

Abstract
Spinal cord injury results in distant pathology around putative locomotor networks that may jeopardize the recovery of locomotion. We previously showed that activated microglia and increased cytokine expression extend at least 10 segments below the injury to influence sensory function. Matrix metalloproteinase-9 (MMP-9) is a potent regulator of acute neuroinflammation. Whether MMP-9 is produced remote to the injury or influences locomotor plasticity remains unexamined. Therefore, we characterized the lumbar enlargement after a T9 spinal cord injury in C57BL/6 (wild-type [WT]) and MMP-9-null (knock-out [KO]) mice. Within 24 h, resident microglia displayed an activated phenotype alongside increased expression of progelatinase MMP-3 in WT mice. By 7 d, increases in active MMP-9 around lumbar vasculature and production of proinflammatory TNF-α were evident. Deletion of MMP-9 attenuated remote microglial activation and restored TNF-α expression to homeostatic levels. To determine whether MMP-9 impedes locomotor plasticity, we delivered lumbar-focused treadmill training in WT and KO mice during early (2–9 d) or late (35–42 d) phases of recovery. Robust behavioral improvements were observed by 7 d, when only trained KO mice stepped in the open field. Locomotor improvements were retained for 4 weeks as identified using state of the art mouse kinematics. Neither training nor MMP-9 depletion alone promoted recovery. The same intervention delivered late was ineffective, suggesting that lesion site sparing is insufficient to facilitate activity-based training and recovery. Our work suggests that by attenuating remote mechanisms of inflammation, acute treadmill training can harness endogenous spinal plasticity to promote robust recovery.
Introduction
A loss in mobility is one of the most noticeable and debilitating consequences of spinal cord injury (SCI). Activity-dependent tasks such as treadmill training can harness endogenous spinal plasticity to promote motor relearning and recovery after injury (Hodgson et al., 1994; Leblond et al., 2003; Basso and Hansen, 2011). However, despite modest improvements with treadmill training in the clinical setting, deficits persist and complete recovery is rare (Buehner et al., 2012; Harkema et al., 2012). The reasons for limited improvements are poorly understood. We theorize that the efficacy of training is related to interactions between the timing of training and the local microenvironment at the site of training-induced neural activity. Previous studies have defined a robust period of plasticity early after injury comprised of structural and synaptic changes throughout the neuroaxis (Fawcett, 2009). Delivering locomotor training when plasticity is primed has the potential to produce greater functional improvement. Surprisingly, some forms of early exercise instead prove detrimental to recovery possibly via mechanisms that disrupt neurovascular integrity (Kozlowski et al., 1996; Griesbach et al., 2007; Maldonado et al., 2008; Smith et al., 2009).
Neuroinflammation is a known impediment to spinal learning and plasticity (Vichaya et al., 2009; Yirmiya and Goshen, 2011; Huie et al., 2012). Glial reactivity and production of inflammatory signaling molecules prevent synaptic plasticity and molecular mechanisms of learning in the hippocampus (Yirmiya and Goshen, 2011). After rat SCI, we showed that activated microglia and cytokine expression extends caudal to the lesion at least 10 segments to the lumbar enlargement and contributes to sensory dysfunction, but the effects on spinal centric learning are unknown (Detloff et al., 2008). Changes in extracellular matrix composition in the lumbar enlargement after SCI also identify an inhibitory microenvironment for plasticity in locomotor interneuron networks (Andrews et al., 2012). Matrix metalloproteinases (MMPs) regulate diverse functions, including tissue remodeling, inflammation, and learning (Ethell and Ethell, 2007; Zhang et al., 2011). In particular, the gelatinase MMP-9 amplifies proinflammatory cytokine production, increases blood spinal cord barrier (BSCB) permeability, and regulates synaptic long-term potentiation (Noble et al., 2002; Nagy et al., 2006; Kawasaki et al., 2008). MMP-9 is produced by various cell types including glial cells, vascular endothelia, and leukocytes at the lesion site in rodent and human SCI (Buss et al., 2007; Zhang et al., 2011). If MMP-9 is produced in remote lumbar regions after SCI, it may contribute to an inhibitory microenvironment and interfere with plasticity and recovery of function even when treadmill training is delivered.
Here we hypothesize that remote production of MMP-9 after T9 SCI impairs the effectiveness of motor relearning and recovery of function. We present the first evidence of MMP-9 upregulation in the lumbar enlargement, which results in remote inflammation during the first week after midthoracic SCI in C57BL/6 mice. Lumbar-focused treadmill training administered during this early period impaired locomotor recovery and resulted in greater deficits in wild-type (WT) mice, whereas robust training-induced recovery occurred in MMP-9-null (KO) mice. Such findings support a time-sensitive adverse interaction between MMP-9 and treadmill training that influences recovery.
Materials and Methods
Subjects and surgeries.
Experiments were conducted in accordance with The Ohio State University Institutional Laboratory Animal Care and Use Committee. Adult (3–4 months of age) female B6.FVB(Cg)-Mmp9tm1Tvu/J KO and C57BL/6J WT mice were obtained from The Jackson Laboratory. The KO mouse shows a mild delay in bone formation (Vu et al., 1998), which was accounted for in kinematic assessments by collecting actual femur and tibia bone lengths postmortem. Laminectomy and spinal cord contusions were performed as described previously (Jakeman et al., 2000; Kigerl et al., 2006). Briefly, mice were anesthetized with a ketamine (138 mg/kg) and xylazine (20 mg/kg) mixture and given prophylactic antibiotics (gentocin, 1 mg/kg). Using aseptic techniques, removal of the spinous process and lamina of T9 exposed the dura. After stabilizing the vertebral column, the Infinite Horizon (Precision Systems and Instrumentation) device delivered 75 kilodynes of force to induce a severe contusion injury. Biomechanics of the injury were screened on day 0 and outlier forces or displacements were excluded (n = 8; Fig. 1). Force and displacement trends were similar to those established by Ghasemlou et al. (2005) (mean force: 77 ± 0.2; mean displacement: 718 ± 19.9). The incision was closed in layers and 2 cc of sterile saline was given subcutaneously to prevent dehydration. During recovery, mice received antibiotics (1 mg/kg gentocin, s.c.) and saline for 5 d and bladders were manually expressed twice per day until tissue harvest (Hoschouer et al., 2010). Based on previous experience, Basso Mouse Scale for Locomotion (BMS) scores were screened on day 1 to be ≤0.5 for training studies and 2.5 or less for ELISA or histology studies. Any outliers were removed from the study (n = 3). Disposition of all mice entered into the study is shown in Figure 1. Randomized group assignment and blinded behavioral assessments occurred.
Figure 1.

Flow diagram of experimental enrollment, screening, and group allocation. To reduce the risk of spontaneous recovery, we applied stringent exclusion criteria to training and behavioral studies. For early assessments of recovery (7 d), pretrained LTT groups were combined with unexercised controls for independent statistical comparisons.
Protein isolation and quantification.
Fresh tissue was harvested from the lumbar enlargement (naive, 1, 2, 3, 7, and 9 d postinjury [dpi]; n = 48) of unexercised KO and WT mice. ELISA was run in replicate groups alongside naive controls (n = 3–5/SCI group, n = 2–3 naives/replicate group). Mice were perfused with sterile saline and spinal cords were quickly dissected, snap-frozen, and stored at −80°C. Segments from L4–L5 were homogenized in RIPA lysis buffer (Pierce) and protease inhibitor mixture (Roche). After centrifugation at 10,000 rpm for 5 min, protein concentrations were determined with a BSA protein assay. Quantification of matrix and cytokine proteins was determined using a custom SearchLight Multiplex ELISA Array and was performed by Aushon Biosystems. Custom arrays were spotted with capture antibodies specific to tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), MMP-2, MMP-3, and MMP-9. The bound proteins were then detected with a biotinylated detection antibody followed by streptavidin-HRP and then visualized with SuperSignal ELISA Femto Chemiluminescent substrate. The luminescent signal produced from the HRP-catalyzed oxidation of the substrate was measured using the SearchLight Imaging System (Pierce) and protein concentrations extrapolated from a standard curve using ArrayVision (Pierce). All protein levels were analyzed relative to uninjured WT controls and are expressed throughout as percent naive.
In situ gelatinase zymography.
In situ zymography was used to detect and localize gelatinase activity in fresh tissue sections (Oh et al., 1999; Noble et al., 2002; Goussev et al., 2003). Segments from L1–L3 from the same mice used for ELISA (n = 48 detailed above) were then blocked and cut in 20 μm transverse cryostat sections for in situ gelatinase zymography. Sections were incubated in 0.05 m Tris-HCl, 0.15 m NaCl, 5 mm CaCl2, and 0.2 mm NaN3, pH 7.6, containing 40 μg of FITC-labeled gelatin (Invitrogen) at 37°C for 1 h. The gelatin is tagged to a peptide that fluoresces when cleaved by gelatinolytic activity. This reaction was then visualized with fluorescent microscopy (Nikon Eclipse E800 B-2E/C FITC filter). This assay localizes gelatinase activity but does not distinguish between members. Therefore, we used ELISA in the same mice to distinguish in situ activity of MMP2 and MMP9. Measurements of gelatinase-positive vascular-like structures were made after identifying the largest stained structure in each section and quantitatively measuring width and length using Stereoinvestigator (MBF Biosciences).
Gelatin zymography.
Gelatin zymography was used to detect MMP-2 and MMP-9 in the remote lumbar cord after T9 SCI. Trained and unexercised WT mice were perfused at 9 dpi (n = 3/group) and segments from L4–L5 were collected and quick-frozen at −80°C. Epicenter tissue at 24 h served as a positive control (n = 3). Each sample was homogenized in lysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS. After centrifugation, supernatants were collected and concentrated with a Microcon filter (50K membrane; Millipore). Equal amounts of protein were loaded on a 10% zymogram gel. After electrophoresis, the gel was incubated with renaturing buffer (Bio-Rad) at room temperature for 30 min to restore the gelatinolytic activity of the proteins and then incubated with developing buffer (Bio-Rad) at 37°C for 48 h. The gel was stained with Coomassie blue and destained until clear bands became evident.
Histology.
WT and KO mice were transcardially perfused with 0.1 m PBS, pH 7.4, followed by 4% paraformaldehyde, pH 7.2, naive (n = 3 WT), 1 dpi (n = 4 WT), and 7 dpi (n = 4 WT, n = 3 KO; Fig. 1). Spinal cord segments at the T9 lesion site and from spinal levels L1–L5 were postfixed for 1 h in 4% paraformaldehyde, rinsed overnight in 0.2 m phosphate buffer, pH 7.4, and cryoprotected in 30% sucrose before being embedded in M-1 Embedding Matrix (Thermo Scientific) and frozen on dry ice (Ma et al., 2001; Ma et al., 2004; Detloff et al., 2008; Basso et al., 2006). Epicenter and lumbar blocks were sectioned in their entirety at 20 μm on a Microm HM505E cryostat and collected in series of equally spaced sections on ColorFrost Plus slides (Fisher Scientific).
Fluorescent immunohistochemistry was performed to identify microglia and macrophages in L1–L2 transverse spinal cord sections of lumbar segments. A 1:200 dilution was used for rabbit anti Iba-1 (Wako 019–19741; MMP-9-null). The antibody was prepared in blocking solution containing 1% BSA, 0.1% Fish Gelatin, 3% NGS, and 0.2% Triton X-100 in PBS. Incubation of the primary antibody occurred overnight at 4°C. A goat anti-rabbit secondary antibody was used at a 1:400 dilution (Alexa Fluor 488, A21103). Final detection of signal was visualized under fluorescence. For all staining, control sections were processed by eliminating the primary antibody and replacing with blocking solution to ensure positive labeling. Sets from L1–L2 were stained for all groups. Sections were imaged using an Olympus FV1000 Filter Confocal Microscope (The Ohio State University Confocal Microscopy Imaging Facility).
To quantify phenotypic changes in microglia and macrophages, digital image analysis of Iba-1 staining was performed on representative sections in the L1–L2 region of the remote spinal cord (Donnelly et al., 2009). Thresholds for positive staining were performed by a blinded investigator. Images were then processed by densitometric scanning of threshold targets using ImageJ software. Proportional area is reported as the average percent area in the positive threshold for all pictures. To quantify morphological changes in microglia cell body sizes, ImageJ was used to measure cell body width in the same sections used for proportional area measurement. Width measurements were made in each section by randomly choosing 10 microglia throughout the gray matter (four dorsal horn, two intermediate laminae, and four ventral horn).
The lesion site was transversely sectioned (20 μm) and every 10th section was stained for myelin using Eriochrome cyanine and differentiated with 5% iron alum and borax ferricyanide solutions (Rabchevsky et al., 2001). The lesion epicenter was identified as the section with the largest central core lesion and least amount of stained myelin. Spared white matter or lesioned tissue areas were measured with point counting methods using Stereoinvestigator software (Microbrightfield). A grid was randomly placed on each spinal cord section and located intersections on spared white matter, spared gray matter, and lesioned tissue. Each intersection counted as a point (p) and point counts for each section were converted to estimated area using the following equation: estimated area = (ΣP) * (a/p) where ΣP is the sum of the points for an individual mouse and a/p is the area of each point. The area of stained white matter at the epicenter was divided by total cross-sectional area to serve as a measure of injury severity.
Training paradigm.
Treadmill training was delivered in subgroups of mice at early time points (early treadmill training [ETT]; 2–9 dpi; n = 9 KO, n = 6 WT) or late time points (late treadmill training [LTT]; 35–42 dpi; n = 6 KO, n = 5 WT) alongside unexercised controls (n = 4 KO, n = 5 WT; Fig. 6). LTT mice served as unexercised (Unex) controls at the 7 d time point (total n = 10 KO Unex, n = 10 WT Unex). An independent group of naive controls (n = 10) were used for kinematic comparisons. Training consisted of 8 consecutive days of manually delivered weight-supported stepping during quadrupedal locomotion on a custom-built treadmill (Columbus Instruments). Hindlimb (HL) stepping was manually assisted as needed using small rounded pestles to achieve toe clearance and plantar placement of the paw on the treadmill belt (Hutchinson et al., 2004). Adjustable harnesses provided partial body weight support while maintaining the trunk in a typical horizontal murine posture (Fig. 6). Each session included two 10 min bouts separated by a 20 min rest interval to prevent delayed onset muscle soreness (McHugh, 2003).
Figure 6.

Mouse kinematics and treadmill training paradigm. Recovery was examined in two treatment paradigms. Quadrupedal treadmill training was delivered early or late after SCI. For ETT, recovery was assessed at 7 dpi and retention was examined at 35 dpi. For LTT, recovery was assessed before (35 dpi) and after (42 dpi) the intervention. Bodyweight support was provided using a harness and manual trunk assistance. A pestle was used to ensure plantar placement of steps. Recovery was quantified using two-dimensional mouse kinematics. Markers were placed on the iliac crest, greater trochanter, femoral condyle, lateral malleolus, and the head of the fifth metatarsal.
Locomotor assessments.
Open Field Locomotor recovery was assessed using the BMS by two raters blind to group assignment (Basso et al., 2006). Rating criteria considered HL joint movement, weight support, plantar stepping, coordination, paw position, and trunk and tail control. Scores range from no HL movement (0) to normal locomotor function (9). The rationale for selection of each behavioral outcome measures is provided in Table 1.
Table 1.
Relevance of behavioral strategies
| Test | Measure | Study-specific use | General properties | Reference |
|---|---|---|---|---|
| Open field | BMS | Differentiate between spontaneous recovery and training induced effects | A sensitive, valid, and reliable test for recovery of locomotion after SCI | Basso et al., 2006 |
| Determine translation to novel task for TT groups | Quantifies a wide range of spontaneous behaviors and movements | |||
| Predicts tissue sparing over a range of severities | ||||
| Gridwalk | Footfalls | More sensitive indicator of motor control than gross locomotion Test of translation to a novel task for all groups |
Test of precise paw placement Neural substrates include corticospinal and propriospinal input |
Ma et al., 2001 Grill et al., 1997 Whishaw et al., 1992 |
| Treadmill kinematics | Trunk instability | Test of proximal motor control under matched locomotor speed conditions for all groups | Quantifies hindquarter sway or collapse | Buehner et al., 2012 |
| Related to clinical measures of balance | ||||
| Toe drags and toe velocity | Test of distal motor control under matched locomotor speed conditions for all groups | Surrogate marker for ankle dorsiflexor strength | Buehner et al., 2012 | |
| Magnuson et al., 2009 | ||||
| Swing time and stance time | Tests the ability to time swing and stance phases to treadmill speed | Measure of phased activation of HL flexor and extensor muscle groups | Buehner et al., 2012 | |
| Measure of speed of transition between swing and stance | Prolonged stance after SCI is an indirect measure of instability | Thompson et al., 2013 |
Gridwalk.
A 2.54 cm square metal grid apparatus (41 × 28 × 36 mm) was used to measure precise paw placement during forward locomotion. Mice were acclimated to the grid preoperatively and then tested at 35 and 42 d. Stepping performance was analyzed for five volitional passes. Passes were recorded with a digital camcorder (Handycam HDR SR11; Sony) and then analyzed in slow motion. Success was determined by a weight-supported step from rung to rung. The number of HL stepping failures are expressed relative to the number of forelimb steps and shown as percent success.
Two-dimensional kinematics.
All mice had two-dimensional kinematic analysis of walking using a novel application for mice based on established criteria for rat and feline models (Basso et al., 1994; Hansen et al., 2012). Kinematic assessments were done for naive animals and at 7, 35, and 42 dpi. Retention of training effects was determined in ETT groups by evaluating locomotor function 4 weeks after training ended (35 dpi). To demarcate the HL joints before injury, mice were briefly anesthetized with 2% isoflurane, the left HLs were shaved, and bony prominences were identified by palpation. Appropriate marker placement was later confirmed during locomotion. Depilatory cream was used to remove hair and to maintain marker locations for the study duration. Marked prominences included the iliac crest, greater trochanter, femoral condyle, lateral malleolus, and head of the fifth metatarsal (Fig. 6). Treadmill locomotion was captured using a Panasonic WV-CL350 camera (60 Hz) with a time code generator. Kinematic markers were digitized using PEAK Motus. To account for movement of the knee joint under the skin during locomotion, a triangulation program was used to estimate its position (Goslow et al., 1973) from actual femur and tibia bone lengths collected postmortem. Trunk instability was determined by calculating maximal change in vertical displacement of the iliac crest during each step. Toe dragging was measured by the difference in time between initial forward movement of the toe and visual confirmation of liftoff from the treadmill belt. Total time in swing and stance was calculated from visually confirmed liftoff and initial contact from video. The relevance and application of kinematic measures are described in Table 1. These assessments were averaged over a total of 20 steps. Animals that failed to produce a minimum of 18 plantar steps on the treadmill were statistically penalized for nonperformance.
Statistics.
All behavioral outcome measures were analyzed using MANOVA with Dunnett's post hoc testing (for ETT) or repeated measures ANOVA with Tukey's post hoc testing (for LTT). Means and SEM are reported throughout. For kinematic data collection, not all mice were able to perform the task (i.e., dragged or dorsal stepped). Nonperformance was corrected by assigning the maximal kinematic value plus 1 SD from the mean or “0.” Group comparisons for open-field plantar stepping and weight support were made using a χ2 analysis. Protein levels are displayed as percent naive and were analyzed using a one-way ANOVA. When appropriate, differences between protein measures were evaluated with a Student's t test. Correlations between white matter sparing and proportional area of iba1 labeling were made using Pearson's correlation analysis. Significance was set at p < 0.05.
Results
Acute injury mechanisms extend to the lumbar enlargement after thoracic SCI
Thoracic spinal cord contusion results in increased TNF-α and metalloproteinase expression in the lumbar enlargement (L4–L5) in WT mice during acute stages after injury (Fig. 2). Within 24 h, progelatinase MMP-3 protein was significantly elevated (423.9% ± 105.9 vs naive; p < 0.001), but returned to basal levels by 2 dpi. In contrast, MMP-9 expression reached a more than a 5-fold increase at 7 dpi and persisted at 9 dpi (526.4 ± 73.3% and 547.2 ± 144.1%, respectively, vs naive; p < 0.001), a time when TNF-α expression is also increased. Gelatin zymograms confirmed lumbar expression of pro-MMP-9 at 9 d (Fig. 7C). Protein expression of the proinflammatory cytokine TNF-α reached a nearly 2-fold increase by 9 dpi (162.1 ± 86.3% vs naive; p < 0.05), whereas the levels of IL-1β remained at baseline in the lumbar enlargement during the first week after SCI (Fig. 2B).
Figure 2.

Injury mechanisms extend to the lumbar enlargement during acute stages of SCI. Protein analyses reveal increased metalloproteinase and cytokine expression in the lumbar enlargement (L4–L5) of WT mice over the first week after SCI (n = 3–5/group). A, Within 24 h, MMP-3 was significantly upregulated (p < 0.001 compared with naive) but returned to baseline by 2 dpi. MMP-9 reached peak values 7–9 dpi (547.2%; p < 0.001 vs naive). MMP-2 protein was unchanged. B, TNF-α expression was increased at 9 dpi (p < 0.002 vs naive), whereas IL-1β was unchanged. In situ gelatinase zymography localized gelatinase activity around microvascular endothelia in lumbar segments. C, Sparse labeling of cellular structures is evident in the naive cord. After SCI, there is pronounced gelatinase activity around vascular-like phenotypes (H) within 24 h (D) and remains apparent at 3, 7, and 9 dpi (E–G). Scale bars: C–G, 130 μm; H, 32 μm.
Figure 7.

Robust locomotor improvements occur with ETT in KO mice. A, By 7 dpi, ETT KO mice plantar stepped in the open field, whereas all other groups primarily had slight and extensive ankle movements (p < 0.01). Weight support and plantar stepping performance in the open field was significantly higher in the treadmill-trained KO group (100% weight support and 66.7% plantar stepping; p < 0.05). B, On the treadmill, early trained KO mice independently plantar stepped and no longer required a supportive harness. Only ETT KO mice were capable of producing quantifiable kinematic metrics. All other groups required bodyweight support and dragged the HLs. C, Gelatin zymograms show that MMP-9 expression persists in the lumbar cord (L4–L5) of both trained and unexercised WT mice.
In situ gelatinase zymography localized gelatinolytic activity in tissue sections from L1–L3 blocks of the same lumbar spinal cords used for ELISA. In the uninjured spinal cord, low gelatinase activity was observed in the meninges and sparse labeling of vascular-like structures corresponding to locations reported previously (Fig. 2C; Noble et al., 2002; Goussev et al., 2003). Within 24 h after SCI, a pronounced increase in gelatinase activity was evident in similar structures in the lumbar cord (Fig. 2D), which remained elevated at 3, 7, and 9 dpi (Fig. 2E–G). The number of branches and width of vessels labeled with gelatinase activity was greatest in the lumbar cord at 3 dpi (mean branches: 5.33 ± 1.86; mean width: 14.5 ± 2.86 μm; p < 0.05 vs naive mean branches: 1.0 ± 0.58; mean width: 6.2 ± 1.1 μm).
WT mice showed robust and rapid microglial activation in the lumbar enlargement after thoracic SCI. Within 24 h, iba1-labeled microglia displayed significantly larger cell bodies (mean width: 19.2 μm ±1.7 at 24 h in WT vs 7.73 ± 0.53 μm in naive; p < 0.001) and a bushy phenotype (Fig. 3) similar to those described previously (Soltys et al., 2001). By 7 dpi, pronounced microglial activation remained with a hypertrophic phenotype (mean width: 14.1 ± 1.1 μm) compared with finely ramified microglia in naive controls (mean width: 7.73 ± 0.53; p < 0.05). Increased microglial activation occurred throughout sectional gray and white matter from L1–L2 (mean area: 1.8 ± 0.5% area in naive and 8.0% ±0.8 area in 7 dpi WT; p < 0.05). The greatest microglial density occurred in the dorsal horn relative to the ventral horn after SCI (Fig. 3). Changes in microglia phenotype occurred alongside a rise in proinflammatory cytokine production.
Figure 3.

Thoracic SCI results in rapid activation of microglia in the lumbar enlargement. Within 24 h, Iba1 labeling shows that resident microglia display an activated bushy phenotype made evident by increased cell body width (p < 0.001 compared with naive; n = 3–4/group). By 7 dpi in WT mice, microglia still exhibit hypertrophy compared with naives and total staining covers a greater proportional area in the dorsal horn (p < 0.01 vs naive, 24 h WT, and 7 d KO) and ventral horn (p < 0.05 vs naive, 24 h WT, and 7 d KO). MMP-9 deficiency resulted in a significant attenuation of microglia phenotype and proportional area. KO mice showed reduced cell body width (10.2 μm; p < 0.01 vs 24 h) and reduced labeling in dorsal horn (p < 0.001 vs 7 d WT) and ventral horn (p < 0.01 vs 7 d WT). Scale bars: A, 150 μm; B, C, 100 μm.
Deletion of MMP-9 improves white matter sparing and reduces inflammation in the lumbar enlargement
Similar to findings of Noble et al. (2002), in a more severe injury model, we show that MMP-9 deficiency results in a modest increase in white matter sparing at the lesion site (Fig. 4). After severe SCI, lesion size was smaller (78.80 ± 1.49% of the area in KO and 87.06 ± 2.32% in WT; p < 0.05) and the amount of white matter sparing was significantly greater in KO mice (21.10 ± 1.49% in KO, and 12.93 ± 2.33% in WT; p < 0.05; Fig. 4B). White matter sparing at the lesion site predicted the extent of microglial activation in the lumbar enlargement. At 7 d, mice with greater sparing at T9 had significantly less cross-sectional iba-1 labeling throughout L1–L2 (r2 = 0.67; p < 0.01; Fig. 4C).
Figure 4.
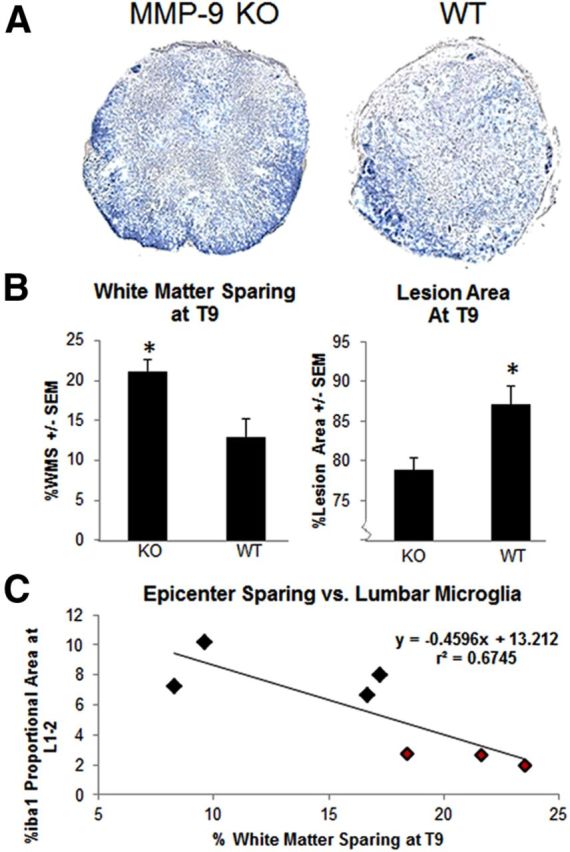
MMP-9 deficiency results in white matter sparing that predicts remote activation of microglia. A, Representative epicenters are shown from KO and WT mice stained for myelin after severe SCI at 7 d. B, MMP-9 KO mice had greater myelin sparing (p < 0.05) and smaller lesion size (p < 0.05; n = 3–4/group). C, The extent of white matter sparing at the lesion epicenter was highly predictive of remote microglial activation. Greater white matter sparing at T9 corresponded to less Iba1 labeling at L1–L2 throughout the entire cross-section at 7 d (WT, black; KO, red; p < 0.05).
Deletion of MMP-9 attenuated inflammation in the lumbar enlargement. MMP-9 deficiency did not alter MMP-3 or MMP-2 expression compared with WT. Progelatinase MMP-3 expression was elevated in KO mice and reached an almost 4-fold increase within 24 h (386.5 ± 57.9%; p < 0.05; Fig. 5A) but returned to baseline by 3 dpi. MMP-2 levels remained at baseline in WT mice and did not show a compensatory increase in KO mice, which confirms work by Hsu et al., 2008 (Fig. 5B). Microglial activation was markedly reduced in MMP-9 KO mice compared with WT at 7 dpi (Fig. 3). In both the dorsal and ventral horn, microglia maintained a hypertrophic phenotype, but over much less area (328.4% reduction in dorsal horn, 193.4% reduction in ventral horn; p < 0.05; Fig. 3). MMP-9 deficiency reduced proinflammatory TNF-α production in the lumbar enlargement over the first week after SCI (Figs. 2B, 5C). In KO mice, TNF-α expression was significantly reduced in L4–L5 at 24 h (52% reduction vs naive; Fig. 5C; p < 0.05). At 7 dpi, deletion of MMP-9 restored TNF-α levels to homeostatic levels (79.7 ± 15.1%; p < 0.05 vs naive; Fig. 5C).
Figure 5.

MMP-9 deficiency attenuates proinflammatory cytokine but not metalloproteinase expression. A, ELISA confirms depletion of MMP-9 in KO mice at 24 h, 3 d, and 7 d. KO mice maintain an acute upregulation of the progelatinase MMP-3 at 24 h (p < 0.05). B, Compensatory increases in MMP-2 protein are not observed at 42 d. Increased expression of TNF-α protein is evident at 7 d in L4–L5 of WT mice (p < 0.01 vs naive; 24 h WT used are same as Fig. 2). C, In KO mice, TNF-α expression is greatly reduced at 24 h (p < 0.01 vs WT naive) but returns to homeostatic levels at 7 dpi.
ETT but not LTT promotes robust recovery only in KO mice
Severe SCI resulted in paresis during overground locomotion with only occasional, slight HL movements at 24 h in all groups (mean BMS = 0.3 ± 0.1 KO, 0.3 ± 0.1 WT). ETT delivered 2–9 dpi (Fig. 6) resulted in substantial locomotor improvements by 7 dpi in MMP-9 KO but not unexercised KO or trained or unexercised WT groups (Fig. 7A). At 7 dpi, recovery was negligible, with plantar stepping occurring in only 9.1% of ETT WT mice, 20% of unexercised WT mice, and 23.1% of unexercised KO mice (Fig. 7A). Conversely, plantar stepping occurred in 66.7% of ETT KO mice in the open field at 7 d (Fig. 7A). This was similarly observed during treadmill locomotion, because ETT MMP-9 KO mice were the only group capable of generating kinematic metrics (Fig. 7B). Unexercised KO and trained and unexercised WT groups required an assistive harness for bodyweight support and consistently dragged HLs on the treadmill. Neither training alone nor MMP-9 depletion alone promoted recovery. MMP-9 expression persisted in the lumbar cord of trained and untrained WT mice at 9 d (Fig. 7C).
Functional improvements in ETT KO were retained 4 weeks after the training period ended. Groups did not differ in open field recovery at 35 dpi (mean BMS = 5.6 ± 0.2 ETT KO, 5.6 ± 0.3 Unex KO, 4.6 ± 0.5 ETT WT, and 5.1 ± 0.2 Unex WT). Kinematic metrics identified significant differences in treadmill performance. At 35 dpi, early trained KO mice stepped with near normal measures of toe dragging, swing time, stance time, and ankle velocity, none of which differed statistically from naive (Fig. 8B–E). In contrast, ETT in WT mice resulted in significant locomotor deficits at 35 d. During treadmill walking, ETT WT mice displayed measures of trunk instability, toe dragging, swing time, stance time, and ankle velocity that showed the greatest mean deviation from naive (Fig. 8A–E; p < 0.05). On the gridwalk, early training resulted in significantly worse stepping precision. Weight supported, rung-to-rung stepping was only successful in 5.49 ± 2.54% of attempts in ETT WT, whereas unexercised WT stepped with a 13.58 ± 1.81% success rate (Fig. 8F).
Figure 8.

Retention of training effects. Four weeks after training ended (35 dpi), motor control was assessed using gridwalk and treadmill kinematics. ETT in KO mice resulted in near-normal measures of trunk displacement (A), toe dragging (B), time spent in swing/stance (C,D), and peak velocity of the ankle during swing (E). Trained WT and unexercised controls displayed significant motor deficits compared with naive in almost all kinematic measures (p < 0.05). ETT resulted in significantly worse rung-rung paw placement on gridwalk in WT mice (F, p < 0.05). n = 6–10/group. *Significantly different from naive for A–E, *Significantly different from KO ETT for F.
The same duration of treadmill intervention delivered in late stages of recovery (35–42 dpi) failed to result in any significant locomotor improvements in either MMP-9 KO or WT mice (Table 2). Because LTT was delivered after spontaneous recovery, when stepping was evident for 100% of mice in the open field, the training intervention differed in that they did not require body weight support or manual assistance of the HLs. The intensity of training, as measured by the number of HL steps performed in the 20 min session, was significantly greater than the early intervention (ETT: 1295.7 ± 27.1; LTT: 3622.2 ± 254.6; p < 0.001). Despite more intensive training, open field scores showed no benefit in recovery, because predominant weight-supported plantar stepping was typical in both groups before and after the delayed intervention. In addition, kinematic metrics of trunk instability, toe dragging, swing time, and ankle velocity showed no significant improvements after training (Table 2). Unlike an early intervention, LTT did not result in motor deficits in KO or WT groups.
Table 2.
Late training is ineffective in KO and WT mice
| Naive (n = 10) | KO Pre (n = 6) | KO Post (n = 6) | WT Pre (n = 5) | WT Post (n = 5) | |
|---|---|---|---|---|---|
| BMS | 9 | 5.7 ± 0.4 | 5.6 ± 0.2 | 4.8 ± 0.2 | 5.0 ± 0.3 |
| Trunk displacement (cm) | 0.54 ± 0.04 | 1.01 ± 0.31 | 0.64 ± 0.11 | 1.24 ± 0.40 | 0.87 ± 0.26 |
| Toe dragging (s) | 0.031 ± 0.001 | 0.067 ± 0.010 | 0.051 ± 0.004 | 0.077 ± 0.013 | 0.078 ± 0.007 |
| Swing time (s) | 0.113 ± 0.005 | 0.060 ± 0.012 | 0.078 ± 0.005 | 0.055 ± 0.023 | 0.068 ± 0.019 |
| Stance time (s) | 0.30 ± 0.01 | 0.69 ± 0.09 | 0.52 ± 0.04 | 0.82 ± 0.14 | 0.91 ± 0.09 |
| Peak ankle velocity (cm/s) | 22.9 ± 1.5 | 15.5 ± 3.3 | 19.3 ± 1.6 | 11.4 ± 4.7 | 12.3 ± 3.7 |
| Peak toe velocity (cm/s) | 30.0 ± 1.8 | 21.8 ± 4.7 | 27.7 ± 2.0 | 16.4 ± 6.8 | 19.6 ± 5.5 |
| Gridwalk (% success) | 84.3 ± 1.7 | 12.3 ± 3.2 | 11.3 ± 3.1 | 13.1 ± 1.2 | 14.6 ± 3.0 |
Recovery was examined before (35 d) and after (42 dpi) LTT in KO and WT mice using a battery of locomotor tests. Open field (BMS), gridwalk, and kinematic measures revealed no improvements or deficits that resulted from training in KO and WT groups. Data are shown as means ± SEM.
Discussion
Spinal cord injury creates dual and conflicting cellular processes along the neuroaxis. In addition to well documented events at the lesion site, mechanisms of both plasticity and inflammation occur remote to the lesion in the lumbar enlargement that influences function (Hains and Waxman, 2006; Detloff et al., 2008; Andrews et al., 2012; Hougland et al., 2012). Given recent evidence that remote neural activity provides an essential substrate for locomotor recovery (Harkema et al., 2011), we propose that remote inflammatory processes alter the cellular microenvironment and dramatically influence retraining and function. We show for the first time that elevated MMP-9 contributes to the failure of motor relearning early after SCI alongside an inflammatory microenvironment in remote lumbar segments. Deletion of MMP-9 reduces the inflammatory signature in the lumbar cord and allows robust locomotor plasticity and improved recovery that is otherwise refractory to early intervention. For the first time, we identify the blunting effect of remote inflammation on activity-dependent plasticity within locomotor interneuron networks. Our work demonstrates a negative, time-dependent interaction between lumbar MMP-9 production and motor relearning.
MMP members display a distinct temporal expression in the lumbar enlargement during the first week after SCI. Numerous cell types, including resident glia, trafficking leukocytes, and vascular endothelia, produce MMPs in response to CNS trauma (Zhang et al., 2011). Progelatinase MMP-3 shows rapid upregulation within 24 h that precedes MMP-9 at 3, 7, and 9 dpi. A similar trend is observed in the brain when an entorhinal cortex lesion results in distant expression of MMP-3 in the dentate gyrus (Kim et al., 2005a; Falo et al., 2006). MMP-3 is considered to be the primary activator of MMP-9 and likely plays a critical role in the transcription and activation of MMP-9 at later time points in the lumbar cord (Vempati et al., 2007). In situ and gelatin zymograms confirm increased MMP-9 expression and localize activity primarily to structures with a vascular phenotype in lumbar segments. Evidence of gelatinase activity around BSCB constituents suggests abnormal permeability or neurovascular reactivity far distant to the site of direct trauma. MMP-9 activity may contribute to demonstrated alterations in gray matter permeability and vascular uptake mechanisms extending to at least L1–L2 in a rat midthoracic contusion model (Popovich et al., 1996).
Midthoracic contusion produced a rapid, proinflammatory shift in resident microglial morphology and increased cytokine expression around putative locomotor networks. At 24 h during peak MMP-3 expression, microglia displayed an exaggerated activation profile, or bushy phenotype, that may indicate a premigratory activation state (Kumar et al., 2013). In culture, MMP-3 functions as a novel signaling protein to activate microglia and may be one of the earliest microglial regulators remote to the injury site (Kim et al., 2005b). Hypertrophic microglia were evident throughout dorsal and ventral regions of the lumbar cord during the first week after SCI. The prolonged hypertrophic phenotype may be part of a paracrine/autocrine loop in which MMP-9 from endothelial cells regulates proinflammatory cytokine availability and increased MMP-9 production in microglia via NF-κB and p38 signaling, as shown in other cell types (Rajashekhar et al., 2011). In addition, the possibility exists that MMP-9 has an indirect influence on lumbar microglia after injury by regulating axonal dieback and demyelination at the injury site (Busch et al., 2009; Liu and Shubayev, 2011). Regardless of the downstream actuators, we demonstrate here that MMP-9 activity is prominent in the lumbar spinal cord and that microglial regulation is MMP-9 dependent, given that deletion of MMP-9 maintained microglia in a resting phenotype and reduced TNF-α expression.
In our training paradigm, repetitive sensorimotor cues delivered to the lumbar enlargement facilitated oscillatory motor output from central pattern generators. Synaptic strengthening and reorganization within interneuronal networks promote locomotor recovery, a process collectively termed motor relearning (Basso and Hansen, 2011). Spinal centric learning is ubiquitous and task dependent (Hodgson et al., 1994). Indeed, the lumbar spinal cord can learn to modulate motor responses, which improves locomotor recovery (Chen et al., 2006). Recently, the same learned modulation was effective in human SCI (Chen et al., 2006; Wolpaw, 2010; Thompson et al., 2013). Neuroinflammation limits synaptic formation and long-term potentiation, two necessary features of motor relearning (Yirmiya and Goshen, 2011). We show here for the first time that MMP-9 and downstream factors interact negatively with treadmill training to impede locomotor recovery after SCI. By eliminating MMP-9 in combination with early training, we restored plantar stepping by 7 d—an extent of recovery unattained by training alone or MMP-9 deficiency alone. Moreover, these training effects extended 4 weeks after the intervention ended. We propose that, after SCI, MMP-9-regulated neuroinflammation is inhibitory to synaptic plasticity and spinal centric learning. The inhibitory role of inflammatory signaling on spinal learning has been well characterized in instrumental conditioning paradigms (Young et al., 2007; Vichaya et al., 2009; Huie et al., 2012).
Surprisingly, white matter sparing at the epicenter was insufficient to produce functional gains in our severe SCI model. Unexercised KO mice did not differ from WT in the open field, which contrasts with the improved locomotor recovery reported by Noble et al. (2002) after reducing or eliminating MMP-9 in a less severe injury. We propose that modest improvement in white matter sparing with this lesion severity is likely too small to support the robust recovery attained with training in KO mice. However, the changes at the epicenter may have influenced lumbar neuroinflammation, because we found a relationship between sparing and lumbar microglial activation. In addition, during the late time point, when inflammatory processes have subsided in the lumbar cord, sparing of descending systems did not facilitate recovery even when combined with training and/or MMP-9 elimination. In rat SCI, we observed that ETT in the presence of moderate or substantial white matter sparing does not produce behavioral benefit (C.N.H. and D.M.B., unpublished data). Therefore, attenuating inflammation throughout the neuroaxis may be a necessary first step to induce activity-dependent plasticity of segmental and spared descending systems.
Treadmill training is not universally effective, but rather must be optimally timed to capture injury-induced plasticity. Despite lower levels of inflammation in KO mice, the same training that produced robust and lasting improvement early after SCI failed to induce recovery when delivered late after SCI. Distinct alterations in extracellular matrix proteins occur in the lumbar enlargement, which may prevent the efficacy of late training (Andrews et al., 2012). Extracellular matrix components such as chondroitin sulfate proteoglycans stabilize and regulate synaptic plasticity. Full-length neurocan and NG2 are known MMP substrates that increase after the first week in the lumbar enlargement and persist to chronic times after injury (Larsen et al., 2003; Rauch, 2004; Andrews et al., 2012). Lasting increases in chondroitin sulfate proteoglycan production around locomotor networks may increase synaptic stability at the cost of reducing plasticity. The inefficacy of LTT identifies a reduction of lumbar plasticity in both KO and WT mice. Therefore, a critical period of spontaneous plasticity acutely after SCI offers a novel opportunity to influence locomotor recovery. In our mouse model, this period is defined by a period of days. However, marked differences in the onset and duration of inflammation in human SCI suggest that an optimal window to create a permissive environment for rehabilitation may be weeks or months in the clinical setting (Fleming et al., 2006; Donnelly and Popovich, 2008).
An important finding of the present study was that intervening with treadmill training early after SCI during a period of marked neuroinflammation exacerbated deficits more than when no exercise was applied. In WT mice, we found that ETT produced substantial impairments. These findings are consistent with work in SCI and other CNS injury models (Kozlowski et al., 1996; Smith et al., 2009). Early after SCI, increasing vascular demand when BSCB integrity is compromised may exacerbate pathology. In fact, a single session of swim training delivered in acute stages of SCI increases lesion site permeability (Smith et al., 2009). Whether MMP-9 facilitates exercise-mediated disruption of BSCB components is unknown, although it is likely given that acute phase BSCB breakdown is reduced in KO mice (Noble et al., 2002). Because early expression of MMP-9 persists in lumbar segments of both trained and unexercised WT mice, mitigation of remote neurovascular reactivity in KO mice likely facilitated plasticity and recovery. In contrast, late after SCI, when reconstitution of the BSCB is evident, MMP-9 inhibition may be insufficient to facilitate lumbar plasticity (Whetstone et al., 2003).
Our results suggest that disrupted neurovascular interactions around locomotor circuitry far distant to the SCI can result in profound functional consequences. We show that locomotor training can be quite effective when administered early after SCI and in combination with reduced inflammation. However, the efficacy of treadmill training is lost when delivered chronically and carries the risk of inducing neurotoxicity in an inflammatory microenvironment. Combinatorial therapies that consider the interaction between spinal centric learning and remote lumbar inflammation will likely permit the most robust synaptic and structural remodeling in locomotor networks for recovery.
Footnotes
This work was supported by the National Institutes of Health (Grants #1RO1NS074882-01A1 to D.M.B. and #1F31NS080512-01 to C.N.H.). We thank Dr. Kristina Kigerl for critical review of the manuscript.
The authors declare no competing financial interests.
References
- Andrews EM, Richards RJ, Yin FQ, Viapiano MS, Jakeman LB. Alterations in chondroitin sulfate proteoglycan expression occur both at and far from the site of spinal contusion injury. Exp Neurol. 2012;235:174–187. doi: 10.1016/j.expneurol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Hansen CN. Biological basis of exercise-based treatments: spinal cord injury. PM R. 2011;3:S73–S77. doi: 10.1016/j.pmrj.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Murray M, Goldberger ME. Differential recovery of bipedal and overground locomotion following complete spinal cord hemisection in cats. Restor Neurol Neurosci. 1994;7:95–110. doi: 10.3233/RNN-1994-7205. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Buehner JJ, Forrest GF, Schmidt-Read M, White S, Tansey K, Basso DM. Relationship between ASIA examination and functional outcomes in the NeuroRecovery Network Locomotor Training Program. Arch Phys Med Rehabil. 2012;93:1530–1540. doi: 10.1016/j.apmr.2012.02.035. [DOI] [PubMed] [Google Scholar]
- Busch SA, Horn KP, Silver DJ, Silver J. Overcoming macrophage-mediated axonal dieback following CNS injury. J Neurosci. 2009;29:9967–9976. doi: 10.1523/JNEUROSCI.1151-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss A, Pech K, Kakulas BA, Martin D, Schoenen J, Noth J, Brook GA. Matrix metalloproteinases and their inhibitors in human traumatic spinal cord injury. BMC Neurology. 2007;7:17. doi: 10.1186/1471-2377-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen XY, Jakeman LB, Chen L, Stokes BT, Wolpaw JR. Operant conditioning of H-reflex can correct a locomotor abnormality after spinal cord injury in rats. J Neurosci. 2006;29:12537–12543. doi: 10.1523/JNEUROSCI.2198-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85:2813–2823. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- Falo MC, Fillmore HL, Reeves TM, Phillips LL. Matrix metalloproteinase-3 expression profile differentiates adaptive and maladaptive synaptic plasticity induced by traumatic brain injury. J Neurosci Res. 2006;84:768–781. doi: 10.1002/jnr.20986. [DOI] [PubMed] [Google Scholar]
- Fawcett J. Molecular control of brain plasticity and repair. Prog Brain Res. 2009;175:501–509. doi: 10.1016/S0079-6123(09)17534-9. [DOI] [PubMed] [Google Scholar]
- Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Kerr BJ, David S. Tissue displacement and impact force are important contributors to outcomes after spinal cord contusion injury. Exp Neurol. 2005;196:9–17. doi: 10.1016/j.expneurol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Goslow GE, Jr, Reinking RM, Stuart DG. The cat step cycle: hind limb joint angles and muscle lengths during unrestrained locomotion. J Morphol. 1973;141:1–41. doi: 10.1002/jmor.1051410102. [DOI] [PubMed] [Google Scholar]
- Goussev S, Hsu JY, Lin Y, Tjoa T, Maida N, Werb Z, Noble-Haeusslein LJ. Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J Neurosurg. 2003;99:188–197. doi: 10.3171/jns.2003.99.1.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Gómez-Pinilla F, Hovda DA. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma. 2007;24:1161–1171. doi: 10.1089/neu.2006.0255. [DOI] [PubMed] [Google Scholar]
- Grill R, Murai K, Blesch A, Gage FH, Tuszynski MH. Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth and partial functional recovery after spinal cord injury. J Neurosci. 1997;17:5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CN, Linklater W, Santiago R, Fisher LC, Moran S, Buford JA, Basso DM. Characterization of recovered walking patterns and motor control after contusive spinal cord injury in rats. Brain Behav. 2012;2:541–552. doi: 10.1002/brb3.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkema SJ, Schmidt-Read M, Lorenz DJ, Edgerton VR, Behrman AL. Balance and ambulation improvements in individuals with chronic incomplete spinal cord injury using locomotor training based rehabilitation. Arch Phys Med Rehabil. 2012;93:1508–1517. doi: 10.1016/j.apmr.2011.01.024. [DOI] [PubMed] [Google Scholar]
- Harkema S, Gerasimenko Y, Hodes J, Burdick J, Angeli C, Chen Y, Ferreira C, Willhite A, Rejc E, Grossman RG, Edgerton VR. Effect of epidural stimulation of the lumbosacral spinal cord on voluntary movement, standing, and assisted stepping after motor complete paraplegia: a case study. Lancet. 2011;377:1938–1947. doi: 10.1016/S0140-6736(11)60547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JA, de Leon R, Dobkin B, Edgerton VR. Can the mammalian lumbar spinal cord learn a motor task? Med Sci Sports Exerc. 1994;26:1491–1497. [PubMed] [Google Scholar]
- Hoschouer EL, Basso DM, Jakeman LB. Aberrant sensory responses are dependent on lesion severity after spinal cord contusion injury in mice. Pain. 2010;148:328–342. doi: 10.1016/j.pain.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougland MT, Harrison BJ, Magnuson DS, Rouchka EC, Petruska JC. The transcriptional response of neurotrophins and their tyrosine kinase receptors in lumbar sensorimotor circuits to spinal cord contusion is affected by injury severity and survival time. Front Physiol. 2012;3:478. doi: 10.3389/fphys.2012.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, Bourguignon LY, Adams CM, Peyrollier K, Zhang H, Fandel T, Cun CL, Werb Z, Noble-Haeusslein LJ. Matrix metalloproteinase-9 facilitates glial scar formation in the injured spinal cord. J Neurosci. 2008;28:13467–13477. doi: 10.1523/JNEUROSCI.2287-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie JR, Baumbauer KM, Lee KH, Bresnahan JC, Beattie MS, Ferguson AR, Grau JW. Glial tumor necrosis factor alpha (TNFa) generates metaplastic inhibition of spinal learning. PLoS One. 2012;7:e39751. doi: 10.1371/journal.pone.0039751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KJ, Gómez-Pinilla F, Crowe MJ, Ying Z, Basso DM. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain. 2004;127:1403–1414. doi: 10.1093/brain/awh160. [DOI] [PubMed] [Google Scholar]
- Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT. Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma. 2000;17:299–319. doi: 10.1089/neu.2000.17.299. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Fillmore HL, Reeves TM, Phillips LL. Elevation of hippocampal MMP-3 expression and activity during trauma-induced synaptogenesis. Exp Neurol. 2005a;192:60–72. doi: 10.1016/j.expneurol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kim SS, Cho JJ, Choi DH, Hwang O, Shin DH, Chun HS, Beal MF, Joh TH. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. J Neurosci. 2005b;25:3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski DA, James DC, Schallert T. Use dependent exaggeration of neuronal injury after unilateral sensorimotor cortex lesions. J Neurosci. 1996;16:4776–4786. doi: 10.1523/JNEUROSCI.16-15-04776.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–1411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NGS proteoglycan. J Neurosci. 2003;23:11127–11135. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond H, L'Esperance M, Orsal D, Rossignol S. Treadmill locomotion in the intact and spinal mouse. J Neurosci. 2003;23:11411–11419. doi: 10.1523/JNEUROSCI.23-36-11411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Shubayev VI. Matrix metalloproteinase-9 controls proliferation of NG2+ progenitor cells immediately after spinal cord injury. Exp Neurol. 2011;231:236–246. doi: 10.1016/j.expneurol.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Basso DM, Walters P, Stokes BT, Jakeman LB. Behavioral and histological outcomes following graded spinal cord contusion injury in the C57Bl/6 mouse. Exp Neurol. 2001;169:239–254. doi: 10.1006/exnr.2001.7679. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol. 2004;474:469–486. doi: 10.1002/cne.20149. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Smith RR, Brown EH, Enzmann G, Angeli C, Quesada PM, Burke D. Swimming as a model of task-specific locomotor retraining after spinal cord injury in the rat. Neurorehabil Neural Repair. 2009;23:535–545. doi: 10.1177/1545968308331147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado MA, Allred RP, Felthauser EL, Jones TA. Motor skill training, but not voluntary exercise, improves skilled reaching after unilateral ischemic lesions of the sensorimotor cortex in rats. Neurorehabil Neural Repair. 2008;22:250–261. doi: 10.1177/1545968307308551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh MP. Recent advances in the understanding of the repeated bout effect: the protective effect against muscle damage from a single bout of eccentric exercise. Scand J Med Sci Sports. 2003;13:88–97. doi: 10.1034/j.1600-0838.2003.02477.x. [DOI] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Matynia A, Balcerzyk M, Okulski P, Dzwonek J, Costa RM, Silva AJ, Kaczmarek L, Huntley GW. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 2006;26:1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 1996;142:258–275. doi: 10.1006/exnr.1996.0196. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Scheff SW. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J Neurotrauma. 2001;18:513–522. doi: 10.1089/089771501300227314. [DOI] [PubMed] [Google Scholar]
- Rajashekhar G, Kamocka M, Marin A, Suckow MA, Wolter WR, Badve S, Sanjeevaiah AR, Pumiglia K, Rosen E, Clauss M. Pro-inflammatory angiogenesis is mediated by p38 MAP kinase. J Cell Physiol. 2011;226:800–808. doi: 10.1002/jcp.22404. [DOI] [PubMed] [Google Scholar]
- Rauch U. Extracellular matrix components associated with remodeling processes in brain. Cell Mol Life Sci. 2004;61:2031–2045. doi: 10.1007/s00018-004-4043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RR, Brown EH, Shum-Siu A, Whelan A, Burke DA, Benton RL, Magnuson DS. Swim training initiated acutely after spinal cord injury is ineffective and induces extravasation in and around the epicenter. J Neurotrauma. 2009;26:1017–1027. doi: 10.1089/neu.2008.0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys Z, Ziaja M, Pawlínski R, Setkowicz Z, Janeczko K. Morphology of reactive microglia in the injured cerebral cortex–fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–97. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO%3B2-9. [DOI] [PubMed] [Google Scholar]
- Thompson AK, Pomerantz FR, Wolpaw JR. Operant conditioning of a spinal reflex can improve locomotion after spinal cord injury in humans. J Neurosci. 2013;33:2365–2375. doi: 10.1523/JNEUROSCI.3968-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vempati P, Karagiannis ED, Popel AS. A biochemical model of matrix metalloproteinase 9 activation and inhibition. J Biol Chem. 2007;282:37585–37596. doi: 10.1074/jbc.M611500200. [DOI] [PubMed] [Google Scholar]
- Vichaya EG, Baumbauer KM, Carcoba LM, Grau JW, Meagher MW. Spinal glia modulate both adaptive and pathological processes. Brain Behav Immun. 2009;23:969–976. doi: 10.1016/j.bbi.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/S0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. J Neurosci Res. 2003;74:227–239. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whishaw IQ, Pellis SM, Pellis VC. A behavioral study of the contributions of cells and fibers of passage in the red nucleus of the rat to postural righting, skilled movements, and learning. Behav Brian Res. 1992;52:29–44. doi: 10.1016/S0166-4328(05)80322-5. [DOI] [PubMed] [Google Scholar]
- Wolpaw JR. What can the spinal cord teach us about learning and memory? Neuroscientist. 2010;16:532–549. doi: 10.1177/1073858410368314. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Young EE, Baumbauer KM, Elliot A, Joynes RL. Lipopolysaccharide induces a spinal learning deficit that is blocked by IL-1 receptor antagonism. Brain Behav Immun. 2007;21:748–757. doi: 10.1016/j.bbi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chang M, Hansen CN, Basso DM, Noble-Haeusslein LJ. Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurotherapeutics. 2011;8:206–220. doi: 10.1007/s13311-011-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]