual protein typically rescue most mutants of the same protein, likely by stabilizing a core region that makes the protein acceptable to the quality control system of the cell. This observation improves the therapeutic reach of these drugs 5; 8; 12 since each mutant of an individual protein will not require a separate drug.
The assays described in this publication should prove useful in the identification of new pharmacoperone chemotypes for development of novel therapeutics, as well as providing useful tools to further characterize the pharmacoperone phenomena. Furthermore, the general approach used provides a framework for designing phenotypic functional assays to identify pharmacoperones in other systems as well.
Acknowledgements
This work was supported by National Institutes of Health Grants DK85040, OD012220, OD011092 and MH084512. OTRADI is partially funded by grants from the Oregon Business Development Department and the Federal Economic Development Administration. We thank Jo Ann Binkerd for formatting the manuscript.
References
- 1.Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Lonergan M, Arthus MF, Laperriere A, Brouard R, Bouvier M, Bichet DG. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–243. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- 2.Castro-Fernandez C, Maya-Nunez G, Conn PM. Beyond the signal sequence: protein routing in health and disease. Endocr Rev. 2005;26:479–503. doi: 10.1210/er.2004-0010. [DOI] [PubMed] [Google Scholar]
- 3.Conn PM, Janovick JA. Drug development and the cellular quality control system. Trends Pharmacol Sci. 2009;30:228–233. doi: 10.1016/j.tips.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2010;21:190–197. doi: 10.1016/j.tem.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- 6.Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford ND, Bayly C, Suto C, Varney M, Strulovici B. Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem. 2003;313:246–254. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 7.Janovick JA, Brothers SP, Cornea A, Bush E, Goulet MT, Ashton WT, Sauer DR, Haviv F, Greer J, Conn PM. Refolding of misfolded mutant GPCR: post-translational pharmacoperone action in vitro. Mol Cell Endocrinol. 2007;272:77–85. doi: 10.1016/j.mce.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003;305:608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- 9.Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- 10.Janovick JA, Park BS, Conn PM. Therapeutic rescue of misfolded mutants: validation of primary high throughput screens for identification of pharmacoperone drugs. PLoS One. 2011;6:e22784. doi: 10.1371/journal.pone.0022784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar M, Hsiao K, Vidugiriene J, Goueli SA. A bioluminescent-based, HTS-compatible assay to monitor G-protein-coupled receptor modulation of cellular cyclic AMP. Assay Drug Dev Technol. 2007;5:237–245. doi: 10.1089/adt.2006.055. [DOI] [PubMed] [Google Scholar]
- 12.Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serradeil-Le Gal C. An overview of SR121463, a selective non-peptide vasopressin V(2) receptor antagonist. Cardiovascular drug reviews. 2001;19:201–214. doi: 10.1111/j.1527-3466.2001.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 14.Smithson DC, Janovick JA, Conn PM. Therapeutic rescue of misfolded/mistrafficked mutants: automation-friendly high throughput assays for identification of pharmacoperone drugs of GPCRs. Methods Enzymol. 2012 doi: 10.1016/B978-0-12-391862-8.00001-6. In Press. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama K, Yamauchi A, Izumi M, Itoh T, Ando A, Imai E, Kamada T, Ueda N. A low-affinity vasopressin V2-receptor gene in a kindred with X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 1996;7:410–414. doi: 10.1681/ASN.V73410. [DOI] [PubMed] [Google Scholar]