

Abstract
Huntington’s disease (HD) is a neurodegenerative disorder of the central nervous system characterised by the presence of choreic abnormal movements, behavioural or psychiatric disturbances and dementia. Noteworthy, despite atypical motor symptoms other than chorea have been reported as initial presentation in some patients, a very few number of HD patients, presenting at onset mostly cerebellar dysfunction masquerading dominant spinocerebellar ataxias (SCA), were occasionally reported. We report the case of a 42-year-old man with a 5-year history of gait disturbance, dysarthria and cognitive impairment and familial antecedents of dementia and movement disorders. Initially the clinical picture suggested the diagnosis of a dominant SCA, but finally a diagnosis of HD was made based on the molecular evidence of abnormal 39 Cytosine-Adenine-Guanine (CAG) repeats in exon 1 of Huntingtin gene. The authors highlight the importance of suspecting HD in the aetiology of spinocerebellar ataxias when dementia is a prominent feature in the proband or their family.
Background
Huntington's disease (HD) is a neurodegenerative disorder of the central nervous system characterised by the presence of choreic abnormal movements, behavioural or psychiatric disturbances and dementia.1 It is autosomal-dominant inherited and is caused by an abnormal Cytosine-Adenine-Guanine (CAG) repeat (36 repeats or more) on the short arm of chromosome 4p16.3 in the Huntingtin gene.1 2
The approximate prevalence of HD has been estimated in 2.71/100 000 (95% CI1.55 to 4.72) therefore about 189 700 patients with HD could be assumed to be present worldwide. Noteworthy, despite atypical motor symptoms other than chorea have been reported as initial presentation in some patients,3–10 only nine patients with HD presenting at onset mostly cerebellar dysfunction masquerading dominant spinocerebellar ataxias (SCAs)3 7 were previously reported in the literature.
We report here a patient with HD who presented with the atypical onset of ataxia and cognitive impairment masquerading some of the dominant SCAs.
Case presentation
A 42-year-old man was referred to our neurogenetics clinic because of a 5-year history of progressive impairment of gait characterised by marked postural instability. During the last year, he developed dysarthria, clumsiness in upper limbs and memory complaints along difficulties to perform daily activities, which obliged him to quit his job 5 months before the time of his first consultation.
His family history was remarkable for the antecedent of his father suffering from an unidentified neurodegenerative disorder which included among its symptoms cognitive impairment and movement disorders with onset at the age of 50. His grandmother suffered from a similar condition that was, at her time, diagnosed as dementia (see figure 1).
Figure 1.
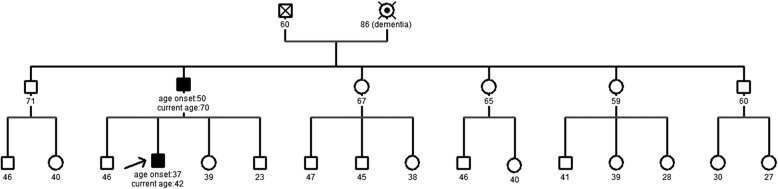
Family pedigree. Filled boxes indicate symptomatic participants. Arrow points to the proband object of this report.
General physical examination was normal. Bedside cognitive assessment showed a Mini Mental State score of 18/30 and a Clock Drawing Test of 3/7. Cranial nerves examination revealed a fractionated smooth-pursuit with slow and long latency saccades, spontaneous and evocated nystagmus was absent, assessment of other cranial nerves was unremarkable. We did not find evidence of pyramidal dysfunction. Primitive reflexes could be elicited: glabellar tap sign was positive and bilateral palmomental reflexes were present. Strength was normal. We found cog-wheel rigidity involving the right hemibody and a bilateral moderate bradykinesia in upper and lower limbs. Sensory examination was unremarkable.
Cerebellar examination revealed a moderate axial and appendicular ataxia. These signs were formally assessed using the Scale for the Assessment and Rating of Ataxia (SARA).11 A global score of 14 was calculated with axial function (gait and stance), dysarthria and appendicular function (finger to nose, finger chase, rapid alternating movements and heel to shin) subscores of 6, 3 and 5, respectively.
Investigations
Routine laboratory tests were normal, levels of vitamin B12, folic acid and thyroid hormones were normal too. Serum Venereal Disease Research Laboratory and HIV serology were negative. Brain MRI scans showed mild cerebellar atrophy without caudate atrophy (see figures 2 and 3). We obtained informed consent for performing genetic studies. Molecular analysis of Huntingtin (HTT) gene showed an abnormally expanded 39 CAG repeats allele in exon 1 of the HTT gene consistent with a molecular diagnosis of HD. Molecular studies of SCA 1, 2, 3, 6, 8, 17, dentatorubropallidoluysian atrophy (DRPLA) and Huntington's disease-like (HDL2) genes did show non-expanded alleles excluding these alternative diagnoses.
Figure 2.
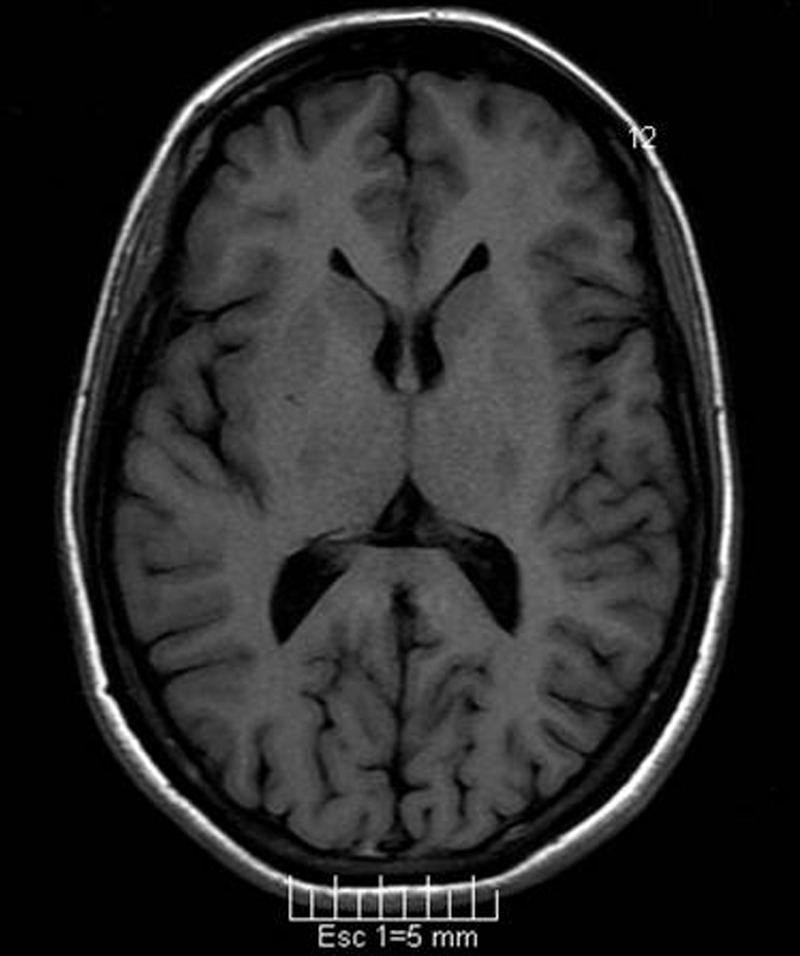
Axial T1-weighted image showing normal size of the head of the caudate nucleus.
Figure 3.
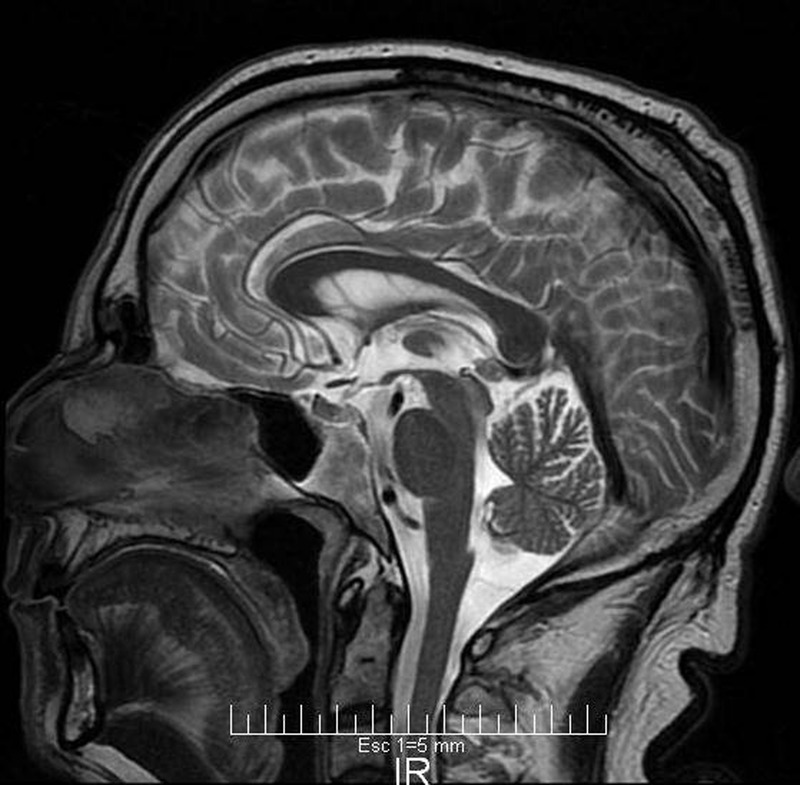
Sagital T2-weighted image showing mild cerebellar atrophy.
Differential diagnosis
The SCAs are a group of clinically, and genetically very heterogeneous, inherited neurological disorders. Their clinical features include, apart of cerebellar ataxia, pyramidal dysfunction, peripheral neuropathy, extra pyramidal signs, cranial nerves affection or cognitive impairment. Even more, these extracerebellar symptoms could be the presenting or dominating ones.12 There have been described more than 30 SCAs.13 There are reports of cognitive impairment in the most prevalent SCAs, for instance: patients with SCA type 1 presenting cognitive impairment, albeit not dementia, have been recognised14 15; different authors have estimated a prevalence of late onset dementia in 19–42% of the patients suffering SCA type 214 16–18; on the other hand, dementia is a highly unlikely feature in the most prevalent SCA type 3.14 18 19; DRPLA is very similar in many aspects to HD; three clinical phenotypes that may show cognitive impairment have been described: one with predominant chorea resembling HD, and two other that present mainly with myoclonus or with ataxia, therefore it must be considered into the differential diagnoses of HD.15 20 HDL3 is an autosomal recessive disorder included in the group of HD phenocopies that may present with mental deterioration, dysarthria, dystonia, pyramidal signs and ataxia;21 however, the early onset of this condition and the pattern of inheritance makes this diagnosis unlikely in our case.
Discussion
Chorea is the predominant motor symptom in HD, which is present in more than 90% of the individuals affected with this disorder.22 However, other abnormal movements, such as dystonia or tics, have been infrequently reported at the onset of HD.4 8–10 Moreover, even less frequently ataxia has been mentioned in a few reports as the first clinical symptom in HD masquerading as SCA. Squitieri found two participants from a cohort of 205 patients with HD that showed limb and gait ataxia as the first clinical manifestation of their disease.3 Dong et al7 found in their group of 82 HD participants that seven patients initially had been diagnosed as suffering from SCA because of the presence of ataxia as the first symptom.
Previous reports have failed to find a relationship between the number of CAG repeats and the presence of atypical motor symptoms3 7 with the exception of parkinsonism in juvenile onset HD caused by very large trinucleotide expansions.1 23 Molecular analysis in our patient showed the abnormal presence of 39 CAG repeats, which is the shortest abnormal allele described in the literature in atypical HD cases highlighting this absence of relationship. Thus, other factors, apart the number of CAG repeats, may be influencing the phenotype and should be further investigated.3
The description of this clinical case illustrates the broad range of clinical presentations that HD can show and the need for considering HD in the differential diagnosis of patients presenting movement disorders and a positive family history for neurological affections. Therefore, HD should also be included in the diagnostic consideration of spinocerebellar ataxias.
Learning points.
Huntington's disease (HD) could have an atypical onset with the presence of movement disorders like parkinsonism, tics, dystonia or ataxia.
Patients with HD can simulate the spinocerebellar ataxias (SCA) at onset, in particular those that showed dementia such as SCA type 17.
In patients with ataxia and a family history suggesting autosomal dominant inheritance, HD should be included among the differential diagnoses.
Footnotes
Contributors: All the authors contributed significantly towards case assessment, manuscript discussion and preparation of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis 2010;2013:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker FO. Huntington's disease. Semin Neurol 2007;2013:143–50 [DOI] [PubMed] [Google Scholar]
- 3.Squitieri F, Berardelli A, Nargi E, et al. Atypical movement disorders in the early stages of Huntington's disease: clinical and genetic analysis. Clin Genet 2000;2013:50–6 [DOI] [PubMed] [Google Scholar]
- 4.Alonso H, Cubo-Delgado E, Mateos-Beato MP, et al. [Huntington's disease mimicking Tourette syndrome]. Rev Neurol 2004;2013:927–9 [PubMed] [Google Scholar]
- 5.Angelini L, Sgro V, Erba A, et al. Tourettism as clinical presentation of Huntington's disease with onset in childhood. Ital J Neurol Sci 1998;2013:383–5 [DOI] [PubMed] [Google Scholar]
- 6.Becker N, Munhoz RP, Raskin S, et al. Non-choreic movement disorders as initial manifestations of Huntington's disease. Arq Neuropsiquiatr 2007;2013:402–5 [DOI] [PubMed] [Google Scholar]
- 7.Dong Y, Sun YM, Liu ZJ, et al. Chinese patients with Huntington's disease initially presenting with spinocerebellar ataxia. Clin Genet 2013;2013:380–3 [DOI] [PubMed] [Google Scholar]
- 8.Hu MT, Chaudhuri KR. Repetitive belching, aerophagia, and torticollis in Huntington's disease: a case report. Mov Disord 1998;2013:363–5 [DOI] [PubMed] [Google Scholar]
- 9.Jankovic J, Ashizawa T. Tourettism associated with Huntington's disease. Mov Disord 1995;2013:103–5 [DOI] [PubMed] [Google Scholar]
- 10.Kerbeshian J, Burd L, Leech C, et al. Huntington disease and childhood-onset Tourette syndrome. Am J Med Genet 1991;2013:1–3 [DOI] [PubMed] [Google Scholar]
- 11.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;2013:1717–20 [DOI] [PubMed] [Google Scholar]
- 12.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;2013:885–94 [DOI] [PubMed] [Google Scholar]
- 13.Klockgether T, Paulson H. Milestones in ataxia. Mov Disord 2011;2013:1134–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang B, Liu C, Shen L, et al. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol 2000;2013:540–4 [DOI] [PubMed] [Google Scholar]
- 15.Kawai Y, Suenaga M, Watanabe H, et al. Cognitive impairment in spinocerebellar degeneration. Eur Neurol 2009;2013:257–68 [DOI] [PubMed] [Google Scholar]
- 16.Geschwind DH, Perlman S, Figueroa CP, et al. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet 1997;2013:842–50 [PMC free article] [PubMed] [Google Scholar]
- 17.Durr A, Smadja D, Cancel G, et al. Autosomal dominant cerebellar ataxia type I in Martinique (French West Indies). Clinical and neuropathological analysis of 53 patients from three unrelated SCA2 families. Brain 1995;2013:1573–81 [DOI] [PubMed] [Google Scholar]
- 18.Schols L, Amoiridis G, Buttner T, et al. Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol 1997;2013:924–32 [DOI] [PubMed] [Google Scholar]
- 19.Lokkegaard T, Nielsen JE, Hasholt L, et al. Machado-Joseph disease in three Scandinavian families. J Neurol Sci 1998;2013:152–7 [DOI] [PubMed] [Google Scholar]
- 20.Schols L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics and pathogenesis. Lancet Neurol 2004;2013:291–304 [DOI] [PubMed] [Google Scholar]
- 21.Schneider SA, Walker RH, Bhatia KP. The Huntington's disease-like syndromes: what to consider in patients with a negative Huntington's disease gene test. Nat Clin Pract Neurol 2007;2013:517–25 [DOI] [PubMed] [Google Scholar]
- 22.Warby SC, Graham RK, Hayden MR. Huntington Disease. In: Pagon RA, Adam MP, Bird TD, et al. eds. GeneReviewstm. Seattle: University of Washington, 23 Oct 1998. (Updated 22 Apr 2010) [Google Scholar]
- 23.Heathfield KW. Huntington's chorea: a centenary review. Postgrad Med J 1973;2013:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]