

Abstract
ATM kinase signals DNA double strand breaks (DSB) to cell cycle arrest via p53 and DNA repair. ATM-defective cells are sensitive to DSB-inducing agents, making ATM an attractive target for anticancer chemo- and radio-sensitisation. KU59403 is an ATM inhibitor with the potency, selectivity and solubility for advanced pre-clinical evaluation.
KU59403 was not cytotoxic to human cancer cell lines (SW620, LoVo, HCT116 and MDA-MB-231) per se but significantly increased the cytotoxicity of topoisomerase I and II poisons: camptothecin, etoposide and doxorubicin. Chemo- and radio-sensitisation by ATM inhibition was not p53-dependent. Following administration to mice, KU59403 distributed to tissues and concentrations exceeding those required for in vitro activity were maintained for at least 4 hr in tumour xenografts. KU59403 significantly enhanced the antitumour activity of topoisomerase poisons in mice bearing human colon cancer xenografts (SW620 and HCT116) at doses that were non-toxic alone and well tolerated in combination. Chemosensitisation was both dose and schedule-dependent.
KU59403 represents a major advance in ATM inhibitor development, being the first compound to demonstrate good tissue distribution and significant chemo-sensitisation in in vivo models of human cancer, without major toxicity. KU59403 provides the first proof-of-principle pre-clinical data to support the future clinical development of ATM inhibitors.
Keywords: ATM inhibitor, p53, ionising radiation, topoisomerase poison, cytotoxicity, pharmacokinetics, xenografts, antitumour activity
INTRODUCTION
DNA is constantly being damaged either from endogenous sources or environmental mutagens and carcinogens. DNA double strand breaks (DSBs) are particularly cytotoxic and cells mount a co-ordinated response of cell cycle arrest and DNA repair in response to these lesions (1). The Ataxia Telangiectasia Mutated (ATM) kinase is a major coordinator of the DSB response and is the product of the ATM gene, which is defective in the disease Ataxia Telangeictasia (A-T) that is characterised by neurodegeneration, immunodeficiency, cancer pre-disposition and an extreme hypersensitivity to ionising radiation (IR) and other DSB-inducing agents (2). In response to DSBs ATM initiates a cascade of phosphorylation events to induce cell cycle arrest via p53 and other checkpoint proteins (reviewed in 3), and promote DNA repair by both homologous recombination and non-homologous end joining (4, 5).
Ionising radiation and topoisomerase poisons are important anticancer agents that induce DNA DSBs. It is estimated that 1 Gy of irradiation induces 1,000 single strand breaks (SSB) and 25-40 double strand DNA breaks per diploid cell (6). Topoisomerase II poisons, by stabilising the topoisomerase II-DNA cleavable complex, cause persistent protein-associated DNA DSBs while topoisomerase I poisons stabilise the topoisomerase I-DNA cleavable complex to cause persistent single strand breaks that are converted to DSB at replication. A-T cells display defective p53 induction and loss of cell cycle arrest; however, lack of ATM also confers radio-sensitivity in some p53-null mouse tissues suggesting the existence of a p53-independent ATM effector pathway (7). ATM inhibition is therefore an attractive approach to anti-cancer chemo- and radio-sensitisation (8) with potential benefits in both p53 functional and dysfunctional cancers.
The C-terminal domain of ATM contains the serine threonine kinase signature motif characteristic of the PI3K family (9). The PI3 Kinase inhibitor LY294002 (Table 1) inhibits other members of the PI3 Kinase family (10) and we previously used scaffold hopping from LY294002 to develop KU55933 as a selective inhibitor of ATM (Table 1) that enhanced the cytotoxicity of ionising radiation (IR) and topoisomerase II poisons in human tumour cell lines (11). Further development identified KU-600019 as a more potent and selective ATM inhibitor that radio-sensitised glioma cells (12). However, neither compound has been evaluated in vivo. In the work presented here we probe the p53-dependency of this class of compound and describe the novel ATM inhibitor KU59403 (Table 1), with increased potency and specificity against ATM, improved pharmacological properties and promising activity in animal models of human cancer.
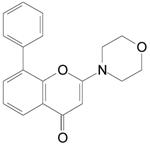
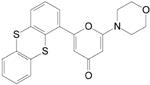
Table 1. Chemical structures and inhibition of ATM and related kinase activity by LY294002, KU55933 and KU59403.
| Name | Chemical structure | Enzyme IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|
| ATM | DNA-PK | PI3K | ATR | PI4K | mTOR | ||
| LY294002 |
|
>100 | 1.5 | 2 | 100 | ND* | 3 |
| KU55933 |
|
0.013 | 2.5 | 1.7 | >10 | >10 | 9.3 |
| KU59403 |
|
0.003 | 9.1 | 10 | >100 | >10 | 14 |
IC50 (μM) against purified enzyme was calculated from sigmoid plots of increasing concentrations of inhibitor versus activity for each of the kinases.
Data represent the mean of five independent experiments.
Data previously reported in (11)
PI4K value was not determined for LY294002 in these experiments
Materials and Methods
Chemicals
The specific ATM inhibitors, KU55933 and KU59403, were kindly provided by KuDOS, Horsham, United Kingdom. Etoposide phosphate and irinotecan (CPT-11) were purchased from the National Health Service (UK). All other chemicals were purchased from Sigma (Poole, UK) unless stated otherwise. KU55933, KU59403, doxorubicin, camptothecin and etoposide were dissolved in dimethyl sulfoxide (DMSO) for in vitro evaluation as 10 mM stocks and stored at −20°C. All drugs were added to cells such that the final concentration of DMSO in culture media was 0.5% (v/v), and results were compared with controls incubated with 0.5% DMSO in media alone. Irinotecan (CPT-11, clinical grade, formulated in saline) and etoposide phosphate (etopophos, clinical grade, formulated in saline) were used in place of camptothecin and etoposide, respectively, for in vivo studies. KU55933 was administered at 10 mg/kg (the maximum administrable dose due to limited solubility) formulated in equimolar phosphoric acid, 5% (v/v) DMSO, 10% (w/v)encapsin pH 4 and KU59403, formulated in equimolar phosphoric acid (Analar, UK) in physiological saline pH 4. All drugs for in vivo evaluation were formulated on the day of the experiment.
Enzyme inhibition
The activity of KU59403 against ATM and other PI3K family members isolated from HeLa cells was determined as previously described (11)
Cell lines and culture
LoVo, HCT116 and SW620 (human colon cancer), and U2OS (human osteosarcoma) and MDA-MB-231 (human breast cancer) cells were purchased from the American Type Culture Collection (Manassas, USA). They were maintained at <30 passages from receipt using separate reagents for each cell line. HCT116 N7 cells (HCT-116 cells stably transfected with a plasmid containing HPV16 E6 cDNA such that p53 protein is degraded through the ubiquitin-proteasome pathway (13)) were a gift from M. D’Incalci (Milan). U2OS p53DN expressing the p53-R248W dominant negative mutant p53 were prepared by transfection of U2OS:PG13-Luc cells (14), and the failure to mount a p53 response to IR was confirmed in these cells (Supplementary Figure 1). All cells were cultured in RPMI 1640 media supplemented with 10% (v/v) fetal bovine serum, penicillin (50 μnits/ml), and streptomycin (50 units/ml) at 37°C in an atmosphere of 5% CO2 in air. Cells were confirmed to be free of mycoplasma contamination and LoVo, SW620, HCT116 and MDA-MB-231 were authenticated by STR profiling (LGC Standards, Teddington, UK). The population doubling time of the cells was approximately 24 hours
Cytotoxicity and growth inhibition studies
We determined the effect of KU55933 and KU59403 on cellular survival following exposure to X-irradiation or the topoisomerase II poisons, etoposide and doxorubicin, and the topoisomerase I poison, camptothecin, (Supplementary Figure 2) by clonogenic assay as described previously (15). Briefly, exponentially growing cells were exposed to the cytotoxic agent with or without KU55933 (10 μM) or KU59403 (1.0 μM) for 16 hours and survival was calculated by comparison to the appropriate control (0.5% DMSO or ATM inhibitor alone). The dose modification ratio (DMR) was calculated as the percentage surviving cells (compared to control) treated with the cytotoxic agent alone, divided by the percentage surviving cells treated with the cytotoxic agent and the ATM inhibitor.
KU55933 and KU59403 pharmacokinetic and tissue distribution studies
All in vivo experiments were reviewed and approved by the relevant institutional animal welfare committees and performed according to national law and published guidelines (16). SW620 colorectal tumour cells (1 × 107 cells in 50 μl culture medium per animal) were injected subcutaneously (s.c.) into the flanks of female athymic nude mice (CD1 nu/nu, Charles River, UK) and tissue distribution studies were performed when tumours had reached a size of approximately 650 mm3. KU59403 was given at 25 mg/kg to non-tumour bearing female Balb/C mice or 50 mg/kg to SW620 tumour-bearing female nude mice. For comparison KU55933 was administered at 10 mg/kg, which was the maximum administrable dose due to the limited solubility of KU55933 (i.e. 1 mg/ml even with the addition of 5% (v/v) DMSO and 10% (w/v) encapsin), to SW620-bearing nude mice. All doses were given at a dosing volume of 10 ml/kg via the intraperitoneal (i.p.) or intravenous (i.v.) route. Mice were bled under terminal anaesthesia via cardiac puncture and the plasma fraction was stored at −20 °C. Tissues were snap frozen in liquid nitrogen and stored at −80 °C until homogenisation in PBS (1:3 w/v), using a stirrer macerator homogenizer (Werke GmbH & Co., KG. Germany) immediately prior to assay.
HPLC analysis of ATM inhibitors in plasma and tissue homogenates
KU55933 and KU59403 were extracted from plasma and tissue homogenates (50 μl) and analysed by HPLC as described previously (15). Plasma samples were quantified using a standard curve, prepared in plasma that was linear over the range: 0.05-10 μg/ml (r2 >0.9) with duplicate QA standards (at 0.1, 1 and 10 μg/ml). Tissue concentrations were calculated using the method of addition (17) to account for the efficiency of recovery and compensate for inter-sample variation.
Anti-tumour efficacy studies
CD-1 nude mice were implanted with SW620 or HCT116-N7 human cancer cell lines at 1×107 cells per animal s.c. (n = 5 per group).Treatment began when tumours were palpable (approximately 5 mm × 5 mm, 8-10 days post implantation) with normal saline (control animals), KU59403 as indicated in the Results section, alone or in combination with etoposide phosphate or irinotecan (CPT-11). For combinations, the first daily dose of KU59403 was administered immediately prior to etoposide phosphate or irinotecan unless otherwise indicated. Tumour volume was calculated from two-dimensional electronic calliper (Mitutoyo, Andover, UK.) measurements using the equation a2 × b/2 where a is the smallest measurement and b the largest. Data are presented as the median relative tumour volume (RTV), where the tumour volume for each animal on the initial day of treatment (day 0) is assigned an RTV value of 1.
Statistical Analysis
Data were analysed data using Graphpad Prism software (GraphPad Software, Inc. San Diego Ca USA). For the in vitro studies significant differences between the effect of cytotoxic agent alone and cytotoxic agent plus KU59403 were determined by Student’s t-test (parametric). For in vivo studies, significant differences between the time take to reach RTV were determined by Mann Whitney test.
Results
In vitro activity of KU59403 and p53 independence of chemo- and radio-sensitisation
KU59403 is a novel ATM inhibitor developed from LY294002 (Table 1), which is more potent against ATM than the previous lead KU55933 (IC50 3 nM vs 13 nM), and has at least 1000 times greater specificity for ATM over other members of the PI3K family tested.
In contrast to the concentrations of 10 μM of KU55933 and 3 μM KU-600019 needed to induce in vitro chemo- and radio-sensitisation (11, 12), KU59403 was an effective chemo-sensitiser at a concentration of 1 μM. At this concentration KU59403 inhibited ATM activity in SW620 cells by >50%, at the higher concentration of 10 μM KU55933 also substantially inhibited ATM activity (Supplementary figure 3). KU59403 alone was not significantly cytotoxic to LoVo or SW620 cells (88± 7% and 91± 6% survival, respectively) but it enhanced camptothecin cytotoxicity (Figure 1A, Table 2) in both cell lines with greater enhancement being observed in the LoVo compared to the SW620 cells (7-fold; p=0.038 versus 4-fold; p=0.014 at 10 nM camptothecin). KU59403 also significantly enhanced the cytotoxicity of fixed concentrations of etoposide (0.1 and 1 μM) or doxorubicin (10 or 100 nM) in these cell lines, with greater enhancement of etoposide in SW620 cells and of doxorubicin in LoVo cells (Table 2).
Figure 1. In vitro chemosensitisation.

A. Sensitisation of SW620 (left panel) and LoVo cells (right panel) to camptothecin by KU59403. Cells were exposed to varying concentrations of camptothecin alone (filled circles, solid line) or in the presence of 1 μM KU59403 (open circles, broken line) for 16 hr prior to seeding for colony formation. Data, normalised to DMSO or KU59403 alone control, are mean ± SD from 3 independent experiments.
B. Survival of wt (circles) and p53 dysfunctional (triangles) HCT116 (left panel) and U2OS cells (right panel) following exposure to increasing concentrations of camptothecin in the presence (open symbols, dashed lines) or absence of KU55933 (filled symbols, solid line). Data, normalised to DMSO or KU55933 alone, are mean ± SD from 3 independent experiments
C. Survival of wt (circles) and p53 dysfunctional (triangles) HCT116 (left panel) and U2OS cells(right panel) to increasing doses of X rays in the presence (open symbols, dashed lines) or absence of KU55933 (filled symbols, solid line). Data, normalised to DMSO or KU55933 alone, are mean ± SD from 3 independent experiments
D. Sensitisation of SW620 HCT116, HCT116-N7 and MDA-MB-231 cells to etoposide by KU59403. Data are 1 μM etoposide alone (white bars) or 1 μM etoposide plus 1 μM KU59403 (black bars) mean ± SD from 3 independent experiments.
Table 2. Cytotoxicity of etoposide, doxorubicin and camptothecin, alone and in combination with KU59403 in LoVo and SW620 cells.
| Cytotoxic drug | SW620 | LoVo | ||||
|---|---|---|---|---|---|---|
| % Survival | Enhancement factor† |
% Survival | Enhancement factor |
|||
| Cytotoxic alone |
Cytotoxic + KU59403 |
Cytotoxic alone |
Cytotoxic + KU59403 |
|||
| Etoposide 100 nM | 89 ± 8.3 | 35 ± 12** | 2.9 ± 1.5 | 58 ± 8.3 | 18 ± 0.5** | 3.3 ± 0.4 |
| Etoposide 1 μM | 3.8 ± 1.1 | 0.41 ± 0.23* | 12 ± 7 | 3.9 ± 0.8 | 1.1 ± 0.2* | 3.8 ± 1.5 |
| Doxorubicin 10 nM | 50 ± 9 | 17 ± 0.2* | 2.9 ± 0.6 | 47 ± 3.1 | 16 ± 6.9* | 3.2 ± 1 |
| Doxorubicin 100 nM | 0.02 ± 0.01 | 0.01 ± .001 | 1.8 ± 0.7 | 2.1 ± 0.5 | 0.35 ± 0.04* | 6.1 ± 2.1 |
| Camptothecin 10 nM | 2.4 ± 0.2 | 0.71 ± 0.35* | 4.3 ± 2.8 | 33 ± 7.9 | 6.2 ± 3. 8* | 6.9 ± 3.8 |
Cells were exposed to etoposide or doxorubicin, at the concentrations indicated, alone or in combination with 1 μM KU59403 in a final concentration of 0.5% (v/v) DMSO for 16 hr. Data, normalised in comparison with DMSO or KU59403 alone control, as appropriate, are the mean ± standard deviation of 3 independent experiments. Significant differences between cytotoxic drug alone versus cytotoxic + KU59404 is given by
p=<0.05
and
p<0.01
Enhancement factor is defined as the survival with the cytotoxic alone ÷ survival with cytotoxic + KU59403 in each individual experiment, and data are the mean ± standard deviation of 3 independent experiments.
There was no consistent difference in the enhancement of cytotoxicity in LoVo cells (wild type p53) compared to SW620 cells (mutant p53) but, as these cells were derived from different tumours, they could harbour other genotypic or phenotypic differences that might mask the effect of p53 status. For this reason we investigated whether chemo- and radio-sensitivity was enhanced by ATM inhibition in a p53-dependent manner using paired cell lines with functional or dysfunctional p53 using KU55933 as a model compound to confirm the data with KU59403 in proof of principle studies. KU55933 (10 μM) sensitised p53 functional and dysfunctional HCT116 and U2OS cells to camptothecin to a similar extent (4 to 5-fold, Fig 1B and Supplementary Table 1). Radiosensitisation by KU55933 was greater in HCT116 than in U20S cells but the p53 status did not affect radiosensitivity or enhancement by KU55933 (Fig 1C, Supplementary Table 1). The p53 status of the cell did not have a consistent effect on chemosensitisation of topoisomerase II poisons by KU55933 either, for example p53 dysfunction conferred reduced sensitisation to etoposide and doxorubicin in U20S cells but had no significant impact in HCT116 cells (Supplementary Table 2). Consistent with the p53 independence of chemo- and radio-sensitisation, KU55933 increased the G2 cell cycle arrest induced by IR, camptothecin, doxorubicin and etoposide to a similar extent in p53 functional and dysfunctional cells and did not affect DNA DSB formation or repair kinetics (Supplementary Figures 4 and 5). These data were confirmed with KU59403 (1 μM) which enhanced etoposide (1 μM) cytotoxicity to a similar extent in HCT116 and HCT116-N7cells by 2.3 ± 1.6-fold (p=0.011) and 3.8 ± 2.5-fold (p=0.019), respectively, and in the p53 mutant SW620 cells and human breast cancer cell line, MDA-MB-231, sensitisation was 11.9 ± 4.7 (p<0.0001) and 3.8 ± 1.8-fold (p= 0.006) respectively (Figure 1 D). Inhibition of IR-induced ATM activity by KU59403 (1 μM) was approximately 50% in MDA-MB231 cells and >50% in HCT116 cells that have low ATM expression and activity (Supplementary Figure 3). These data indicate that p53 status has no major impact on sensitisation by KU59403, and that SW620 cells, where a 12-fold enhancement was observed, are the most susceptible to etoposide sensitisation by ATM inhibition. On the basis of these data, SW620 tumours treated with etoposide were chosen as our primary model system for the evaluation of KU59403 in in vivo studies.
Pharmacokinetics
As part of initial studies, plasma and tumour concentrations of drug were measured at 1 and 4 hours after administration of a single dose of KU59403 at 50 mg/kg i.p. and KU55933 at the maximum administrable dose of 10 mg/kg. The plasma concentration of KU59403 was 5 μM, and maintained for at least 4 hours. In comparison, plasma levels of KU55933 were just over 1 μM, consistent with the 5-fold lower dose administered (Figure 2A). KU59403 accumulated in tumour tissue up to the 4 hour time point with a concentration at this time of 1.9 μM, which is greater than that shown to be necessary for activity in the in vitro studies (Figure 2A). In contrast, the levels of KU55933 in the tumour were below the limit of detection (0.5 μM). To determine the pharmacokinetics of KU59403 in normal tissues, the compound was administered to female Balb/C mice at 25 mg/kg intravenously (i.v.) (Figure 2B). In contrast to the previous experiment, KU59403 was cleared rapidly from the plasma and at 4 hr the plasma concentration was less than 0.1 μM. This difference could be due to different route of administration, different dose or strain specific metabolism. There was, nevertheless, substantial accumulation and retention in the tissues, especially the liver, indicating that hepatic clearance may be the main route of elimination of this compound. At this dose and route of administration KU59403 achieved concentrations in tissues in excess of those required for in vitro chemo-sensitisation.
Figure 2. Tissue distribution of KU59403.

A. Plasma (white bars) and tumour (black bars) concentrations 1 hr and 4 hr after i.p. administration of KU59403 (50 mg/kg) or KU55933 (10 mg/kg) to female CD-1 athymic mice bearing the SW620 tumour xenograft subcutaneously.
B. Tissue concentrations of KU59403 following treatment of BalbC mice with 25mg/kg KU59403 i.v. Concentrations of KU59403 were determined in plasma (white bars) and tissue homogenates (brain; pale grey bars, liver; dark grey bars, spleen; striped bars and lung; black bars) by HPLC at various time points post administration as indicated. Data are the mean of 3 samples from 3 mice per time-point from a single experiment
Antitumour efficacy studies
To investigate whether the marked chemo-sensitisation by KU59403 observed in vitro could be reproduced in vivo we treated mice bearing SW620 tumour xenografts with etoposide phosphate (etopophos) at a fixed dose of 11.35 mg/kg (equivalent to 10 mg/kg free etoposide) i.p. daily for 5 days, or irinotecan (2.5 mg/kg i.p) daily for 5 days alone and in combination with KU59403. We also investigated the dose and schedule dependency of KU59403 administration in combination with etopophos. KU59403 was given at doses of 6, 12.5 and 25 mg/kg i.p. twice daily (0 and 4 hours) and 12.5 mg/kg once daily, either immediately prior to etopophos dosing or 4 hours after etopophos dosing.
Tumours in control mice reached 4 times their starting volume (RTV4) at a median time of 6.5 days (Figure 3 A, Table 3). Treatment with etopophos alone caused a modest tumour growth delay of 4 days (time to RTV4 = 10.5 days). This delay was extended to 8.5 days (time to RTV4 = 15 days, p=0.093) when given with KU59403 at 12.5 mg/kg i.p. twice daily for 5 days and 11.5 days (time to RTV4 = 18 days) when given with KU59403 at 25 mg/kg i.p. twice daily for 5 days. This latter treatment was the most effective dosing schedule for KU59403 identified; increasing etopophos efficacy by 190% (p=0.032, Table 3). In contrast, when KU59403 was administered 4 hours after etopophos administration there was no increase in efficacy compared with etopophos alone. In the above studies, neither KU59403 nor etopophos given as a single agent caused any measurable toxicity (maximum body weight loss <2%) and the combination of drugs did not cause unacceptable toxicity (maximum body weight loss = 7%) (Supplementary Figure 5A).
Figure 3. Anti tumour efficacy of KU59403 in combination with topoisomerase poisons.

A. SW620 xenograft: KU59403 in combination with etoposide phosphate. CD-1 nude mice were treated with vehicle control (filled circles, solid line), KU59403 12.5 mg/kg 2× daily ×5 (open circles, broken line), etoposide phosphate 10 mg/kg daily ×5 alone (filled triangles solid line) or in combination with KU59403 12.5 mg/kg 2× daily ×5 (open triangles, broken line), KU59403 6 mg/kg 2× daily ×5 (open inverted triangles, broken line), KU59403 12.5 mg/k 1× daily ×5 (filled inverted triangles, solid line), or KU59403 25 mg/k 2× daily ×5 (open squares, broken line). Growth of SW620 xenografts is presented as the median relative tumour volume (RTV) of groups of 5 animals
B. HCT116 N7 xenograft: KU59403 in combination with etoposide phosphate. CD-1 nude mice were treated with vehicle control (filled circles, solid line), etopophos 10 mg/kg daily ×5 alone (filled triangles solid line) or in combination with KU59403 25 mg/k 2× daily ×5 (open triangles, broken line). Growth of HCT116-N7 xenografts is presented as the median relative tumour volume (RTV) of groups of 5 animals
C. SW620 xenograft: KU59403 in combination with irinotecan. CD-1 nude mice were treated with vehicle control (filled circles solid line), KU59403 12.5 mg/kg 2× daily ×5 (open circles, broken line), irinotecan 2.5 mg/kg daily ×5 alone (filled triangles, solid line) or in combination with KU59403 25 mg/k 2× daily ×5 (open triangles, broken line). Growth of SW620 xenografts is presented as the median relative tumour volume (RTV) of groups of 5 animals
Table 3. Chemosensitisation of antitumour activity determined in mice bearing SW620 xenografts.
| Treatment | Nadir % Starting Body weight |
Time to RTV4 |
Delay (days)§ |
Enhancement (%)† |
|---|---|---|---|---|
| Vehicle control | 96.3 | 6.5 | ||
| KU59403 12.5 mg/kg 2× daily ×5 | 100 | 7 | 0.5 | |
| Etopohos (equivalent to 10 mg/kg etoposide) daily ×5 |
98.6 | 10.5 | 4 | |
| Etophos + KU59403 6 mg/kg 2× daily ×5 |
96.9 | 12 | 5.5 | 38 |
| Etophos + KU59403 12.5 mg/kg 2× daily ×5 |
94.9 | 15 | 8.5 | 113§ |
| Etophos + KU59403 25 mg/kg 2× daily ×5 |
93.3 | 18 | 11.5 | 190* |
| Etophos + KU59403 12.5 mg/kg 1× daily ×5 concurrent |
96.5 | 16.5 | 10 | 150 |
| Etophos + KU59403 12.5 mg/kg 1× daily ×5 5 hr post |
94.3 | 10 | 3.5 | 0 |
| Irinotecan 2.5 mg/kg daily ×5 | 93.6 | 14.5 | 8 | |
| KU59403 25 mg/kg daily ×5 | 99.7 | 12 | 5.5 | |
| Irinotecan + KU59403 25 mg/kg daily ×5 |
100 | 26 | 19.5 | 144* |
Delay (days) is the tumour growth delay, which is calculated as the time to median relative tumour volume 4 (median RTV4) following relevant treatment minus time to median RTV4 in control mice
Enhancement (%) is calculated as (100*(delay combination/delay cytotoxic alone))-100
indicates statistically significant enhancement (p<0.05)
indicates marginally statistically significant enhancement (p<0.1)
In order to investigate the enhancement of etopophos by KU59403 in a different xenograft model, mice bearing HCT116-N7 tumours were treated with vehicle alone, etopophos 11.35 mg/kg i.p. daily for 5 days and/or KU59403 25 mg/kg i.p. twice daily for 5 days (Figure 4B). These tumours grew rapidly with tumours reaching RTV4 at 4.5 days, and HCT116-N7 tumours were resistant to etopophos alone (median time to RTV4 = 5.5 days). However, the etopophos-induced tumour growth delay was extended to 8.5 days by co-administration of KU59403, representing a 300% enhancement of etopophos activity that was statistically significant (Mann Whitney test p=0.037). Toxicity, as measured by body weight loss, was tolerable and transient (Supplementary Figure 5B).
Irinotecan, a member of the camptothecin group of compounds. is commonly used in the treatment of colon cancer. In this study irinotecan alone caused an initial modest regression of the SW620 tumour followed by rapid re-growth resulting in a tumour growth delay of 7.5 days, which was extended to 19.5 days by the co-administration of KU59403. This represents a 144% enhancement of irinotecan-induced tumour growth delay, which was significantly different from irinotecan alone (p=0.032) (Figure 3C, Table 3). There were no unacceptable adverse effects on animal body weights at any of the doses given in this study (Supplementary Figure 5C). As is clearly indicated from these data, enhancement of the efficacy of etopophos can be obtained in the SW620 and HCT116 xenograft models, and of irinotecan in the SW620 model, by combination with KU59403 with little enhancement of toxicity (measured by body weight loss).
Discussion
We had previously identified KU55933 as a potent and selective inhibitor of ATM (11), and subsequently KU-600019 has been identified as a more potent ATM inhibitor (12) Unfortunately, although these compounds provided in vitro evidence that inhibiting ATM induced chemo- and radio-sensitisation in tumour cell lines, to date there have been no in vivo investigations with small molecule ATM inhibitors. Here we described KU59403, a novel inhibitor of the ATM kinase that is more potent (IC50 = 3 nM) and specific (at least 1000-fold selective for ATM compared with the other members of the PIKK family tested) than previously described compounds of this class. As well as improved potency over KU55933, KU59403 also exhibits improved solubility, enabling us to determine the effect of ATM inhibition in animal models of human cancer for the first time.
KU59403 had no inherent cytotoxicity in vitro at a concentration (1 μM) sufficient to cause marked chemopotentiation of topoisomerase I and II poisons, making it the most potent ATM inhibitor described to date. Enhancement of etoposide cytotoxicity, ranging from 3 to 12-fold, was observed in a panel of human tumour cell lines with the greatest sensitisation observed in SW620 cells. Interestingly, KU59403 only induced 2 to 3-fold sensitisation of etoposide in HCT116 cells, which have have been reported to have reduced ATM expression due to promoter methylation (18) and defects in MRE11 (19). Sensitisation in MDA-MB-231 cells, which are reported to have mutated ATM (20) was also relatively modest. Both of these cell lines had reduced ATM activation by IR (approximately 4-fold) in comparison to SW620 cells (6 to 7-fold). Studies in matched p53 proficient and deficient cell lines showed that p53 status had no impact on chemo-sensitisation by KU59403 or KU55933, and p53 status was not a determinant of the effect of KU55933 on cell cycle arrest or DNA DSB repair. Cytotoxic drug or IR exposure resulted in G2 arrest in cells with both wild type and dysfunctional p53 suggesting that G1 checkpoints were compromised in these cells irrespective of p53 status (21). The G2 arrest was enhanced by KU49403 independently of p53 status but whether this reflects further impairment of the G1 checkpoint, or that ATR signalling to the G2 checkpoint is increased when ATM is inhibited, remains to be determined.
Pharmacokinetic investigation of KU59403 revealed a more rapid clearance in BalbC mice after an intravenous dose but that plasma concentrations were maintained for at least 4 hr in tumour-bearing CD1 nude mice after intraperitoneal administration. Whether this difference in clearance reflects the route of administration, dose or strain effects was not determined. The pharmacokinetic studies in tumour bearing mice indicated that levels of KU59403 sufficient for chemosensitisation in vitro could be maintained in the tumour for at least 4 hr. Although levels of KU59403 in excess of those required for chemo- and radio-sensitisation in vitro were also detected in normal tissues for at least 4 hr following a dose of 25 mg/kg i.p., which could potentially have toxic consequences KU59403 was non-toxic alone and did not cause a profound increase in either etoposide or irinotecan toxicity.
Similar to the in vitro studied KU59403 alone had no impact on tumour growth rate, However it did enhance the antitumour activity of etoposide against SW620 xenografts in a dose- and schedule-dependent manner. Significant sensitisation was seen with a single daily dose of KU59403 at 12.5 mg/kg; administration but splitting the same total dose into 2 separate injections of 6 mg/kg was not as effective. Increasing the dose of KU59403 to 25 mg/kg given twice daily resulted in the greatest chemo-sensitisation with a 3-fold increase in etopophos-induced tumour growth delay in both SW620 and HCT116-N7 xenografts, in the absence of a significantly increased toxicity This is in contrast to the in vitro data where KU59403 enhanced etoposide cytotoxicity to a greater extent (3- to 12-fold) in SW620 cells than HCT116-N7 cells (2 to 4-fold) and suggests that in vitro data do not entirely predict in vivo results. It is possible that the tumour microenvironment may influence the efficacy of the combination as we have previously observed with chemosensitisation studies (22) Interestingly, it would seem that it is necessary to have KU59403 present at the time of etoposide dosing to have an effect, as delaying the administration of KU59403 by only 4 hr completely abolished chemosensitisation. Since ATM signalling is proposed to be an early response to DNA DSB, these data confirm the need to inhibit ATM whilst DNA DSB are being induced.
We have also shown that KU59403 can be used to enhance the sensitivity of human colon cancer cell lines to topoisomerase I poisons both in vitro and in vivo. KU59403 was shown to enhance the activity of camptothecan in both SW620 and LoVo cells in vitro (4 and 7-fold, respectively), and gave a 144% enhancement of irinotecan efficacy in a SW620 human xenograft model. These data as a whole are very encouraging and support the further development of this class of compound.
In summary, our studies have shown that ATM is valid target for the development of drugs designed to improve the activity of certain cytotoxic anticancer therapies. KU59403 is a potent and selective inhibitor of ATM, which is without intrinsic cytotoxicity but is a potent enhancer of topoisomerase I and II poison cytotoxicity in vitro. We have demonstrated that KU59403, increases the efficacy of topoisomerase I and topoisomerase II poisons in vivo without intrinsic toxicity despite normal tissue exposure. These data provide further proof of principle evidence for the strategy of inhibiting ATM as a therapeutic manoeuvre for anticancer therapy.
Supplementary Material
Acknowledgements
Grant Support
This work was supported by AstraZeneca and KuDOS Pharmaceuticals (grant number AZ305642, to N.J. Curtin and D.R. Newell) and Cancer Research UK (grant number C240/A7409, to D.R. Newell and N.J. Curtin).We thank Graeme Smith for helpful discussions throughout the course of this work
Footnotes
CoI: NJC and DRN have received research funding from KuDOS Pharmaceuticals Ltd and AstraZeneca, the purchasers of KuDOS. AL, NMBM, CR, GCMS and AS are former employees of KuDOS Pharmaceuticals
References
- 1.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Chun HH, Gatti RA. Ataxia Telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–96. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y. The ATM-mediated DNA damage response: taking shape. TIBS. 2006;21:402–10. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–6. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 5.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Olive PL. The role of DNA single- and double-strand breaks in cell killing by ionising radiation. Radiation Research. 1998;150:S42–S51. [PubMed] [Google Scholar]
- 7.Westphal CH, Hoyes KP, Canman CE, Huang X, Kastan MB, Hendry JH, et al. Loss of ATM radiosensitises multiple p53 null tissues. Cancer Res. 1998;58:5637–9. [PubMed] [Google Scholar]
- 8.Sarkaria JN, Eshleman JS. ATM as a target for novel radiosensitizers. Semin Radiat Oncol 2001. 2001;11:316–27. doi: 10.1053/srao.2001.26030. [DOI] [PubMed] [Google Scholar]
- 9.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Current opinion in Cell Biology. 2001;13:225–31. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 10.Izzard RA, Jackson SP, Smith GCM. Competitive and non-competitive inhibition of the DNA dependent protein kinase. Cancer Res. 1999;59:2581–6. [PubMed] [Google Scholar]
- 11.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 12.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009:2894–902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vikhanskaya F, Colella G, Valenti M, Parodi S, D’Incalci M, Broggini M. Cooperation between p53 and hMLH1 in a human colocarcinoma cell line in response to DNA damage. Clin. Cancer Res. 1999;5:937–41. [PubMed] [Google Scholar]
- 14.Moumen A, Masterson P, O’Connor M, Jackson SJ. hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–78. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase (DNA-PK) inhibitor, NU7441. Cancer Res. 2006;66:5354–62. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 16.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Committee of the National Cancer Research Institute Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–77. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potter GWH. Analysis of biological molecules: an introduction to principles, instrumentation and techniques. 1st Ed. Chapman and Hall; London: 1995. p. 202. [Google Scholar]
- 18.Kim WJ, Vo QN, Shrivastar M, Lataxes TA, Brown KD. Aberrent methylation of the ATM promoter correlates with increased radiosensitivity in human colorectal tumour cell line. Oncogene. 2002;21:3864–71. doi: 10.1038/sj.onc.1205485. [DOI] [PubMed] [Google Scholar]
- 19.Takemura H, Rao VA, Sordet O, Furuta T, Miao ZH, Meng L, et al. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J Biol Chem. 2006;281:30814–23. doi: 10.1074/jbc.M603747200. [DOI] [PubMed] [Google Scholar]
- 20.Lu Y, Condie A, Bennett JD, Fry MJ, Yuille MR, Shipley J. Disruption of the ATM gene in breast cancer. Cancer Genet Cytogenet. 2001;126:97–101. doi: 10.1016/s0165-4608(00)00401-5. [DOI] [PubMed] [Google Scholar]
- 21.Massague J. G1 cell cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 22.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Preclinical evaluation of a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, AG14361, with significant anticancer chemo- and radio-sensitization activity. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.