

Abstract
Inhibition of kynurenine 3-monooxygenase (KMO), an enzyme in the eukaryotic tryptophan catabolic pathway (i.e. kynurenine pathway), leads to amelioration of Huntington’s disease-relevant phenotypes in yeast, fruit fly, and mouse models1–5, as well as a mouse model of Alzheimer’s disease3. KMO is a FAD-dependent monooxygenase, and is located in the outer mitochondrial membrane where it converts L-kynurenine to 3-hydroxykynurenine. Perturbations in the levels of kynurenine pathway metabolites have been linked to the pathogenesis of a spectrum of brain disorders6, as well as cancer7,8, and several peripheral inflammatory conditions9. Despite the importance of KMO as a target for neurodegenerative disease, the molecular basis of KMO inhibition by available lead compounds has remained hitherto unknown. Here we report the first crystal structure of KMO, in the free form and in complex with the tight-binding inhibitor UPF 648. UPF 648 binds close to the FAD cofactor and perturbs the local active site structure, preventing productive binding of the substrate kynurenine. Functional assays and targeted mutagenesis revealed that the active site architecture and UPF 648 binding are essentially identical in human KMO, validating the yeast KMO:UPF 648 structure as a template for structure-based drug design. This will inform the search for new KMO inhibitors that are able to cross the blood-brain barrier in targeted therapies against neurodegenerative diseases such as Huntington’s, Alzheimer’s, and Parkinson’s diseases.
There is great interest in the causative role of kynurenine pathway (KP) metabolites in neurodegenerative disorders such as Huntington’s (HD) and Alzheimer’s diseases (AD)6. Several of these metabolites are neuroactive: quinolinic acid (QUIN) is an excitotoxin10,11, 3-hydroxykynurenine (3-HK) generates free-radicals12, xanthurenic and cinnabarinic acids activate metabotropic glutamate receptors13,14 and kynurenic acid (KYNA) is a neuroprotectant6. KMO lies at a critical branching point in the pathway between the synthesis of 3-HK\QUIN and KYNA (Figure 1a) and its activity plays a role in the neurotoxic and neuroprotective potential of the pathway. In the brain, KMO is expressed at low levels in neurons15 and is predominantly expressed in microglia1,16, the resident immune cells of the CNS, suggesting a link between KMO function and inflammatory processes in the brain.
Figure 1.

Panel A. Schematic overview of kynurenine metabolism. The KMO inhibitor UPF 648 is shown in blue. The hydroxyl moiety introduced by KMO is shown in red. Panel B. Fractional velocity of 3-HK formation as a function of UPF 648 concentration with human and S. cerevisiae KMO (blue circles, human KMO; red squares, Saccharomyces cerevisiae KMO). Error bars are standard deviation of three replica points. Inset: HPLC elution curves of product (3-HK) and substrate (L-KYN) at varied UPF 648 concentrations.
Inhibition of KMO activity leads to amelioration of several disease-relevant phenotypes in yeast, fruit fly, and mouse models1–5. Increased levels of KYNA relative to neurotoxic metabolites appear critical for this protection. Restoring endogenous levels of 3-HK to fruit flies lacking KMO activity eliminates this neuroprotection4, highlighting beneficial effects of 3-HK reduction due to KMO inhibition. Additionally, pharmacological inhibition of KMO is neuroprotective in animal models of cerebral ischemia17,18, reduces dystonia in a genetic model of paroxysmal dyskinesia19, improves levodopa-induced dyskinesia in parkinsonian monkeys20, and extends lifespan in a mouse model of cerebral malaria21. Therefore, inhibition of KMO activity is an attractive therapeutic strategy for several acute and chronic neurological diseases6.
Despite interest in targeting KMO only a few potent inhibitors are available, and none appreciably penetrate the blood-brain barrier in adult animals3,22. One of these, UPF 648, has an IC50 of 20 nM and provides protection against intrastriatal QUIN injections in kynurenine aminotransferase (KAT II) deficient mice23. UPF 648 treatment also shifts KP metabolism towards enhanced neuroprotective KYNA formation4,24, and ameliorates disease-relevant phenotypes in a fruit fly model of HD4. That known inhibitors do not cross the blood-brain barrier is an impediment to KMO-targeted drug discovery. KMO structures in complex with tight-binding inhibitors are required to design small molecule inhibitors that can penetrate the blood-brain barrier. With this in mind, we determined the crystal structure of yeast KMO complexed with UPF 648. This enzyme-inhibitor structure can now be used to develop new inhibitors of highly related human KMO.
We expressed full-length human KMO using the insect cell baculovirus system which yielded small quantities (0.5 mg/L culture) of detergent-solubilised active KMO. The recombinant form had similar kinetic constants to native KMO from pig liver mitochondria25. UPF 648 binds tightly to recombinant KMO (Ki 56.7 nM). Poor stability and low expression yields however, prevented crystallisation. We therefore turned to Saccharomyces cerevisiae KMO, which is related to human KMO (38 % identity and 51 % similarity). Expression of full-length Saccharomyces cerevisiae KMO yielded a protein fragment (ΔKMO-396Prot) with a lower molecular weight than anticipated. Electrospray ionisation mass spectrometry indicated proteolytic cleavage at residue 396. Subsequently, we isolated a ΔKMO-394 (deleted in residues 394 to 460) version of the enzyme engineered by site-directed mutagenesis (Supplementary Methods) to define the cleavage point prior to crystallization (Figure S1; Table S1). The ΔKMO-394 enzyme was active (Figure S2, S3), generated authentic 3HK in HPLC-based assays (Figure 1b) and was inhibited by UPF 648 (Ki 74 nM) with potency similar to that with human KMO (Figure 1b).
The structure of the proteolysed form of yeast KMO (ΔKMO-396Prot) was determined using selenomethionine single anomalous diffraction (PDB codes 4J2W and 4J31). We also solved structures of ΔKMO-394 to 1.85 Α̊ resolution. The final model contains residues 1-97 and 101-390 and the bound FAD cofactor. Both crystal forms contain a putative KMO dimer in the asymmetric unit (Figure 2a). The KMO fold is similar to other flavin-dependent hydroxylase structures26,27 with highest structural similarity to 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase28 (rmsd 2.3 Α̊ over 310 Cα, overall sequence identity 16%, Q score 0.43, Z score 15.0). An overlay of individual KMO monomers reveals significant variation in the position of the C-terminal α-helix, with most monomers showing disorder beyond residue 380. The linker region following the second strand of the antiparallel β-sheet involved in substrate binding is disordered with large variations in the positions of residues 96-97 and 101-104. The relative position of the FAD-binding domain and six-stranded antiparallel β-sheet domain is subject to minor variation, reminiscent of domain motion coupled to substrate binding in other members of this family26.
Figure 2.
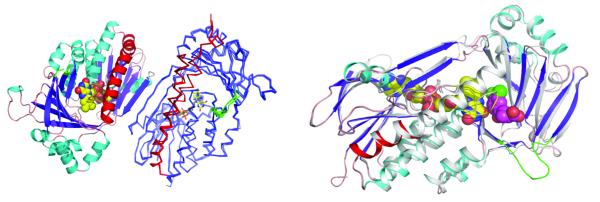
Panel A. Depiction of the Saccharomyces cerevisiae KMO dimer. One monomer is represented in cartoon format coloured according to secondary structure and the second monomer is displayed in ribbon format, with an overlay of the 5 ligand-free KMO monomers. For the second monomer, the C-terminal region (residue 348 onwards) is coloured red, the flexible linker region (residue 96-104) is depicted in green. Panel B. Depiction of changes induced by binding of the KMO inhibitor UPF 648. An overlay of both monomers from the KMO:UPF 648 structure is presented (PDB code 4J36), the unbound monomer coloured in grey. The UPF-bound monomer is coloured as in panel A. UPF 648 and FAD are shown as atom coloured spheres.
In absence of substrate, flexibility in relative positioning of both domains flanking the KMO active site is reflected also in distinct conformations observed for residues lining the active site (Figure 2c). The re-face of the FAD is connected to solvent by a narrow water-filled cavity that runs perpendicular to the active site cleft (Figure S4). A structural water is located above the FAD C4a, mimicking the position of the C4a-peroxide intermediate formed upon reaction with oxygen. The dimethylbenzene moiety of the FAD isoalloxazine is protected from solvent by Lys 48 and the conserved residue Tyr 195 (Figure S4). In absence of large protein rearrangements, this suggests a “waving flavin” motion as demonstrated in other FAD-dependent mono-oxygenases29 is unlikely to occur in KMO during turnover.
We were unable to obtain a KMO complex with kynurenine but succeeded in co-crystallising with UPF 648 (Table S3, PDB code 4J36). The asymmetric unit contains a putative KMO dimer with one monomer containing UPF 648 bound in the active site, adjacent to the FAD re-face (Figure 2b, Figure 3). The UPF 648 carboxylate is bound by conserved residues R83 and Y97 while the aromatic dichlorobenzene moiety is flanked by several hydrophobic residues (L221, M230, I232, L234, F246, P321, and F322), which are conserved in many KMO enzymes. UPF 648 binding induced structural changes in the enzyme, notably reorientation of the 321-325 loop flanking the re-side of the FAD. A minor reorientation in the position of the six-stranded antiparallel β-sheet domain with respect to the flavin binding domain is also evident. These changes result in increased disorder of the C-terminal alpha-helix, which is only visible up to R359 (Figure 2b). Reorientation of the 321-325 loop is a consequence of the active site adapting to the presence of vicinal chloride substituents in UPF 648, neither of which have a counterpart in kynurenine. To provide sufficient space for both chlorides, F322 moves away from the active site, effectively occupying a position previously taken by Y323. The P321-Q325 loop reorients to compensate for the altered F322 position. This loop lines the postulated oxygen binding site above the re-side of the FAD, which is effectively destroyed on binding UPF 648. Binding UPF 648 was found to accelerate hydrogen peroxide formation by a factor of ~20-fold compared to reactions in absence of UPF 648 (Supplementary Methods). This indicates a destabilisation of the flavin C4a hydroperoxide intermediate formed in the natural catalytic cycle of flavin monooxygenases in the presence of UPF 648 (Table S2).
Figure 3.

The Saccharomyces cerevisiae KMO active site. The left view shows an overlay of free enzyme structures obtained from various crystal forms; the right view depicts KMO:UPF 648 interactions. The middle view is a model of the KMO:kynurenine complex. Electron density is shown for UPF 648 (2Fo-Fc contoured at 1σ).
The chemical similarity of UPF 648 and kynurenine allowed modelling of kynurenine in the KMO active site (Supplementary Methods). Modelling suggests kynurenine is bound similarly, but without effect on the P321-Q325 loop. The aromatic substrate moiety is located in the conserved hydrophobic pocket (residues L221, M230, I232, L234, F246, P321, and F322) on the re-face of the flavin (Figure 3). Additional polar contacts are formed between the conserved Q325 and the kynurenine carbonyl group, and between the substrate aniline nitrogen and the FAD O4. While the amino acid carboxylate is bound by R83 and Y97, the amino group is devoid of direct interactions with protein in the model. An additional salt bridge may be made between the kynurenine amine and the sidechain of Glu 102, which is located in a highly flexible region of KMO. However, this residue is often replaced by a Gln in other KMOs (Figure S5), suggesting that this interaction is not critical for enzyme activity. The model places the substrate C3 atom adjacent to the flavin C4a, where it is poised to attack the flavin C4a peroxide intermediate (Figure 4a).
Figure 4.

Panel A. The proposed KMO kynurenine hydroxylation mechanism based on the Saccharomyces cerevisiae KMO:kynurenine model. Following hydroxylation, the intermediate rearranges to form the 3-hydroxyl kynurenine product. Panel B. Enzymatic activity comparison between wild type KMO and R83 mutants. Enzyme activity is significantly reduced following mutation (< 29% and < 1% of wild-type activity for Ala-83 and Met-83 KMOs). Inset: Measurement of binding constant for KMO inhibitor UPF 648 by ligand perturbation of flavin fluorescence emission. Titration of UPF 648 to 5 μM wild type KMO (closed squares), R83M (closed triangles) and R83A (closed circles) mutants resulted in perturbation of 520 nm fluorescence emission. Fluorescence changes at 520 nm as function of UPF 648 concentration fitted to the Morrison equation yields an observed KD= 137.8 ± 8 nMfor wild type, KD= 3.1± 0.2 μM for R83M and KD= 3.2 ± 0.2 μM for R83A enzymes. Error bars represent the standard deviation of three replica points.
All residues implicated by the KMO:kynurenine model as being involved in kynurenine binding are conserved across KMOs (Figure S5). We validated this model by mutating residue Arg-83 (replaced by Ala-83 and Met-83) and performing inhibitor binding/kinetic assays (Figure 4b). Enzyme activity is compromised following mutation (25% and <3% of wild-type activity for Ala-83 and Met-83 KMOs, respectively) as predicted by the KMO:kynurenine model. Mutation led to ~20-fold increase in Kd for the KMO-UPF 648 inhibitor complex, which implicates Arg-83 in inhibitor binding.
Elucidation of the KMO crystal structure in free form and in complex with an established inhibitor is a major breakthrough for new KMO inhibitor design. This will permit docking screens using virtual compound libraries that may ultimately identify novel inhibitor scaffolds. Such studies will enable the design of new inhibitors that possess the selectivity and affinity to open up new opportunities for therapeutic intervention. Critically, this should inform the development of brain-penetrant KMO inhibitors. The KMO inhibitor Ro 61-8048 and prodrug JM6 have shown preclinical promise in animal models of neurodegeneration2–4,16–20, but they do not appreciably penetrate the blood-brain barrier. These compounds likely confer neuroprotection by inhibiting KMO in the blood, and consequent active transport of kynurenine into the brain, ultimately yielding increased levels of the neuroprotective metabolite KYNA3,30. As this neuroprotection appears to be independent of KMO inhibition in the brain, and reduction of downstream neurotoxic metabolite levels3, delivery of KMO inhibitors directly to the CNS should provide critical additional therapeutic efficacy and broaden the scope of disorders amenable to targeting.
Crystallization and diffraction data collection
Crystals of Saccharomyes cerevisiae KMO were obtained using the sitting-drop vapour diffusion technique. Details describing the crystallogenesis can be found in Supplementary Methods. Diffraction data from Saccharomyes cerevisiae KMO crystals were collected from single cryofrozen samples at the Diamond Light Source, Harwell, UK. A single-wavelength anomalous dispersion (SAD) data set at the selenium edge was used for initial phase determination. Data collection and refinement statistics are present in Table S3.
Methods
Cloning, expression and purification of human full-length kynurenine 3-monooxygenase
The gene encoding full length human kynurenine-3-monooxygense was synthesised (Genescript, Piscataway, NJ, USA) and ‘codon optimised’ for over-expression in mammalian cells. The gene was sub-cloned into the baculovirus transfer vector pAcGHLT-A GST as a Nde I – Eco RI fragment and transfected into Hi5 cells along with linearised baculovirus using the following sense and anti-sense primers: 5’-GGC ATA TGC ATG GAC AGC AGC GTG ATC CAG CGG AAG-3′; 5′-CCC GAA TTC CTA CCG GCT GAT CAG GTT GCT G-3′. Hi-5 cells (1.5 × 106) were infected with recombinant virus for 72 h at 28 °C. A WAVE Bioreactor System (GE Healthcare Life Sciences) was used to grow batches of 5 L cell culture. Cultured cells were lysed in 20 mM potassium phosphate buffer, pH 7.5, 10 % glycerol, 0.5 % n-dodecyl β-D-maltoside (DDM), 150 mM NaCl, 7 mM 2-mercaptoethanol, 50 μM FAD supplemented with protease inhibitors (Sigma-Aldrich Corporation, Dorset, UK). Soluble lysate was incubated with 3 mL pre-equilibrated glutathione uniflow resin (Clontech, Saint Germain-en-Laye, France). The resin was then packed and washed with buffer B (20 mM potassium phosphate buffer, pH 7.5, 10 % glycerol, 0.012 % DDM, 150 mM NaCl, 7 mM 2-mercaptoethanol, 50 μM FAD). Fractions (0.5 mL) were eluted with buffer C (buffer B + 33 mM glutathione) and fractions containing KMO pooled, concentrated and loaded onto a Superdex 200 (10/30) size exclusion chromatography column. Pure KMO was pooled and stored at −80 °C.
Cloning, expression and purification of Saccharomyces cerevisiae kynurenine 3-monooxygenase
A synthetic gene (BNA4 UniProtKB accession number P38169) encoding Saccharomyces cerevisiae kynurenine 3-monooxygenase was ‘codon optimised’ for overexpression in E. coli (Genescript, Piscataway, NJ, USA). The gene was sub-cloned into pET15b and pET24b (Merck, Whitehouse Station, NJ, USA) as a Nde I - Xba I fragment for over-expression in E. coli strain BL21(DE3). A deletion variant of kynurenine 3-monooxygenase (Δ394BNA4) and single amino acid variants of this deletion (Δ394BNA4-R83A and Δ394BNA4-R83M) were generated by site-directed mutagenesis (Quikchange, Strategene, Agilent Technologies, USA). Sense and anti-sense primers are shown in Table S1. Amplification conditions were: denaturation at 95 °C for 30 s, followed by 16 cycles of amplification at 95 °C (30 s), 55 °C (60 s), 68 °C (7 min). PCR products were incubated for 1 h to digest methylated template DNA and then transformed into XL1-Blue supercompetent cells (Stratagene, Agilent Technologies, USA). Transformed bacterial colonies were isolated and mutated genes were identified by DNA sequencing (MWG Eurofins) to verify presence of desired, and lack of spurious changes, to the DNA sequence.
Transformed bacterial cells were grown overnight at 37 °C in Lysogeny Broth (LB; 0.75 L) containing 100 μg/ml ampicillin. Cultures were grown at 37 °C to optical density ~0.3, and the temperature was then reduced to 27 °C. Cells were induced (0.1 mM isopropyl β-D-thiogalactopyranoside; IPTG) and grown (16-20 h) at 27 °C. Cells were lysed in buffer A (20 mM potassium phosphate, buffer pH 7.5, 10% glycerol, 300 mM NaCl, 50 μM FAD) containing protease inhibitors. Soluble lysate was loaded onto a 5 mL HisTrap FF Ni Sepharose 6 Fast Flow column (GE Healthcare) pre-equilibrated with buffer A. Protein was eluted using a 50 – 250 mM imidazole linear gradient. Yellow fractions containing partially purified KMO were pooled and diluted 10 times in buffer B (20 mM potassium phosphate buffer, pH 7.5, 10% glycerol, 7mM 2-mercaptoethanol) to reduce salt and imidazole concentration. Protein was subsequently loaded onto 10 mL blue Sepharose 6 fast flow column (GE Healthcare) equilibrated with buffer B. The protein was eluted using a linear gradient to 2.5 M NaCl prepared in buffer B. Fractions containing KMO were pooled and diluted 10-fold in buffer B to reduce the NaCl concentration. Protein was concentrated and chromatographed using a Superdex 200 10/300 GL column (GE Healthcare) equilibrated with buffer C (25 mM ammonium acetate buffer, pH 7.0, 150 mM NaCl, 7 mM 2-mercaptoethanol). Pure fractions were collected and stored at −80 °C.
Incorporation of selenomethionine into yeast Saccharomyces cerevisiae KMO
Expression of selenomethione labelled protein was achieved by inhibiting methionine biosynthesis shortly before induction of KMO expression by adding high concentrations of isoleucine, leucine, phenylalanine, lysine and threonine to the cell culture. Transformed E. coli cells containing the Saccharomyces cerevisiae KMO expression plasmid were grown in LB media at 37 °C to late exponential phase, harvested and then re-suspended in M9 minimal media and grown until mid-log phase. At that point lysine, phenylalanine, threonine (100 mg/l each), and selenomethionine, isoleucine, leucine, and valine (50 mg/l each) were added to the culture and induced 15 min after addition of amino acids with 0.1 mM IPTG. The culture was grown for 12-16 hours. Purification of selenomethionine incorporated Saccharomyces cerevisiae KMO was as described for conventional Saccharomyces cerevisiae KMO.
Enzyme Assays
Steady-state kinetic parameters for kynurenine 3-monooxygenase were obtained by initial rate measurements of enzyme activity. KMO catalyses the NADPH-dependent hydroxylation of L-kynurenine to 3-hydroxykynurenine and the enzymatic reaction can be monitored by following the decrease in absorbance of NADPH at 340 nm. Rate assays were carried out with 1 cm path length quartz microcuvette at 37 °C for human KMO and 30 °C for Saccharomyces cereviseae KMO. Pure enzyme (0.2 - 1μM) was added to 200 μl reaction buffer (20 mM potassium phosphate buffer, pH 8.0, 7 mM 2-mercaptoethanol) containing different concentrations of NADPH and L-kynurenine (Sigma-Aldrich Corporation) and the time-dependent absorbance change at 340 nm was recorded using a using a UV-vis Cary Eclipse spectrophotometer (Agilent Technologies). Assays at each reaction condition were performed in triplicate. Apparent Michaelis constants, Km, for L-kynurenine and NADPH were determined by varying the concentration of the first substrate at a constant concentration of the second substrate and vice versa. Reaction data were fitted to the standard Michaelis-Menten equation using Origin Software (OriginLab, Northampton, MA).
KMO inhibition by UPF 648 was measured by following formation of the product 3 hydroxykynurenine by C18 reversed-phase HPLC. Enzyme (100 nM) was incubated at 37 °C in 1 mL reaction buffer (20 mM potassium phosphate buffer, pH 8.5, 7 mM 2-mercaptoethanol) with 2 mM NADPH, 500 μM L-kynurenine and different concentrations (0 – 2 μM) of UPF 468. The reaction was stopped with 8 % trifluoracetic acid added at different time points during the assay. Precipitated protein was removed by centrifugation, and the supernatant was analysed by C18 reversed-phase HPLC equilibrated with 50 mM ammonium acetate, pH 3.0, containing 1% methanol and 0.1% heptanesulfonic acid; elution from the column was with 50 mM ammonium acetate, pH 4.5, containing 5% methanol and 0.5% heptanesulfonic acid, and monitored at 229 nm. 3-hydroxykynurenine was quantified by comparison with a 3-hydroxykynurenine calibration curve. Each experiment was performed in triplicate. Data were fitted to the Morrison equation (Equation 1):
| (1) |
where ʋ0 is the enzyme activity without inhibitor, ʋi is the enzyme activity at ligand concentration I, [E] is the enzyme concentration, [I] is the inhibitor concentration and Ki is the inhibition constant.
Ligand binding assays
Fluorescence emission measurements were performed using a Cary Eclipse Fluorimeter (Agilent Technologies) to determine enzyme-ligand dissociation constants (Kd) for KMO ligands, exploiting ligand perturbation of flavin (enzyme-bound FAD) fluorescence. Excitation light was provided from a xenon light source and excitation and emission slit widths were 5 nm. The perturbation in FAD fluorescence emission was followed at 520 nm using an excitation wavelength of 450 nm and excitation spectra were recorded from 470 to 700 nm. L-kyn and UPF 648 were titrated into a 5 μM solution of KMO (20 mM potassium phosphate buffer, pH 8, 50 mM NaCl and 7 mM 2-mercaptoethanol) at 25 °C. Dissociation constants, Kd, were calculated by fitting data for the difference in fluorescence emission measured for KMO-ligand complexes and free KMO at 520 nm divided by the maximum emission versus the ligand concentration (Equation 2). This equation is a variation of Morrison equation (Equation 1) and is used for tight binding ligands or when Kd values are similar in magnitude to the concentration of enzyme used, in which case substantial amounts of both enzyme (E) and ligand (X) are consumed in forming the EX complex as the titration progresses. All data fitting was done using Origin software (OriginLab, Northampton, MA).
| (2) |
In Equation 2, Eobs represents the fluorescence emission difference at each ligand concentration, X, Emax is the maximum emission change at saturating ligand concentrations, [E] is the enzyme concentration, and Kd the dissociation constant for the ligand bound to the enzyme.
Hydrogen peroxide quantification
Measurements to observe the formation of hydrogen peroxide from the ΔKMO-394-UPF inhibitor complex were performed using a horseradish peroxidase (HRP) assay. Steady-state assays were performed as described above to calculate the NADPH consumption rate in the presence and absence of an excess of UPF 648 (30 μM). KMO (5 μM) was incubated with 150 μM NADPH, 150 μM L-KYN, 400 μM o-dianisidine and ~5 units HRP in the presence and absence of excess UPF 648. The rate of oxidation of o-dianisidine by H2O2, catalysed by HRP, was monitored at 440 nm (Δε =11.600M−1cm−1 440 ). The rate of hydrogen peroxide production was compared to the rate of NADPH consumption.
Crystallisation of Saccharomyces cerevisiae KMO
Initial crystals of Saccharomyces cerevisiae KMO (ΔKMO-396Prot & ΔKMO-396Prot-Se) were obtained by mixing 200 nl of 14 mg/ml protein in 20 mM ammonium acetate, pH 7.0, 150 mM NaCl and 7 mM 2-mercaptoethanol (buffer A) with 200 nl of a reservoir solution containing 0.1 M sodium acetate, pH 5.5, and 35 % isopropanol. A second, more readily reproducible crystal form was obtained with ΔKMO-394 and ΔKMO–394-HIS−. In both cases crystals were grown by mixing 200 nl of protein (buffer A) with 200 nl of a reservoir solution containing 0.1 M imidazole, pH 7.8, and 11 % w/v polyethylene glycol 8K. The UPF complex (ΔKMO-394-UPF) was also obtained as described above; however, prior to setting the trays the protein was pre-incubated with 1 mM UPF for ~30 minutes. All trays were incubated at 277 K, with crystals forming over a period of ~72 h.
Diffraction data processing, structure determination and refinement
X-ray diffraction data were collected at Diamond Light Source and subsequently integrated and scaled using the program XDS31. Initial phases were obtained from a single SAD data set (S1) collected at the selenium edge. Selenium sites were located using Phenix AutoSol32 yielding an electron density map that could be auto traced using Phenix AutoBuild33. The resulting model was completed through iterative rounds of rebuilding in COOT34 and refinement in Phenix35. All subsequent structures were solved by molecular replacement in PHASER36 using this initial SeMet derived structure as the template. Structure validation with MOLPROBITY37 was integrated as part of the iterative rebuild and refinement procedure.
KMO-kynurenine modeling
NAMD software38 was used to perform all molecular dynamics (MD) simulations of the kynurenine-KMO complex. Topology and parameters files for substrate and FAD were obtained using Antechamber program39 using AM1-BCC charges40. The complex model was then immersed in a periodic water box (TIP3) and neutralized by adding Na+ ions. Several cycles of minimizations (steepest descent, 10 000 steps) and MD simulations (50K, 20 ps) were performed to equilibrate the model (backbone protein atoms were kept fixed). Then MD simulations were performed (310 K, 1 ns) at a time step of 2 ps, with the protein backbone restrained on the X-ray structure conformation. Individual snapshots were extracted and minimized to assess the KMO-kynurenine interactions.
Supplementary Material
Table 1.
Kinetic and dissociation constants for H. sapiens, S. cerevisiae and active-site variants of KMO
| Enzyme | Ki app UPF648 (nM) | KD UPF648 (μM) |
|---|---|---|
| hKMO1 | 56.7 ± 8 | - |
| ScΔKMO-394 | 74 ± 14 | 0.14 ± 0.01 |
| ScΔKMO-394-R83A 2 | - | 3.1 ± 0.2 |
| ScΔLKO–394–P83M 2 | - | 3.2 ± 0.2 |
Dissociation constant (KD) for the human KMO-UPF 648 complex could not be determined by fluorescence emission due to low KMO expression yield.
Kinetic assays to calculate the inhibition (Ki app) of ScKMO variants by UPF648 could not be carried out because enzyme activity was substantially compromised following mutagenesis. As an alternative, fluorescence emission measurements were performed to determine dissociation constants (KD) by ligand perturbation of flavin fluorescence.
Acknowledgements
We thank Dr Robert Schwarcz (University of Maryland School of Medicine, Baltimore, USA) for supplying UPF 648. We also thank Dr. E. McKenzie (Protein Expression Facility, The University of Manchester) for expressing human KMO. We also thank Diamond Light Source for access to MX beamlines.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author contributions. NSS, FG, DL, and TFO initiated the project, designed experiments, analysed data and wrote manuscript; MA cloned purified and crystallised proteins and performed biochemical assays; CL crystallised proteins, collected and processed diffraction data; DH developed and analysed some of the biochemical assays; PL perfromed molecular modelling of kynurenine binding.
Data deposition. Atomic coordinates and structure factors have been deposited in the protein data bank (www.rcsb.org/pdb/home/home.do) under accession codes 4J2W, 4J31, 4J33, 4J36 & 4J34.
Reprints and permissions information is available at www.nature.com/reprints
Competing financial interests. The authors declare there are no compting financial interests.
References
- 1.Giorgini F, et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem. 2008;283:7390–7400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]
- 2.Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zwilling D, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145:863–874. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campesan S, et al. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr Biol. 2011;21:961–966. doi: 10.1016/j.cub.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green EW, et al. Drosophila eye color mutants as therapeutic tools for Huntington disease. Fly (Austin) 2012;6:117–120. doi: 10.4161/fly.19999. [DOI] [PubMed] [Google Scholar]
- 6.Schwarcz R, Bruno JP, Muchowski PJ, Wu H-Q. Kynurenines in the mammalian brain: when physiology meets pathology. Nature reviews Neuroscience. 2012;13:1–13. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Platten M, Litzenburger U, Wick W. The aryl hydrocarbon receptor in tumor immunity. Oncoimmunology. 2012;1:396–397. doi: 10.4161/onci.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Newton RC, Friedman SM, Scherle PA. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9:938–952. doi: 10.2174/156800909790192374. [DOI] [PubMed] [Google Scholar]
- 9.Filippini P, et al. Emerging Concepts on Inhibitors of Indoleamine 2,3-Dioxygenase in Rheumatic Diseases. Curr Med Chem. 2012;19:5381–5393. doi: 10.2174/092986712803833353. [DOI] [PubMed] [Google Scholar]
- 10.Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- 11.Schwarcz R, Whetsell WO, Jr., Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- 12.Okuda S, Nishiyama N, Saito H, Katsuki H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci U S A. 1996;93:12553–12558. doi: 10.1073/pnas.93.22.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copeland CS, Neale S. a, Salt TE. Actions of Xanthurenic Acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology. 2013;66:133–42. doi: 10.1016/j.neuropharm.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Fazio F, et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol Pharmacol. 2012;81:643–56. doi: 10.1124/mol.111.074765. [DOI] [PubMed] [Google Scholar]
- 15.Guillemin GJ, et al. Characterization of the kynurenine pathway in human neurons. J Neurosci. 2007;27:12884–92. doi: 10.1523/JNEUROSCI.4101-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew BJ. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol. 2003;527:105–112. doi: 10.1007/978-1-4615-0135-0_12. [DOI] [PubMed] [Google Scholar]
- 17.Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61-8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab. 1999;19:771–777. doi: 10.1097/00004647-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Moroni F, et al. Studies on the neuroprotective action of kynurenine mono-oxygenase inhibitors in post-ischemic brain damage. Adv Exp Med Biol. 2003;527:127–136. doi: 10.1007/978-1-4615-0135-0_15. [DOI] [PubMed] [Google Scholar]
- 19.Richter A, Hamann M. The kynurenine 3-hydroxylase inhibitor Ro 61-8048 improves dystonia in a genetic model of paroxysmal dyskinesia. Eur J Pharmacol. 2003;478:47–52. doi: 10.1016/j.ejphar.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 20.Samadi P, et al. Effect of kynurenine 3-hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in Parkinsonian monkeys. Mov Disord. 2005;20:792–802. doi: 10.1002/mds.20596. [DOI] [PubMed] [Google Scholar]
- 21.Clark CJ, et al. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect Immun. 2005;73:5249–5251. doi: 10.1128/IAI.73.8.5249-5251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinhart PH, Kelly JW. Treating the periphery to ameliorate neurodegenerative diseases. Cell. 2011;145:813–814. doi: 10.1016/j.cell.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 23.Sapko MT, et al. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington ’ s disease. Exp Neurol. 2006;197:31–40. doi: 10.1016/j.expneurol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Ceresoli-Borroni G, Guidetti P, Amori L, Pellicciari R, Schwarcz R. Perinatal kynurenine 3-hydroxylase inhibition in rodents: pathophysiological implications. J Neurosci Res. 2007;85:845–854. doi: 10.1002/jnr.21183. [DOI] [PubMed] [Google Scholar]
- 25.Uemura T, Hirai K. L-kynurenine 3-monooxygenase from mitochondrial outer membrane of pig liver: purification, some properties, and monoclonal antibodies directed to the enzyme. J Biochem. 1998;123:253–62. doi: 10.1093/oxfordjournals.jbchem.a021930. [DOI] [PubMed] [Google Scholar]
- 26.Palfey B, McDonald C. Control of catalysis in flavin-dependent monooxygenases. Arch Biochem Biophys. 2010;493:26–36. doi: 10.1016/j.abb.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 27.Van Berkel WJH, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J biotechnol. 2006;124:670–89. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 28.McCulloch M, Mukherjee T, Begley TP, Ealick SE. Structure of the PLP degradative enzyme 2-methyl-3- hydroxypyridine-5-carboxylic acid oxygenase from Mesorhizobium loti MAFF303099 and its mechanistic implications. Biochemistry. 2010;48:4139–4149. doi: 10.1021/bi900149f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortiz-Maldonado M, Ballou DP, Massey V. A rate-limiting conformational change of the flavin in p-hydroxybenzoate hydroxylase is necessary for ligand exchange and catalysis: studies with 8-mercapto- and 8-hydroxy-flavins. Biochemistry. 2001;40:1091–101. doi: 10.1021/bi002139s. [DOI] [PubMed] [Google Scholar]
- 30.Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
References (methods)
- 31.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, Storoni LC, Read RJ. Simple algorithm for a maximum-likelihood SAD function. Acta Crystallogr D Biol Crystallogr. 2004;60:1220–1228. doi: 10.1107/S0907444904009990. [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mccoy AJ, et al. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips JC, et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornell WD, et al. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 40.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.