

Abstract
Background
Studies of mechanisms mediating resistance to chemotherapy led to the discovery of the multidrug transporter ABCB1, often expressed in leukemic cells of patients with acute myeloid leukemia (AML). Most clinical trials evaluating the strategy of inhibiting efflux-mediated chemotherapeutic resistance have been unsuccessful, clearly indicating the need for a better approach.
Methods
Here, we investigated the clinical relevance of 380 genes whose expression has been shown to affect the response to chemotherapy, mostly through in vitro studies, in 11 paired samples obtained at AML diagnosis and at relapse. The expression profiling of these 380 genes was performed using TaqMan-based qRT-PCR. Patients had a median age of 58 years at diagnosis, a median duration of complete remission (CR) of 284.5 days, and a median overall survival (OS) of 563 days. Cytogenetic abnormalities were detected at diagnosis in four patients, while five displayed a normal karyotype and two were not investigated.
Results
Hierarchical clustering shows that samples taken at diagnosis and relapse clustered in pairs for six patients out of the eleven studied, suggesting recurrence of the same leukemic blast, while for the other five patients, the data indicate their relapse blasts arose from different origins. A patient-by-patient analysis of the paired samples led to the striking observation that each had a unique gene signature representing different mechanisms of resistance.
Conclusions
The data underline the need for personalized molecular analysis to tailor treatment for patients with AML.
Keywords: acute myeloid leukemia, acquired drug resistance, therapy-related drug resistance, multidrug resistance, leukemia cell lines
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by clonal proliferation of malignant precursors of a myeloid lineage with impaired differentiation of normal hematopoietic progenitors1. While the majority of AML patients achieve remission, most eventually relapse, ultimately succumbing to the disease. Recent studies revealed the clonal architecture of secondary AML, which is a dynamic process shaped by multiple cycles of mutation acquisition and clonal selection2. However, the underlying alterations in gene transcription that allow relapse, after chemotherapy remain poorly understood.
ABCB1 (MDR1/P-gp) exports a wide variety of drugs that are mechanistically and structurally unrelated,3 and is often found to be highly expressed in leukemic blasts 4–6. Although inhibition of ABCB1 to increase chemosensitivity has been successful in vitro, translating this strategy to clinical settings has failed7, 8. Indeed, trials evaluating ABCB1 inhibitors in over 2,000 patients with AML have failed to yield positive results9. Since the discovery of ABCB1 and the emergence of the genomic era, numerous genes have been revealed to mediate drug resistance10. Yet the benefits of this research for patients who develop resistance to chemotherapy are minimal.
The current analytical methods for high-throughput gene expression profiling are based upon mean values to find differentially expressed genes, with the goal of identifying a common gene signature that defines a trend among patients. However, such a strategy is clearly insufficient, as the variability among patients is totally ignored. For such an analysis to identify rare signatures in individuals that are associated with drug resistance in which the prediction is based on a Boolean rule, the analysis of several hundred samples would be needed to avoid overfitting problems. Nevertheless, in a heterogeneous disease such as AML, one effective way to identify rare resistance mechanisms is to do a patient-by-patient analysis of paired samples taken at diagnosis and after relapse, thereby, minimizing false hits due to interpersonal variability11.
We have conducted a unique study using paired samples to discern the mechanisms involved in the acquisition of MDR in patients with AML, using highly sensitive and specific TaqMan-based qRT-PCR12. The 380 genes involved in phase I and II metabolism, signal transduction, DNA repair, stress response, tumor suppressor activity, oncogenic transformation, apoptosis, and drug uptake or efflux were chosen based on their potential role in MDR, as indicated by reports published over the past three decades13. Here, we extend to the transcriptional level the recent genetic discoveries in AML showing great heterogeneity of the tumor2, as the data demonstrate that the mechanisms of resistance are highly heterogeneous.
MATERIALS AND METHODS
Patient Samples
The collection of tumors for research and specifically molecular analysis was first approved by the ethical review board of the Karolinska Institutet, Stockholm, Sweden (ethical permit number: KI Dnr 03-600) and written informed consent was obtained from the patients. Peripheral blood was then collected from eleven patients with AML at diagnosis and at relapse after treatment, and separated with Ficoll-Paque (GE Healthcare, Piscataway, NJ, USA) according to the manufacturer’s instructions. Blasts were cryopreserved at the biobank at the Department of Hematology, Karolinska University Hospital. Pathological analysis at the Karolinska Institutet confirmed that each sample contained at least 80% leukemic blasts. Patients had undergone treatment with cytarabine combined with daunorubicin, idarubicin, etoposide, mitoxantrone, and/or thioguanine. Data on clinical outcomes were obtained from patient records. Duration of complete remission was the number of days between CR (bone marrow aspirates that showed less than 5% blasts) and first documented relapse in blood or bone marrow. Patients had a median age of 58 years at diagnosis, ranging from age 28 to 72. The median duration of complete remission (CR) was 284.5 days, with a range of 48 to 1166 days and the median overall survival (OS) was 563 days, ranging from a low of 193 to a high of 1664 days (Table 1)14. Cytogenetic aberrations were found in four patients at the time of diagnosis, five displayed a normal karyotype while two were not investigated. Regarding mutations of individual genes, e.g. FLT3 or NPM1, no such assessments were made at diagnosis, since the collection of samples took place during the time period 1987–2001.
Table 1.
Characteristics of 11 adult patients with AML who had undergone conventional chemotherapy and contributed paired samples (at diagnosis and after relapse)
| Patient | FABa | Sex | Age | CRb (days) | Treatmentc | OSd (days) | Karyotype according to chromosome banding analysis | Chrom scoree | WBCf | Platelets | SCTg | AHDh |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M4 | F | 62 | 56 | A/E/N | 264 | 45,XX,del(5)(q?),der(11)r(11;?) (p15q25?;?)-18 | 3 | 19.6 | 44 | n | n |
| 2 | M1 | M | 67 | 588 | A/E/N | 1252 | del (7q)(q22) 6 of 8 metafases | 2 | 9 | 65 | n | n |
| 3 | M1 | F | 63 | 190 | A/E/N | 440 | 46,XX,Ph+/46,XX,Ph+, 18,mar/46,XX | 3 | 83 | 303 | n | n |
| 4 | M5b | M | 72 | 1166 | A/T | 1664 | 46,XX | 2 | 9.6 | 165 | n | n |
| 5 | M4 | F | 72 | 532 | A/D/T | 686 | 46,XX | 2 | 32.2 | 36 | n | y |
| 6 | M5A | F | 29 | 48 | A/E/I | 193 | 46,XX | 2 | 35 | 87 | n | n |
| 7 | M1 | F | 54 | 113 | A/E/I | 225 | 46,XX | 2 | 45 | 48 | n | n |
| 8 | M4 | F | 53 | 368 | A/D/T | 1165 | Unknown | 9 | 12.4 | 47 | y | n |
| 9 | M5B | F | 53 | 893 | A/E/N | 1495 | Unknown | 9 | 15.6 | 33 | n | n |
| 10 | M2 | F | 43 | 201 | A/E/N | 384 | 46,XX | 2 | 34 | 96 | y | n |
| 11 | M0 | M | 23 | 757 | A/E/I | 1174 | 46,XY,add,(10)(p13),del(11)(q13),add(17)(q25)/46,XY | 2 | 37 | 150 | y | n |
FAB type: French-American-British classification system;
CR: complete remission;
Treatment: A/E/N=Ara-C/Etoposide/Novantrone, A/D/T=Ara-C/Daunorubicin/Thioguanin, A/D=Ara-C/Daunorubicin; A/E/I=Ara-C/Etoposide/Idarubicin; A/I=Ara-C/Idarubicin;
OS: overall survival;
Chrom score: riskgroups according to WHO: 9 = unknown, 1 = good, 2 = intermediate, 3 = bad, 4 = very bad;
WBC: white blood cell count, at diagnose, x 10(9)/L;
SCT: allogeneic stem cell transplantation performed, y/n;
AHD: associated hematological disease, y/n
Preparation of Total RNA
Total RNA was prepared using the Trizol method (Invitrogen, Carlsbad, CA, USA). RNA was quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA). The integrity of the RNA samples was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA, USA) and then stored at −80°C. RIN > 7 for each RNA sample analyzed.
Reverse Transcription
Synthesis of cDNA from 1 μg total RNA in a 20 μl reaction volume was carried out using the High Capacity cDNA kit with RNAse inhibitor (Applied Biosystems, Foster City, CA, USA) as per the manufacturer’s instructions. The reverse transcription conditions were as follows: 10 minutes at 25°C, 120 minutes at 37°C, 5 minutes at 85°C. Following reverse transcription, cDNA was stored at 4°C.
TaqMan Low Density Arrays (TLDAs)
Expression levels of 380 MDR-associated genes were measured using custom-made TaqMan Low Density Arrays in addition to the ribosomal RNA 18s being arrayed in four replicates (Applied Biosystems, Foster City, CA, USA)13, 15. cDNA was mixed with 2X Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), loaded on a TLDA card, and run on an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) as per the manufacturer’s instructions.
Data Analysis
Data from the TLDAs was collected for each of the eleven paired AML samples (available from the GEO Database, accession number: GSE33787). The normalization of the data was performed in subtracting the median expression of each sample from all gene expression data for that sample. The expressions from replicate probes were averaged together. This analysis was carried out using BRB ArrayTools, a microarray-data statistical analysis tool (http://linus.nci.nih.gov/BRB-ArrayTools.html)16. In order to remove genes that are not likely to be informative, those expressed by fewer than 50% of the samples were filtered out, resulting in analysis of 331 genes. Hierarchical clustering was done on the normalized, filtered data, using an average linkage algorithm with 1-Pearson correlation as the distance. A Spearman rank test was used to assess the correlation between FAB and the expression of each gene (threshold: Spearman Rank p-value <0.05). The Cox proportional Hazards test was used to study the correlation of gene expression with the duration of complete response. The False Discovery Rate (FDR) for each gene was calculated using the Benjamini-Hochberg method for both methods. Pairwise comparisons were manually performed using the delta delta Ct method 17.
RESULTS
Comparison of MDR-linked Gene Expression Profiles of Paired AML Samples Taken at Diagnosis and After Relapse
Unsupervised hierarchical clustering was performed on all paired samples (Fig. 1). For six out of eleven patients, the pair of samples (at diagnosis and after relapse) clustered together, indicating that the development of resistance did not involve a major change in pattern of gene expression. This also supports that at least for this set of patients, diagnosis and relapse leukemias had similar origins. For the other five pairs of samples that clustered apart, for which large changes in the gene expression profile were observed between the paired samples, no distinguishing trend could be found for age, the subtype (based on the cell type from which the leukemia developed and its degree of maturation-i.e. French-American-British (FAB) classification18, the CR duration, the treatment, or the overall survival time Table 1.
Figure 1.

Unsupervised hierarchical clustering on 11 paired samples. The X-axis shows clusters of samples, while the Y-axis shows the expression levels according to the filtering criteria (red and green represent up- and down-regulation, respectively. The data suggest the role of differentiation in contributing to relapse, as three specimens of M5a and b subtypes had diagnosis and relapse samples that clustered apart, while all three patients with acute myeloblastic leukemia with minimal maturation (M1 subtype) had diagnosis and relapse samples that clustered together.
Genes that significantly differ in expression between diagnosis and relapse blast samples from the same patient may give insight into what contributes to relapse and why relapsed AML may be more resistant to treatment. Supervised comparison of samples taken at diagnosis and after relapse, paired according to the patient, revealed 27 genes that are differentially expressed, in most cases only to a small extent, between these two groups (p<0.05; Table 2). None of these genes fulfilled the stringent criteria of false discovery rate (FDR) <0.05, but this may be due to the low number of samples analyzed.
Table 2.
Differentially expressed genes in samples taken at diagnosis compared with samples taken at relapse
| Genes | Parametric p-value | FDRa | Geometric meanb (Level of expression at diagnosis/level of expression at relapse) |
|---|---|---|---|
| MSH3 | 0.002 | 0.508 | 0.81 |
| GSS | 0.007 | 0.508 | 0.82 |
| ERCC5 | 0.010 | 0.508 | 0.82 |
| RAD50 | 0.010 | 0.508 | 0.71 |
| MLH1 | 0.013 | 0.508 | 0.88 |
| EHBP1 | 0.014 | 0.508 | 0.65 |
| ERCC4 | 0.014 | 0.508 | 0.82 |
| BIRC6 | 0.016 | 0.508 | 0.8 |
| SLC19A2 | 0.016 | 0.508 | 0.73 |
| ERCC8 | 0.018 | 0.508 | 0.7 |
| RAD1 | 0.020 | 0.508 | 0.79 |
| RAF1 | 0.021 | 0.508 | 1.27 |
| GSTM5 | 0.021 | 0.508 | 0.13 |
| ERBB2 | 0.022 | 0.508 | 0.66 |
| MLH3 | 0.023 | 0.508 | 0.72 |
| BID | 0.026 | 0.536 | 1.44 |
| RAD17 | 0.028 | 0.556 | 0.75 |
| MNAT1 | 0.031 | 0.556 | 0.78 |
| POLI | 0.033 | 0.556 | 0.66 |
| GSTZ1 | 0.035 | 0.556 | 0.73 |
| SP1 | 0.039 | 0.556 | 1.13 |
| TNF | 0.041 | 0.556 | 2.81 |
| ABCF1 | 0.042 | 0.556 | 1.19 |
| DOT1L | 0.043 | 0.556 | 1.29 |
| MAPK8 | 0.046 | 0.556 | 0.82 |
| GSTM3 | 0.046 | 0.556 | 0.54 |
| POLH | 0.046 | 0.556 | 0.79 |
False discovery rate (FDR) is measured using the Benjamini-Hochberg method and is an estimate of the proportion of the genes with a p-value < 0.05 that represent false positives. For example, there is 50.8% of chance that MSH3 is a false positive. One can consider that genes with a FDR > 15% are not statistically significant.
The geometric mean denotes the average of the logarithmic values (base 2), converted back to linear values.
Gene Correlation with French-American-British (FAB) Classification Reveals That the Expression Level of BCL2A1 And GSR Increases Along with FAB Subtypes
We expected that some of the MDR genes measured in this study would reflect the state of differentiation of AML cells and would therefore correlate with FAB classification. In fact, 52 genes out of the 331 genes that passed the filtering criteria (see data analysis section in Experimental Procedures) were found to correlate with FAB classification for samples taken at diagnosis (p<0.05). Among these genes, two, encoding the anti-apoptotic bcl2-related protein a1 (BCL2A1) and glutathione reductase (GSR), fulfilled a false discovery rate (FDR) lower than 15% and were found to be positively correlated, implying that the expression level of these genes increases along with FAB (from 0 to 5) (Fig. 2, Table 3). More precisely, the probability of finding these genes by chance at p<0.05 is 7.1 and 14.3%, respectively. Four additional genes fulfilled a FDR<15% and were negatively correlated with FAB classes, and were therefore up-regulated in immature cells compared with differentiated cells. These genes were POLH, NOLA2, ABCD4, and MNAT1 (Table 3). Fifteen genes were found to be correlated with FAB at p<0.05 for samples taken at relapse. However, none of these genes fulfilled a FDR<15% (data not shown).
Figure 2.

Transcript level of the anti-apoptotic BCL2-related protein A1 (BCL2A1) and glutathione reductase (GSR) correlated with FAB classification in samples taken at diagnosis. The transcript level values are presented as -ΔCT. (A) BCL2A1 and (B) GSR were found to correlate with FAB classification for samples taken at diagnosis (p<0.05). These genes fulfilled a false discovery rate (FDR) lower than 15% and were found to be positively correlated, implying that the expression level of these genes increases along with FAB (from 0 to 5) (See Table S3). More precisely, the probability of finding these genes by chance at p<0.05 is 7.1 and 14.3%, respectively.
Table 3.
Genes correlated with FAB for samples taken at diagnosis
| Gene symbol | Correlation coefficient | FDR | Parametric p-value |
|---|---|---|---|
| BCL2A1 | 0.886 | 0.0715 | 0.0004 |
| GSR | 0.834 | 0.143 | 0.0026 |
| KLF1 | 0.794 | 0.255 | 0.0156 |
| TP73 | 0.784 | 0.237 | 0.0070 |
| ABCC3 | 0.774 | 0.255 | 0.0137 |
| VEGFA | 0.763 | 0.237 | 0.0092 |
| TIMP1 | 0.739 | 0.255 | 0.0134 |
| SLC28A3 | 0.727 | 0.271 | 0.0324 |
| AQP9 | 0.724 | 0.258 | 0.0242 |
| ATP6V0C | 0.699 | 0.257 | 0.0208 |
| FKBP1A | 0.695 | 0.257 | 0.0231 |
| TP53BP2 | 0.694 | 0.257 | 0.0231 |
| MVP | 0.693 | 0.257 | 0.0231 |
| GSTK1 | 0.685 | 0.258 | 0.0255 |
| SLC29A1 | 0.674 | 0.26 | 0.0281 |
| ASAH1 | 0.67 | 0.26 | 0.0281 |
| TNFSF10 | 0.645 | 0.281 | 0.0370 |
| MAP2K1 | 0.645 | 0.281 | 0.0370 |
| XRCC4 | 0.624 | 0.3 | 0.0440 |
| ACTB | 0.622 | 0.307 | 0.0478 |
| PARP2 | −0.622 | 0.307 | 0.0478 |
| SIRT5 | −0.622 | 0.307 | 0.0478 |
| C8orf33 | −0.627 | 0.3 | 0.0440 |
| MSH2 | −0.633 | 0.287 | 0.0404 |
| ERCC5 | −0.635 | 0.287 | 0.0404 |
| XRCC5 | −0.638 | 0.287 | 0.0404 |
| MSH6 | −0.645 | 0.281 | 0.0370 |
| RAD17 | −0.648 | 0.281 | 0.0370 |
| PARP1 | −0.66 | 0.271 | 0.0309 |
| ABCE1 | −0.666 | 0.271 | 0.0309 |
| ERCC8 | −0.669 | 0.26 | 0.0281 |
| HSPB1 | −0.682 | 0.258 | 0.0255 |
| PIK3CA | −0.688 | 0.257 | 0.0231 |
| SLC5A6 | −0.69 | 0.257 | 0.0231 |
| TOP2B | −0.696 | 0.257 | 0.0208 |
| RAD1 | −0.703 | 0.257 | 0.0208 |
| HSF1 | −0.718 | 0.255 | 0.0168 |
| BCL2 | −0.719 | 0.255 | 0.0168 |
| APEX1 | −0.721 | 0.255 | 0.0168 |
| ABCA2 | −0.723 | 0.255 | 0.0168 |
| SIRT4 | −0.727 | 0.271 | 0.0324 |
| SLC7A1 | −0.736 | 0.255 | 0.0134 |
| ABCF3 | −0.749 | 0.255 | 0.0119 |
| ABL1 | −0.761 | 0.237 | 0.0092 |
| SLC25A30 | −0.762 | 0.237 | 0.0092 |
| ERCC3 | −0.766 | 0.237 | 0.0092 |
| GLO1 | −0.79 | 0.237 | 0.0061 |
| CDK2 | −0.807 | 0.211 | 0.0044 |
| POLH | −0.832 | 0.143 | 0.0026 |
| NOLA2 | −0.833 | 0.143 | 0.0026 |
| ABCD4 | −0.861 | 0.142 | 0.0013 |
| MNAT1 | −0.928 | < 1e-07 | < 1e-07 |
Gene Correlation with Duration of Complete Response Does Not Highlight Any Trend Among Patients
Genes that correlate with the duration of the first and second complete remissions (CRs) may be important predictors of relapse. This would hold true unless a small population of cells present at diagnosis is responsible for relapse or there is a fundamental change in the biology of AML between diagnosis and relapse19. We found 38 genes to be correlated with CR in samples taken at diagnosis at p<0.05 (data not shown). None fulfilled a FDR<15%. Seven were found in recurrent samples but none were statistically significant (data not shown).
Patient-By-Patient Analysis of Paired Diagnosis and Relapse Samples
Given that there were no striking correlations between expression of specific genes and overall resistance to chemotherapy, we hypothesized that each patient may have a unique set of genes within his/her AML blasts that contribute to the observed drug resistance, which would have been missed by statistical analysis highlighting trends of gene patterns observed “on average” across samples20. This issue was addressed by performing a patient-by-patient analysis of paired diagnosis and relapse samples (Table 4), which led to the striking observation that when each of these patients relapsed their AML cells expressed genes representing different potential mechanisms of resistance. Table 5 shows the expression profile of nine ABC transporters that have been shown to mediate multidrug resistance (MDR) (data extracted from Table 4). Although no trend was revealed based upon FAB classification, as mentioned above, it is observed that the leukemic cells of all patients except patient 1 (FAB: M4) showed increase at relapse of at least one ABC transporter capable of transporting either anthracyclines, vinca-alkaloids, or both. The analysis of samples taken from patient 9 (FAB: M5b) reveals that four ABC transporters, ABCB1 (MDR1/P-gp), ABCC1 (MRP1), ABCC5 (MRP5) and ABCG2 (BCRP) are over-expressed in leukemic cells. This indicates that conventional chemotherapy (anthracyclines/vinca-alkaloids) isn’t likely to benefit this patient and will probably only have a negative effect by causing deterioration of the patient’s condition through the side effects of these compounds.
Table 4a.
Patient-by-patient analysis showing genes with greater than 4-fold up- or down-regulation
| Patient 1 (M4) | |
| Gene | Change |
| MMP9 | 6.5 |
| CLDN5 | 6.2 |
| TGFA | −5.1 |
| SLC7A11 | −5.6 |
| PDGFRB | −6.8 |
| CLDN4 | −7.4 |
| CDH1 | −10.0 |
| ABCB4 | −10.7 |
| AQP9 | −16.5 |
| CCL2 | −19.2 |
| ABCD2 | −21.2 |
| SLC7A9 | −28.7 |
| Patient 2 (M1) | |
| Gene | Change |
| F3 | −5.0 |
| SLC29A2 | −5.3 |
| BIRC5 | −5.3 |
| ATP7B | −5.9 |
| AQP9 | −6.2 |
| MT2A | −7.0 |
| PTEN | −7.0 |
| CYP2D6 | −7.1 |
| ABCA6 | −7.1 |
| NR1I2 | −7.6 |
| SGPP1 | −7.6 |
| APOE | −7.6 |
| STARD4 | −8.1 |
| ABCB1 | −9.4 |
| CCNE1 | −10.3 |
| ATP8B1 | −10.9 |
| MT1F | −11.1 |
| MMP2 | −13.4 |
| SLCO4A1 | −13.5 |
| TNF | −13.6 |
| IGF1R | −14.7 |
| ABCB4 | −14.9 |
| MYC | −17.1 |
| SLC2A5 | −33.4 |
| GJA1 | −90.0 |
| CLU | −127.3 |
| Patient 3 (M1) | |
| Gene | Change |
| IL6 | 19.7 |
| FN1 | 14.0 |
| ABCC3 | 13.3 |
| CLDN7 | 10.8 |
| GPR177 | 9.6 |
| SLC7A8 | 7.7 |
| CLDN2 | 7.6 |
| FOS | 6.8 |
| SLC28A3 | 6.5 |
| SLC22A1 | 6.3 |
| CYP2D6 | 5.3 |
| GSTA4 | 5.3 |
| BCL2A1 | 5.0 |
| SGPP1 | 5.0 |
| SFN | −5.9 |
| ABCG2 | −7.4 |
| CDKN2A | −8.4 |
| CLDN5 | −9.5 |
| TP73 | −15.3 |
| Patient 4 (M5b) | |
| Gene | Change |
| MMP2 | 122.9 |
| KIT | 74.1 |
| SLCO4A1 | 69.0 |
| ATP1B1 | 27.0 |
| NR1I2 | 21.5 |
| SLC7A8 | 16.9 |
| GSTM5 | 11.1 |
| APOE | 10.6 |
| ITGAE | 10.5 |
| TCEAL4 | 9.3 |
| ABCB6 | 6.9 |
| MYC | 6.4 |
| XRCC2 | 5.9 |
| NR1H3 | 5.8 |
| ATP7B | 5.3 |
| ABCC3 | −5.1 |
| TP73 | −5.2 |
| ATP6V0C | −5.7 |
| BIRC3 | −5.8 |
| GSTT2 | −9.8 |
| SLC28A3 | −10.0 |
| TGFA | −10.5 |
| HIF1A | −11.6 |
| NFKBIA | −11.6 |
| MMP9 | −14.6 |
| CLDN7 | −17.6 |
| SLC22A1 | −18.0 |
| CDKN1A | −20.3 |
| TNF | −21.6 |
| BCL2A1 | −26.0 |
| FN1 | −27.1 |
| VEGFA | −32.7 |
| AQP9 | −71.4 |
| CCL2 | −119.4 |
| IL6 | −235.8 |
| Patient 5 (M4) | |
| Gene | Change |
| CYP2D6 | 15099.5 |
| FASLG | 34.6 |
| MMP2 | 12.8 |
| KIT | 12.4 |
| PDGFRB | 11.4 |
| OCLN | 10.2 |
| ATP1B1 | 7.9 |
| SLC16A2 | 7.4 |
| GSTM5 | 6.7 |
| ABCB1 | 5.7 |
| APOE | 5.5 |
| F3 | 5.4 |
| NR1I2 | 5.1 |
| AKR1C1* | 4.2 |
| PTEN | −4.0 |
| TNF | −4.9 |
| ABCC3 | −6.4 |
| KCNMA1 | −6.5 |
| SFN | −6.8 |
| MMP9 | −9.8 |
| CCL2 | −46.4 |
| CYP2E1 | −145.8 |
| Patient 6 (M5a) | |
| Gene | Change |
| SIRT4 | 1246.9 |
| CASP3 | 68.2 |
| GPX3 | 37.5 |
| GSTM5 | 36.6 |
| ASAH3 | 23.5 |
| MMP9 | 12.1 |
| HSPB1 | 12.0 |
| CYP2C9 | 10.7 |
| CLDN4 | 9.0 |
| SFN | 6.0 |
| GBP1 | −5.1 |
| KLF1 | −8.1 |
| GSTA4 | −12.1 |
| FN1 | −12.2 |
| SLC28A3 | −26.4 |
| AQP9 | −136.9 |
AKR1C1 assay also detects AKR1C2; CYP2C8 assay also detects CYP2C19; CYP2A6 also detects CYP2A7 and CYP2A13
Table 5.
ABC transporter gene expression profiles*
| Genes | P11 | P 2 | P 3 | P 7 | P 10 | P 1 | P 5 | P 8 | P 6 | P 4 | P 9 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M0 | M1 | M1 | M1 | M2 | M4 | M4 | M4 | M5a | M5b | M5b | |
| ABCA3 | −2.2 | NA | NA | 2.1 | NA | NA | NA | NA | 2.6 | NA | NA |
| ABCB1 | 3.5 | −9.4 | 2.5 | −6.0 | −2.2 | NA | 5.7 | 3.8 | −2.4 | −4.6 | 24.6 |
| ABCB4 | 4.6 | −14.9 | NA | NA | NA | −10.7 | 2.5 | NA | NA | NA | −13.3 |
| ABCC1 | NA | NA | NA | NA | NA | NA | 2.0 | −2.5 | NA | 4.3 | 5.4 |
| ABCC2 | NA | 3.8 | −2.9 | −2.0 | NA | NA | NA | NA | NA | NA | −2.9 |
| ABCC3 | 25.9 | NA | 13.3 | NA | NA | −3.8 | −6.4 | NA | NA | −5.1 | −43.5 |
| ABCC4 | NA | −3.0 | NA | NA | NA | NA | NA | −10.0 | −3.2 | 3.4 | 3.2 |
| ABCC5 | NA | NA | NA | NA | NA | NA | NA | NA | −2.4 | NA | NA |
| ABCG2 | NA | 3.8 | −7.4 | NA | 3.3 | NA | NA | 2.5 | 2.0 | NA | 8.3 |
Fold-change of 9 ABC transporters known to be involved in chemotherapy resistance, in samples taken after relapse compared with their counterparts taken at diagnosis
Abbreviations: P = patient; NA = not applicable (no detection of the measured genes)
ABCB1, which has been evaluated in patients with AML as a potential target for pharmacologic down-regulation of efflux-mediated chemotherapy resistance, was found to be over-expressed in the leukemic cells of five patients out of the eleven (Fig. 3). A similar observation was made for ABCG2, although that transporter may actually be associated with the intrinsic biology of the leukemia rather than with drug efflux mediating resistance per se (Fig. 3).
Figure 3.
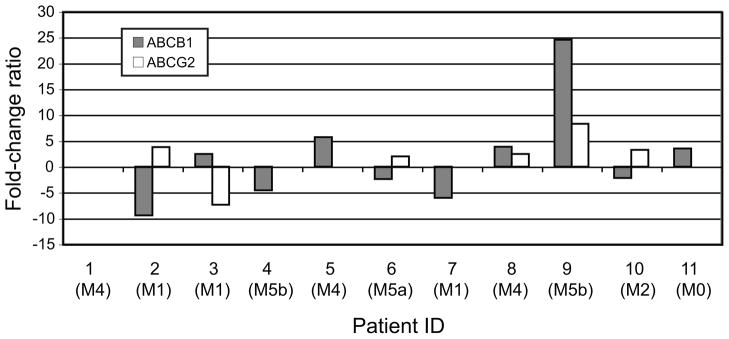
ABC transporter-mediated multidrug resistance. Histogram presenting the expression levels of ABCB1 and ABCG2, two ABC transporters intensively studied in patients with AML, in relapse samples compared with their paired samples taken at diagnosis. No bar appears when the measured gene is not expressed.
DISCUSSION
Genome-wide analyses have revealed the molecular genetic heterogeneity of AML21–23. A number of reports have established that AML cases can be classified into various groups based on their chromosomal abnormalities, somatic acquired mutations, gene and microRNA expression profiles and methylation status24. However, our understanding of the mechanisms causing the relapse of patients is limited. Although most patients with AML achieve CR, a large fraction of them will relapse and have a dismal prognosis, demonstrating the dire need to specifically address the reason for relapse. Pairs of samples taken at diagnosis and after relapse, combined with a reliable gene expression profiling assay, are prerequisites to understanding mechanisms mediating MDR.
In this study based on paired samples obtained at AML diagnosis and at relapse, for the first time, we assessed 380 MDR-related genes using TaqMan-based qRT-PCR, a state-of-the-art gene expression profiling assay. An unsupervised hierarchical clustering revealed that samples taken at diagnosis and relapse clustered in pairs for six patients out of the eleven studied, indicating return of the same leukemic blast, while the other five relapses had different transcription patterns and may have originated from different blasts. None of the gene signatures found to be correlated with either relapse or duration of remission were statistically significant. The observed enormous heterogeneity in gene expression across study patients remained true when we examined AML by subtype (M1, M4 and M5 subtypes are represented by three samples each) (Table 4).
The present investigation extends to the transcriptional level the recent findings highlighting the dynamic clonal evolution during AML relapse2. Importantly, these data also demonstrate that systemic chemotherapy has a substantial effect on the increased number of new mutations. Identifying new targets whose expression is altered after chemotherapy is critical to limit the occurrence of relapse. However, a personalized approach presents multiple challenges, such as pinpointing the clinically relevant targets from among numerous candidates in each individual gene signature, and eventually designing multiply-targeted treatment regimens to limit the survival of tumor cells through alternative drug resistance pathways. Although by definition the individual changes we see in this study are not “statistically significant”, each patient acts as his/her own control. By evaluating only genes known to be capable of conferring resistance to drugs such as those used to treat AML, we can generate specific hypothesis for each patient about how best to treat their relapsed AML. These hypotheses could be tested in short term ex vivo cultures of their cells using drugs known not to be subject to resistance mechanisms expressed in their AML cells, or by using inhibitors of demonstrated resistance mechanisms.
Table 4b.
Patient-by-patient analysis showing genes with greater than 4-fold up- or down-regulation
| Patient 7 (M1) | |
| Gene | Change |
| TP73 | 82.1 |
| CDKN2A | 52.5 |
| SGPP1 | 43.0 |
| SLCO1B3 | 26.5 |
| CCL2 | 23.1 |
| MKI67 | 17.1 |
| CDKN1A | 14.0 |
| TOP2A | 13.1 |
| BIRC5 | 9.4 |
| SLC9A3R2 | 9.0 |
| GPX3 | 6.7 |
| HSPB1 | 6.0 |
| ABCB5 | −5.3 |
| GBP1 | −6.0 |
| ABCB1 | −6.0 |
| SFN | −6.5 |
| CLDN3 | −6.8 |
| ATP1B1 | −7.3 |
| CLDN5 | −7.7 |
| KLF1 | −11.2 |
| CYP2A6* | −11.7 |
| IGF1R | −28.1 |
| CLDN4 | −43.1 |
| GJA1 | −176.7 |
| Patient 8 (M4) | |
| Gene | Change |
| FASLG | 24.4 |
| GBP1 | 17.7 |
| KCNMA1 | 14.5 |
| GPR177 | 10.3 |
| PDGFRB | 9.6 |
| NTRK2 | 7.2 |
| MT1H | 6.3 |
| CLDN7 | 5.8 |
| STAT1 | 5.6 |
| TAP1 | 5.6 |
| ABCD2 | 5.1 |
| IGF1R | −5.9 |
| F3 | −6.2 |
| ITGAE | −6.5 |
| ABCB6 | −6.5 |
| TCEAL4 | −6.7 |
| TP73 | −6.7 |
| SLC7A8 | −7.5 |
| CLU | −8.8 |
| ABCC4 | −10.0 |
| SLC2A5 | −11.9 |
| CLDN3 | −14.2 |
| CCND1 | −24.6 |
| SLC7A11 | −27.6 |
| KIT | −34.8 |
| MMP2 | −37.0 |
| Patient 9 (M5b) | |
| Gene | Change |
| GSTM5 | 135.4 |
| GPX3 | 91.3 |
| ABCB1 | 24.6 |
| ITGAE | 21.1 |
| SLC9A3R2 | 20.9 |
| ATP7B | 14.3 |
| MMP2 | 14.2 |
| ATP1B1 | 13.0 |
| ABCG2 | 8.3 |
| CASP3 | 7.7 |
| SIRT4 | 7.7 |
| GSTM3 | 6.7 |
| CLDN4 | 6.6 |
| KIT | 6.4 |
| SLC7A11 | 6.2 |
| APEX1 | 5.7 |
| MGMT | 5.7 |
| ABCC1 | 5.4 |
| CHEK1 | 5.0 |
| IGF1R | −5.3 |
| MT1X | −5.3 |
| ABCA6 | −5.7 |
| GJA1 | −5.8 |
| ABCA9 | −5.8 |
| ASAH2 | −6.3 |
| CDKN1A | −7.0 |
| TNF | −7.5 |
| GSTT1 | −8.3 |
| BIRC3 | −8.6 |
| SLC22A4 | −9.8 |
| ABCD2 | −12.2 |
| TNFSF10 | −13.1 |
| ABCB4 | −13.3 |
| NFKBIA | −13.7 |
| TP73 | −20.6 |
| BCL2A1 | −22.8 |
| CLDN5 | −24.1 |
| SLC28A3 | −28.8 |
| MT2A | −37.3 |
| ABCC3 | −43.5 |
| BAG3 | −55.6 |
| S100A10 | −80.4 |
| SLC25A5 | −132.9 |
| GBP1 | −148.4 |
| MT1H | −209.5 |
| SLC22A1 | −227.5 |
| IL6 | −1115.3 |
| AQP9 | −1642.9 |
| Patient 10 (M2) | |
| Gene | Change |
| FASLG | 41.7 |
| ATP8B1 | 10.2 |
| CLDN4 | 7.2 |
| MT1A | 6.1 |
| AURKA | −11.1 |
| TNF | −13.3 |
| IL6 | −14.9 |
| Patient 11 (M0) | |
| Gene | Change |
| ATP7B | 148.1 |
| TP73 | 90.5 |
| VEGFA | 44.9 |
| AQP9 | 35.8 |
| ABCC3 | 25.9 |
| BCL2A1 | 19.9 |
| GPR177 | 16.3 |
| CDKN1B | 12.7 |
| LIG4 | 11.5 |
| ABCC6 | 11.4 |
| S100A10 | 11.0 |
| CIAPIN1 | 10.5 |
| TGFA | 9.4 |
| MMP9 | 9.1 |
| ABCA9 | 8.2 |
| FZD1 | 7.5 |
| JUN | 7.2 |
| ABCA6 | 6.0 |
| CLDN7 | 5.6 |
| ETS1 | 5.5 |
| TNFSF10 | 5.1 |
| AKR1C1* | −5.1 |
| KCNMA1 | −5.4 |
| PDK1 | −5.7 |
| NR1I2 | −6.4 |
| BRCA2 | −6.8 |
| ABCB9 | −7.0 |
| MMP2 | −7.4 |
| ITGAE | −7.8 |
| CLDN4 | −7.9 |
| XRCC2 | −8.0 |
| KIT | −37.2 |
AKR1C1 assay also detects AKR1C2; CYP2C8 assay also detects CYP2C19; CYP2A6 also detects CYP2A7 and CYP2A13
Acknowledgments
FUNDING SUPPORT
The HHMI-NIH Research Scholars Program supported the research training of Chirayu Patel. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, the Swedish National Board of Health and Welfare, and the Adolf H. Lundin Charitable Foundation.
Footnotes
DISCLOSURES
The authors make no disclosures.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
References
- 1.Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009;37:649–658. doi: 10.1016/j.exphem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calcagno AM, Kim IW, Wu CP, Shukla S, Ambudkar SV. ABC drug transporters as molecular targets for the prevention of multidrug resistance and drug-drug interactions. Curr Drug Deliv. 2007;4:324–333. doi: 10.2174/156720107782151241. [DOI] [PubMed] [Google Scholar]
- 4.Leith C. Multidrug resistance in leukemia. Curr Opin Hematol. 1998;5:287–291. doi: 10.1097/00062752-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Marie JP, Legrand O. MDR1/P-GP expression as a prognostic factor in acute leukemias. Adv Exp Med Biol. 1999;457:1–9. doi: 10.1007/978-1-4615-4811-9_1. [DOI] [PubMed] [Google Scholar]
- 6.Wuchter C, Leonid K, Ruppert V, et al. Clinical significance of P-glycoprotein expression and function for response to induction chemotherapy, relapse rate and overall survival in acute leukemia. Haematologica. 2000;85:711–721. [PubMed] [Google Scholar]
- 7.Gruber A, Bjorkholm M, Brinch L, et al. A phase I/II study of the MDR modulator Valspodar (PSC 833) combined with daunorubicin and cytarabine in patients with relapsed and primary refractory acute myeloid leukemia. Leuk Res. 2003;27:323–328. doi: 10.1016/s0145-2126(02)00181-9. [DOI] [PubMed] [Google Scholar]
- 8.Libby E, Hromas R. Dismounting the MDR horse. Blood. 2010;116:4037–4038. doi: 10.1182/blood-2010-09-304311. [DOI] [PubMed] [Google Scholar]
- 9.Shaffer B, Gillet J-P, Patel C, Baer M, Bates S, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat. 2012;15:62–69. doi: 10.1016/j.drup.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 11.van der Kolk DM, de Vries EG, Noordhoek L, et al. Activity and expression of the multidrug resistance proteins P-glycoprotein, MRP1, MRP2, MRP3 and MRP5 in de novo and relapsed acute myeloid leukemia. Leukemia. 2001;15:1544–1553. doi: 10.1038/sj.leu.2402236. [DOI] [PubMed] [Google Scholar]
- 12.Gillet JP, Gottesman MM. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr Pharm Biotechnol. 2011;12:686–692. doi: 10.2174/138920111795163931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillet JP, Wang J, Calcagno AM, et al. Clinical Relevance of Multidrug Resistance Gene Expression in Ovarian Serous Carcinoma Effusions. Mol Pharm. 2011 doi: 10.1021/mp200240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derolf AR. Medicine. Stockholm: Karolinska University Hospital Solna and Institutet; 2010. Predictors of prognosis in acute myeloid leukemia : A clinical and epidemiological study; p. 56. [Google Scholar]
- 15.Calcagno AM, Salcido CD, Gillet JP, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 17.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620–625. doi: 10.7326/0003-4819-103-4-620. [DOI] [PubMed] [Google Scholar]
- 19.Wang ES, Sait SN, Gold D, et al. Genomic, immunophenotypic, and NPM1/FLT3 mutational studies on 17 patients with normal karyotype acute myeloid leukemia (AML) followed by aberrant karyotype AML at relapse. Cancer Genet Cytogenet. 2010;202:101–107. doi: 10.1016/j.cancergencyto.2010.07.117. [DOI] [PubMed] [Google Scholar]
- 20.Rottenberg S, Vollebergh MA, de Hoon B, et al. Impact of Intertumoral Heterogeneity on Predicting Chemotherapy Response of BRCA1-Deficient Mammary Tumors. Cancer Res. 2012;72:2350–2361. doi: 10.1158/0008-5472.CAN-11-4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 22.Haferlach T, Kohlmann A, Wieczorek L, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theilgaard-Monch K, Boultwood J, Ferrari S, et al. Gene expression profiling in MDS and AML: potential and future avenues. Leukemia. 2011;25:909–920. doi: 10.1038/leu.2011.48. [DOI] [PubMed] [Google Scholar]
- 24.Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]