

Abstract
Background
Pulmonary hypertension (PH) is an enigmatic vascular syndrome characterized by increased pulmonary arterial pressure and adverse remodeling of the pulmonary arterioles and often of the right ventricle. Drawing parallels with tumorigenesis, recent endeavors have explored the relationship between metabolic dysregulation and PH pathogenesis.
Design
We will discuss the general mechanisms by which cellular stressors such as hypoxia and inflammation alter cellular metabolism. Based on those principles, we will explore the development of a corresponding metabolic pathophenotype in PH, with a focus on WHO groups I and III, and the implications that these alterations may have for future treatment of this disease.
Results
Investigation of metabolic dysregulation in both the pulmonary vasculature and right ventricle during PH pathogenesis has provided a more unifying understanding of how disparate disease triggers coordinate end-stage disease manifestations. Namely, as defined originally in various cancers, the Warburg effect describes a chronic shift in energy production from mitochondrial oxidative phosphorylation to glycolysis. In many cases, this Warburg phenotype may serve as a central causative mechanism for PH progression, largely driving cellular hyperproliferation and resistance to apoptosis. Consequently, new therapeutic strategies have been increasingly pursued that target the Warburg phenotype. Finally, new technologies are increasingly becoming available to probe more completely the complexities of metabolic cellular reprogramming and may reveal distinct metabolic pathways beyond the Warburg effect that drive PH.
Conclusion
Studies of metabolic dysregulation in PH are just emerging but may offer powerful therapeutic means to prevent or even reverse disease progression at the molecular level.
Keywords: pulmonary hypertension, metabolism, hypoxia, Warburg effect, mitochondria, glycolysis
Introduction
Pulmonary hypertension (PH) is a serious and often fatal pulmonary vascular condition that is becoming increasingly prevalent worldwide. It is characterized by a complex panvasculopathy involving excessive proliferation and dysregulation of multiple cell types, as well as inflammation and fibrosis throughout the vasculature. Specific and severe forms of the disease, such as pulmonary arterial hypertension (PAH), display especially excessive remodeling and obliteration of the lumen of small arterioles, often histologically marked by the presence of complex vascular lesions known as plexogenic lesions. Increased resistance in the pulmonary arteries places a substantial hemodynamic burden on the right ventricle, leading to right ventricular hypertrophy and, in many cases, progression to frank heart failure.
Etiologies of PH are numerous, and five types of PH have been defined in an official classification system developed at the World Health Organization (WHO) symposium on PH in 2008 1, 2. WHO Group 1 consists of individuals suffering from PAH, stemming either from idiopathic and hereditary forms or secondarily from co-morbidities such as congenital heart disease, autoimmune disease, HIV infection, and others. WHO Groups 2-5 can be triggered by a wide range of other conditions, such as left heart disease, chronic hypoxia, and thromboembolic disease among others 1, 2. Especially in PAH, mortality rates have remained persistently high. Without treatment, the median survival rate of PAH is estimated at less than three years after first diagnosis 1. Even with the advent of specific pulmonary vasodilator therapies, the three-year mortality rate is high, estimated at roughly 33% 1. Beyond PAH, regardless of the etiology, the presence of PH with or without right ventricular failure portends a worse prognosis in any clinical context. Yet, epidemiological data are imperfect, because precise and non-invasive methods for the detection of PH are lacking. For example, up to 6.4 million Americans with chronic obstructive pulmonary disease (COPD) may suffer from PH 3, with many more cases potentially linked with other chronic illnesses. Yet, the diagnosis is confirmed in only a small percentage of these patients when severe manifestations develop. As a result, there exist substantial populations of undiagnosed and at-risk patients who remain entirely unaccounted for and unprotected from this disease.
Similarly, molecular studies of the pathogenesis of PH have not been equitably distributed among all forms of this disease. Consequently, because most of our mechanistic insights into PH have originated from study of PAH (WHO Group 1) and hypoxia-dependent PH (WHO group III), this review will focus primarily on disease from these two groups. Importantly, despite some key differences, there also exists substantial overlap at the molecular level, with both disease types displaying pronounced imbalance of cell growth and cell death, leading to remodeling of the pulmonary vasculature as well as changes in vasomotor tone and thrombotic potential. It remains unproven if similar mechanisms of disease primarily contribute to the burden of vascular disease seen in Groups 2 and 4. However, the functional importance of these similarities in all groups has been suggested by clinical data demonstrating that pulmonary vasodilator therapy can be effective across Groups 1-4 4. At the cellular level, PAH and hypoxia-induced PH are often characterized by multiple coordinated levels of dysregulation, including down-regulation of voltage-gated potassium (Kv) channels, membrane depolarization, high [Ca2+]i currents, activation of HIF-1, and a general shift involving a multitude of factors that favor cell growth and proliferation over apoptosis (Fig. 1). In particular, the proproliferative and antiapoptotic features of PAH and hypoxia-induced PH have led to a comparison between the underlying characteristics of PH and those of cancer 5. Importantly, like many cancers, PH of these types presents with such symptoms as dysregulated angiogenesis 6, high expression of cellular growth factors 7, and, as mentioned, a strong resistance to apoptosis 7.
Figure 1.
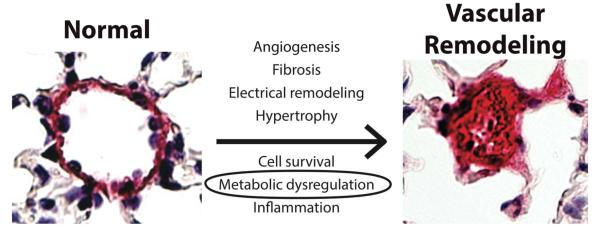
Vascular cell types in the pulmonary arteries undergo dramatic remodeling during PH, especially noted in PAH and hypoxia-induced PH. Up-regulation of growth factors, electrical remodeling, and persistent inflammation produce abnormal PASMCs that proliferate excessively, occluding the arteries and restricting blood flow. Metabolic and mitochondrial dysregulation are crucial to several of these processes, as mitochondrial hyperpolarization and the glycolytic shift lead to the up-regulation of stress factors and impaired release of apoptotic factors. Adapted with permission from 81.
Establishing a link between cancer and PH has revealed recently a common reliance upon mitochondrial dysregulation (Fig. 1). Specifically, early PH research focused primarily on the role of hypoxia, or low oxygen availability, and the phenomenon of hypoxic vasoconstriction (HPV) 8. Although the exact mechanisms of HPV remain unclear, mitochondria have been theorized to act as mediators of this effect via increased production of reactive oxygen species (ROS) by the mitochondrial electron transport chain (ETC) 9. Further study of the role of mitochondrial dysfunction beyond HPV has led to the understanding that the mitochondria have a much broader role in the course of PH, and can be implicated in the induction of pulmonary vascular pathology even when hypoxia is not present as a trigger 9.
Inherent to the broad importance of mitochondrial function in the pulmonary vasculature is a global metabolic shift, displayed by all metazoan and mammalian cells in response to hypoxia to favor the production of ATP via glycolysis and lactic acid fermentation at the expense of oxidative phosphorylation 10. In the setting of hypoxia, this is known as the “Pasteur effect,” but in the setting of normal oxygen levels, this is termed the “Warburg effect,” and was first described in tumor cells in the 1950’s by Otto Warburg. This shift is associated with the internalization of oxygen-sensing potassium channels 11, increased influx of calcium into the cytosol 12, and hyperpolarization of the mitochondrial membrane 11, 13, all of which enforce a cellular phenotype that is resistant to apoptosis and allows the cell to survive periods of short-term stress 14. However, pulmonary vascular cell types derived from patients with PAH display this glycolytic predominance even in the presence of adequate oxygen 15. A similar phenotype is seen in a variety of PH models, most notably PH induced by monocrotaline or exposure to hypoxia 15. Such reliance on aerobic glycolysis favors cell growth over apoptosis and has been proposed as a key pathogenic mechanism in both PH and cancer 9, 16, 17. Together, these findings have led to what has been termed the metabolic theory of PAH, which suggests that a series of self-propagating mitochondrial abnormalities are responsible for key steps in pathogenesis, a theory which could be applicable to other forms of the disease as well, most notably hypoxia-induced PH 9. This review will focus on the known molecular mechanisms by which mitochondrial metabolism is altered in PAH and hypoxia-induced PH as well as future directions for research in this emerging area.
HIF Acts As a “Master Regulator” of the Glycolytic Shift
The hypoxia-stabilized transcription factor—hypoxia-inducible factor or HIF—is a master transcriptional regulator of the hypoxic response and has long been known to play a predominant role in the induction of PH. Importantly, mice that lack important subunits of HIF, such as HIF-1α 18 or HIF-2α 19, are resistant to hypoxia-induced PH. Even in the absence of overt hypoxic stress, abnormal HIF activation is seen in animal models such as the fawn-hooded rat—a strain predisposed towards the spontaneous development of PH—and in the PASMCs of human PAH patients 20. This section will explore primarily on the oxygen-sensing role of HIF and its relationship to alterations in metabolism in healthy tissue during acute hypoxia, in order to lay groundwork for the discussion of these systems in the chronic disease state of PH (as summarized in Fig. 2).
Figure 2.

Role of HIF and hypoxia in the Pasteur/Warburg effects. During hypoxic conditions, newly stabilized HIF up-regulates an array of glycolytic enzymes that activate anaerobic glycolysis and suppress the the electron transport chain (ETC) though inhibition of glucose oxidation and iron sulfur cluster assembly.
HIF is a heterodimeric transcription factor consisting either of HIF-1α or HIF-2α and HIF-1β. Under normoxia, HIF-α subunits are prolyl-hydroxylated, leading to their recognition by the von Hippel-Lindau tumor suppressor protein (VHL), subsequent ubiquitination, and rapid degradation by the 26S proteasome 21. In hypoxia, prolyl hydroxylation of HIF-α is repressed, leading to its accumulation and translocation to the nucleus where it binds HIF-1β. The HIF heterodimer then binds to a consensus hypoxia responsive element (HRE) in the promoters of a variety of target genes to activate global gene programs that include a switch from oxidative phosphorylation to glycolysis 22.
Recently, HIF-dependent molecular mechanisms have been described that link HIF to the glycolytic shift. While not itself a direct target of HIF, central to this process is the mitochondrial enzyme, pyruvate dehydrogenase (PDH), often considered the mitochondrial “gate-keeping” enzyme for its role in promoting pyruvate entry into the mitochondria 23. Pyruvate produced in the early stages of glucose metabolism is converted either into lactate, via anaerobic glycolysis, or acetyl-CoA, via glucose oxidation. The conversion of pyruvate into lactate is mediated by lactate dehydrogenase A (LDHA), while the conversion into acetyl-CoA is mediated by PDH 24. PDH is inhibited by the activity of pyruvate dehydrogenase kinases (PDKs), which are in turn directly induced by HIF 23. The up-regulation of PDK1 by HIF-1 is essential to the suppression of glucose oxidation during hypoxia, as indicated by a repression of the glycolytic phenotype, as well as PDK1 expression, when HIF-1α is absent. Moreover, Kim and colleagues demonstrated that mouse embryo fibroblasts lacking HIF-1αdisplayed increased mitochondrial production of ROS in hypoxia, indicating persistent activation of glucose oxidation. Accordingly, ROS levels decreased when these cells were transfected with an expression vector encoding PDK1 25. Thus PDH and PDK1 are critical enzymes that mediate the causal relationship between intracellular hypoxia and the metabolic shift towards glycolysis.
HIF also intersects with the glycolysis pathway at several other points via the induction of LDHA 26, which mediates the conversion of pyruvate into lactate; the up-regulation of glucose transporters GLUT1 and GLUT3 27; and the alteration of the fourth subunit of cytochrome c oxidase (COX) to better favor hypoxic conditions 28. This last step is accomplished via a two-part mechanism. Of the two COX4 isoforms found in mammals, the COX4-1 isoform is predominant and more favorable during normoxia, but ill-suited to hypoxia 29. Fukuda and colleagues demonstrated that O2 consumption, COX activity, and ATP generation were compromised when increased COX4-1 expression was forced during hypoxia. Conversely, decreased COX4-1 activity during normoxia was equally damaging, with forced expression of the alternative COX4-2 isoform resulting in excessive ROS production and caspase activation, leading to cell death 28. HIF is responsible both for inducing COX4-2 expression directly, and for mediating the suppression of COX4-1 via up-regulation of the mitochondrial protease, LON, which is required for COX4-1 degradation 28.
Recent work in the area of microRNA (miRNA) biology has suggested that the role of HIF in the glycolytic shift is not limited to regulation of protein-coding genes alone. MicroRNA-210 (miR-210), a known transcriptional target of HIF 30 and one of several endogenous miRNA now understood to have an essential role in a variety of cellular processes 31, has been implicated in the down-regulation of iron-sulfur cluster assembly proteins, ISCU1 and ISCU2 32. These proteins are responsible for the assembly of [4Fe-4S] and [2F3-2s] iron-sulfur clusters, prosthetic groups known to be integral to the tricyclic acid (TCA) cycle and to the activity of mitochondrial respiratory complexes I, II, and III, which facilitate electron transport 33. Suppression of the ISCU proteins by miR-210 prevents the assembly of these groups, thereby inhibiting mitochondrial respiration 32. Defects in iron-sulfur cluster assembly may be linked to PH in vivo, as individuals carrying mutations in NFU1, an essential iron-sulfur cluster assembly protein, often develop a fatal form of mitochondrial disease involving metabolic acidosis, lactic acidemia, and hyperglycinemia. In nine of the ten patients studied, histological evidence of pulmonary vascular remodeling was noted 34.
These mechanisms illustrate the central role of HIF in the induction of anaerobic glucose metabolism. When conducted in healthy tissue, these mechanisms can be described as an adaptive response to short-term hypoxic stress, maximizing cellular energy production and protection from adverse accumulation of ROS. Yet, once dysregulated, these alterations may represent a major contributor to PH pathogenesis 35.
Induction of the Warburg Phenotype: Chronic Hypoxia, Inflammation, and Other Stressors
The HIF-induced shift in glucose metabolism is beneficial for the generation of ATP and the prevention of mitochondrial ROS production. Such a shift serves to prolong survival during acute hypoxic stress. Yet, a persistent metabolic shift over a long period of time—as in the case of chronic hypoxia or inflammation in the pulmonary vasculature—can result in permanent electrical remodeling of the mitochondria and the cell, and can prolong the glycolytic shift well after any acute stressor such as hypoxia has been removed (e.g., the Warburg effect displaying “aerobic glycolysis”) 20. Indeed, positron emission tomography (PET) has suggested that diseased pulmonary vasculature in human PAH patients often display chronic induction of the Warburg phenotype as shown by a robust increase in glucose uptake 36. This effect has been replicated in animal models, and recently traced back to HIF-1 mediated up-regulation of the glucose transporter, GLUT1, in the vasculature of PAH-afflicted Sprague-Dawley rats 37. Yet, the complete mechanisms controlling the Warburg effect in vascular tissue are just emerging (as summarized in Fig. 3).
Figure 3.

Electrical remodeling induced by metabolic reprogramming in diseased pulmonary vasculature. Down-regulation of voltage-gated potassium (Kv) channels prevents K+ efflux, leading to a high concentration of K+ in the cytosol. The resulting membrane depolarization triggers the opening of L-type calcium channels, which allows for Ca2+ influx. Calcium suppresses GSK-3, thus preventing inhibition of HK-II. HK-II then freely inhibits the voltage-dependent anion channel (VDAC) of the mitochondrial permeability transition pore (MPTP), causing mitochondrial hyperpolarization and preventing the release of pro-apoptotic factors through the MPTP. Ca2+ also promotes NFAT entry into the cell. NFAT further inhibits Kv channels, creating a feedback loop and augmenting the above responses.
In the case of hypoxic PH, PDH inhibition by HIF-induced PDK1 in remodeled pulmonary vasculature represses mitochondrial function and ROS production. This has been demonstrated by the reversal of the glycolytic shift in monocrotaline-induced PH in rats when PDH inhibition is blocked by dichloroacetate (DCA), an agent which represses the activity of PDKs 13. As a consequence of this low ROS production, components of the oxygen-sensitive voltage-gated potassium channel (Kv channel), such as the pore-forming-subunit, Kv1.5, are inhibited 14, 38. These potassium channels, located on the cell surface, are up-regulated by intracellular ROS, and thus serve as an additional oxygen-sensor for the cell 14. Inhibition of these channels results in a buildup of cytosolic K+, and as the cell becomes depolarized, voltage-gated L-type calcium channels open, allowing for an influx of Ca2+ and the induction of pulmonary vasoconstriction 12. If HIF up-regulation persists, this change in cytosolic ionic composition further dictates cellular function. High intracellular levels of Ca2+ and K+ boost cell proliferation and inhibit pro-apoptotic caspases, respectively 13, 14, and the increasingly low density of Kv channels inhibits cellular ability to detect increases in available O2. In support of this latter notion, Pozeg and colleagues have demonstrated that chronically hypoxic rats are less prone to develop PH, as measured by vascular resistance and cardiac output, upon receiving an in vivo gene transfer of Kv1.5 11. Mutations in Kv1.5 have also been discovered in PASMCs derived from human patients suffering from idiopathic PAH, further implicating Kv1.5 dysfunction as a causative feature of this disease 38. The inhibition of this oxygen-sensor thus has the capacity to trap the cell in the hypoxia-induced metabolic state, even after normal oxygen conditions have returned.
Another consequence of this electrical remodeling involves dysregulation of the mitochondrial permeability transition pore (MPTP), a large voltage-gated channel through which pro-apoptotic mediators pass from the mitochondria into the cytoplasm 39. Hyperpolarization of the mitochondrial membrane, a typical feature of the Warburg phenotype, is associated with a proportionately high threshold for the opening of the MPTP, making it more difficult for apoptotic factors to be released through the channel 40. This hyperpolarization is, in part, due to the activity of the glycolytic enzyme hexokinase-II (HK-II), which inhibits the voltage-dependent anion channel (VDAC), a key component of the MPTP, and prevents the diffusion of anions across the mitochondrial membrane 39. Also relevant to this situation is glycogen synthase kinase-3 (GSK3) which ordinarily promotes mitochondrial depolarization by phosphorylation of VDAC, thereby preventing its inhibition by HK-II 39. GSK3 also opposes the influx of NFAT into the nucleus induced by high levels of intracellular calcium ([Ca2+]i) 40. Notably, GSK3 is inhibited in the Warburg phenotype, allowing both NFAT and HK-II to operate largely unhindered within the cell 41. The final step in this self-perpetuating system is the further down-regulation of membrane Kv1.5 channels by NFATc2, further increasing cell depolarization and insensitivity to O2 16.
The TCA cycle itself plays a role in the stabilization of HIF during hypoxia, as the prolyl hydroxylases (PHDs) responsible for catalyzing the degradation of HIF-1 under normoxic conditions are highly sensitive to the relative concentrations of TCA cycle intermediates. Several intermediates just upstream of acetyl-CoA’s entrance into the cycle (succinate, fumarate, and oxaloacetate) have been shown to inhibit PHD activity, thereby sparing HIF-1 when their concentrations are high 42. Conversely, the earlier intermediate, α-ketoglutarate, is required for the PHD-induced hydroxylation of HIF-1 to take place. Mutations in TCA cycle enzymes have accordingly been shown to have a direct impact on HIF activity. Isocitrate dehydrogenase (IDH) activity was recently found to be abnormally high both in the serum of PAH patients, and in cultured human pulmonary microvascular endothelial cells with disease-causing BMPR2 mutations 43. IDH is responsible for catalyzing the conversion of α-ketoglutarate to isocitrate, a reaction that counters the normal progression of the TCA cycle. Thus, the increased activity of IDH would have the dual effect of depriving the PHDs of α-ketoglutarate substrate while also obstructing further TCA cycle function. In the case of PAH patients, elevated IDH activity has been correlated with poor clinical prognosis 43. Similarly, fumarate hydratase, the enzyme responsible for catalyzing the conversion of fumarate to malate, may also be relevant, as it has been shown to be mutated in renal cell carcinoma cells experiencing a glycolytic shift 44. Defective fumarate hydratase fails to produce malate, allowing for the accumulation of fumarate, which also has the effect of stabilizing HIF 44.
These TCA cycle mutations illustrate the point that hypoxia is not a prerequisite for the HIF-mediated induction of the Warburg phenotype, both within the domain of PH and without. Recently, Sutendra and colleagues provided another alternative trigger by demonstrating that tumor necrosis factor alpha (TNF-α) isolated from activated human CD8+ T cells induces a glycolytic shift in otherwise healthy human PASMCs 41. Cells treated in this way display low levels of mitochondrial ROS, high [Ca2+]i +, activated NFAT in the cell nucleus, mitochondrial hyperpolarization, and decreased K+ flux through the cell membrane 41. This study thus suggests a link between PH and certain inflammatory conditions that may act as triggers for the disease, such as scleroderma and mixed connective tissue disease 41. The notion that a diverse array of cellular stress factors, beyond the well-described case of HIF, may influence the development of PH is further supported by the recent discovery that Nogo-B and endoplasmic reticulum stress play a large role in undermining mitochondria-dependent apoptosis, as described below 45.
Endoplasmic Reticulum Stress and the Role of Nogo-B
A strong relationship exists between endoplasmic reticulum (ER) function and mitochondria-dependent apoptosis. Mitochondrial depolarization, a prerequisite for the release of pro-apoptotic factors through the MPTP, is promoted by the presence of high [Ca2+] within the mitochondria. The ER contains the largest Ca2+ reserve in the cell, and under normal cellular conditions, Ca2+ transfer between the ER and the mitochondria plays a crucial role both in mitochondrial depolarization and in activating certain mitochondrial enzymes such as PDH 46. This transfer is largely mediated by the presence of Ca2+ microdomains in the ER, creating a concentration gradient through which calcium can be transported between the two structures 47. Recently, Sutendra and colleagues found that Nogo-B, a protein responsible for regulating the tubular structure of the endoplasmic reticulum (ER), plays a role in the development of PH during exposure to hypoxia 45. Of the three isoforms of Nogo (Nogo-A, B, and C), only Nogo-B is found in the pulmonary vasculature and promotes endothelial cell migration 48. Nogo expression is increased via activating transcription factor 6 (ATF6) during periods of ER stress, a condition triggered by the accumulation of newly synthesized unfolded proteins within the organelle 49. Such accumulation will typically lead to the unfolded protein response (UPR), a highly conserved response to a wide range of cellular stress conditions, particularly those that result in an increased demand for protein synthesis or else increased retention of new proteins within the ER 23. ER stress has been associated with hypoxia and inflammation, as well as BMRP2 mutations, viral infections, and Notch3 signaling 45, and has been shown to be highly active in the pulmonary vasculature, both in rat models with monocrotaline-induced PH and in human PAH patients 50.
Regardless of how the UPR is triggered, is thought that the resulting up-regulation of Nogo-B can cause remodeling of the ER and disrupt communication between the ER and the mitochondria 45. Once depleted of calcium, the mitochondria become hyperpolarized, thus raising the threshold for the opening of the MPTP and rendering the cell more resistant to apoptosis. Accordingly, mice lacking Nogo-B expression demonstrate resistance to PH when exposed to chronic hypoxia, a typical cause of ER stress. Conversely, in normoxic PASMCs exposed to high levels of exogenous Nogo-B, symptoms of PH develop even in the absence of hypoxia exposure 45. This response to ER activity is notable in that it is not inherently dependent on hypoxia, and while oxidative stress was responsible for Nogo-B up-regulation in the experimental model used by Sutendra and colleagues, they suggest that it may be possible to generalize the finding to other conditions that have been tied to both ER stress and vascular pathology. Examples of this could include HIV infection and mutations in bone morphogenic protein receptor II (BMPR2), two conditions which have been implicated in the clinical development of PH 45.
Adding to the notion that ER stress contributes to metabolic dysregulation in PH is the recent work by Yeager and colleagues, implicating endothelin-1 (ET-1) as another trigger of the unfolded protein response (UPR) in pulmonary vascular dysregulation 51. ET-1 is a vasoactive peptide that is known to be up-regulated in the plasma of PH patients under a variety of PH etiologies, and is thought to contribute to elevated pulmonary vascular resistance 50. In this disease context, robust up-regulation of ET-1 generates an inflammatory and pro-survival form of the UPR 51. Taken together, these two studies indicate that the ER, with its heavy involvement both in mitochondrial function and in several cellular stress programs, may serve as a point of convergence for the seemingly disparate triggers of metabolic dysfunction in this disease.
Metabolic Dysregulation in the Right Ventricle
While pathological changes in the pulmonary vasculature have been the traditional subject of most intensive study in PH, structural remodeling to the RV portends a much worse prognosis, thus suggesting its distinct importance to this disease (Fig. 4) 52. As the disease syndrome progresses, the RV is forced to maintain cardiac output in the face of mounting vascular resistance. This leads to right ventricular hypertrophy (RVH) which, in turn, carries a greater oxygen requirement that cannot be met by coronary blood flow 52. In cases of adaptive remodeling, RVH does not lead to RV failure; but in many PH patients, termed maladaptive remodelers, RVH is closely followed by a loss of RV function 52. Current understanding of the mechanisms involved in RVH remains limited, and, perhaps erroneously, relies heavily on data extrapolated from studies of the left ventricle (LV). Evidence of a Warburg phenotype specifically in the RV, however, has emerged, and is consistent with prior knowledge of suppressed oxidative phosphorylation during LV overload 53. Thus, these molecular events represent the first available clues that may help to elucidate the potential importance of metabolic dysfunction in the RV as it controls the clinical course of PH.
Figure 4.

End-diastolic (A) and end-systolic (B) cine short-axis images from a patient with PH. Hypertrophy and dilation of the right ventricle in PH is the result of the increased pressure placed on the ventricle to maintain blood flow in the face of high vascular resistance. Increased mass in the right ventricle leads to a higher oxygen requirement. Without the appropriate blood supply, this imbalance can lead to myocardial hypoxia and a variety of cellular abnormalities. Adapted with permission from 82.
In contrast with the pulmonary vasculature, 60-90% of ATP produced in the myocardium is derived from fatty acid oxidation (FAO). The other 10-40% is accounted for by glucose oxidation 52. FAO and glucose oxidation share a mutually inhibitory relationship in tissue, as defined by the Randle cycle. This relationship is common to most muscle and adipose tissues, and occurs without any form of hormonal mediation 15. Faced with an increased oxygen requirement and a decreased oxygen supply, it is thought that hypertrophic RV tissue experiences higher degrees of hypoxia, resulting in the activation of transcription factors HIF-1 and Myc 54, 55. As in the pulmonary vasculature, the activation of HIF-1 produces a glycolytic phenotype via up-regulation of glycolytic enzymes and suppression of glucose oxidation. This has been demonstrated by evidence of a glycolytic shift in several experimental models of RV dysfunction. For example, via positron emission tomography (PET) studies, Oikawa and colleagues reported that glucose uptake is unusually high in the RV in both human RVH patients and in animal disease models 56. It has also been demonstrated that the glucose transporters GLUT1 and GLUT4, as well as the rate-limiting glycolytic enzyme HK-II are all up-regulated in a variety of RVH models 52, 57. Moreover, Piao and colleagues confirmed that rats experiencing monocrotaline-induced RVH display increased activation of PDK1 52. FAO has also been shown to be impaired in severe cases of hypoxic PH and PAH, though this effect was not seen in milder forms of either disease. Importantly FAO requires more oxygen per unit of ATP than glucose oxidation by about 12% 52. It is likely that myocardial FAO is unable to compensate for the ATP production lost when glucose oxidation is suppressed, even in cases where FAO itself is relatively spared.
Because anaerobic glycolysis is inherently less efficient for ATP generation than either form of oxidative phosphorylation, it has been proposed that insufficient energy production induces RV hibernation as seen in late-stage RVH 52. This notion is supported by studies that contrast adaptive and maladaptive RV remodeling, such as those demonstrating that p38-MAPK, a factor known to promote glycolysis, is increasingly activated by maladaptive, but not the adaptive, RV alterations 58. Thus, as discussed below, recent therapies have been developed to reverse the glycolytic phenotype in the remodeled RV and thus ameliorate the clinical progression of disease. Furthermore, given the nascency of these research endeavors, we anticipate that future studies will point to additional critical metabolic pathways driving RV dysfunction beyond even glycolysis and oxidative phosphorylation.
Recent Therapies and Potential Therapeutic Targets
Until recently, therapeutic agents developed to treat PH have largely focused on targeting end-stage disease phenotypes such as vasoconstriction and thrombosis. As a result, the use of many PH-specific therapies has not made a significant contribution to reverse the molecular origins of this disease 59. We will briefly summarize recent approaches that attempt to target upstream metabolic pathways controlling the Warburg phenotype in PAH, and other PH etiologies that exhibit this effect. By reversing the causative metabolic changes that occur even at early stages of PH, it may be possible to prevent or regress vascular remodeling, extend life-expectancy, and even reverse the course of the disease.
A potentially promising metabolic agent for the treatment of PH is dichloroacetate (DCA), which acts by inhibiting PDKs 60, 61. As previously mentioned, PDK-1 promotes glycolysis via inhibitory phosphorylation of PDH, thus preventing the conversion of pyruvate into acetyl-CoA. Notably, DCA has been highly successful in animal models to ameliorate PH manifestations. In both monocrotaline and hypoxia-induced PH in rodents, as well as in the fawn hooded rat, DCA has been able to reduce PDH phosphorylation, increase Kv1.5 channel expression, and improve RV function, as evidenced by shortened action potential duration (APD) and electrocardiographic QTc intervals 13, 57. DCA has been tested in several human clinical trials distinct from PH and RV failure 60, 62, 63. Notably, one study was terminated early due to the presentation of peripheral nerve toxicity, a side effect that likely will need to be addressed before large scale pharmacologic utilization 62. Nonetheless, direct investigation of the utility of DCA in human PH is currently underway 5.
Another key feature of the Warburg phenotype is the down-regulation of oxygen-sensing Kv channels on the cell surface, leading to myriad abnormalities that stem from the resulting electrical remodeling of the cell. The actions of NFAT predominate in this effect. High levels of [Ca2+] activate NFAT via the activation of calcineurin, and consequently, NFAT promotes apoptosis resistance in addition to suppressing Kv channel expression. The peptide, VIVIT, and the immunosuppressant, cyclosporin A, have both been used experimentally to inhibit NFAT activation. VIVIT acts by interrupting the binding between calcineurin and NFAT, and cyclosporin by direct inhibition of calcineurin 16. Both agents have been effective at reversing membrane depolarization and abnormalities in cytosolic ion concentrations in PASMCs derived from human PH 16. Cyclosporin A displayed some degree of success in animal models, reducing pulmonary vascular resistance and pulmonary arterial pressure, and increasing cardiac output in rats experiencing monocrotaline-induced PH 16. Interestingly, cyclosporin A also acts as an inhibitor of cyclophilin D, a known regulator of the MPTP. Thus cyclosporin A may also be used to prevent cell death due to MPTP opening during reperfusion following a period of hypoxia or ischemia 64, 65.
A third aspect of metabolism that may be targeted in PH is the balance between fatty acid oxidation and glucose oxidation. Since these two processes share a competitive relationship, thus it is believed that the suppression of myocardial FAO may allow for a potential recovery of glucose oxidation in RVH 66. Both trimetazidine and ranolazine have been used to inhibit fatty acid oxidation in animal models, resulting in increased glucose oxidation that serves to combat the glycolytic shift 67, 68. Trimetazidine has also seen success in clinical trials, where it improved both left and right ventricular function in patients experiencing heart failure 69, 70. It should also be noted that both compounds also possess marked anti-ischemic properties, and are approved for the treatment of refractory ischemia, ranolazine in America 71 and trimetazidine in Europe 72, where they are well tolerated. Ranalozine, in particular, has been shown to decrease late inward sodium current, which is increased during oxidative stress as well as during cardiac hypertrophy 68. Thus, clinical trials have been proposed to “re-purpose” these medications for treatment specifically in PH and RV dysfunction.
Finally, a fourth aspect of metabolic dysfunction that may be targeted in the treatment of PH involves microRNA (miRNA) biology. While not yet targeted by any established medications, miRNA have been shown to play a wide variety of roles in the pathology of PH, and as such, represent attractive new alternatives for therapeutic intervention. Notably, both miR-204 and miR-328 influence metabolic phenotypes at the cellular level, and they have been shown to play protective roles in pulmonary vascular biology in PAH and hypoxic PH, respectively 73, 74. miR-328 has been shown to inhibit L-type calcium channel expression in the pulmonary arteries of mice with hypoxia-induced PH, with a corresponding decrease in pulmonary arterial vasoconstriction 74. Similarly, miR-204 has been shown to indirectly suppress activation of NFATc2, a factor which is, as previously mentioned, responsible for downregulating oxygen-sensitive Kv1.5, contributing to cellular depolarization and O2 insensitivity 73. With the list of hypoxia- and PH-relevant miRNA growing longer, these factors represent a new treatment avenue worthy of exploration.
Conclusions
The Warburg shift from mitochondrial oxidative phosphorylation to glycolysis is a dominant vascular feature in several PH etiologies, including the clinically severe cases of PAH and RV failure as well as hypoxia-induced PH, and provides a useful parallel to cancer and other chronic pathological conditions. Its association with numerous forms of cellular stress also suggests its importance in mechanistically linking the seemingly unrelated triggers of PH to some of the stereotyped features of vascular and myocardial remodeling in this disease. Metabolic drug targets have also been identified aimed at reversing the Warburg phenotype, and a number of clinical trials are underway to determine their utility in the management of PH.
Yet, the Warburg effect may only represent a small portion of the overall metabolic alterations important in PH. For example, inhibition of methylarginine metabolism has been shown to suppress nitric oxide synthase (NOS), a known vasodilator 75; impaired iron metabolism leads to insufficient heme formation and is associated with poor clinical prognosis in PH 76; and inhibition of the metabolic enzyme, malonyl-CoA, is associated with the suppression of fatty acid oxidation and improved glucose oxidation in diseased vasculature 77. Furthermore, new technologies may offer more efficient methods to elucidate the importance of these and other metabolic pathways in this emerging area. For instance, in recent years, metabolomic screening has been used to identify novel metabolites, such as 2-hydroxyglutarate 78 and glycine 79, as causative factors in cancer progression. Similar metabolomic studies could prove useful in shedding light on many of the missing links in the induction of the metabolic phenotype in PH. Alternatively, the emerging field of network medicine may allow for systems-level insight into how metabolic factors interface with the complex and overlapping pathways associated with this complex vascular disease. As a result, hypotheses generated from computational analyses of relevant network topology and/or dynamics could then be interrogated in vivo, thus maximizing our ability to utilize the full potential of available high-throughput data 80.
Taken together, the study of metabolic dysregulation in PH has bolstered substantially our fundamental understanding of this enigmatic disease. In combination with rapidly developing technologies to interrogate more effectively the pulmonary vasculature and RV, we expect that even greater clinical and therapeutic insights based on metabolic reprogramming will be realized in the coming years.
Acknowledgements
We thank S. Tribuna for expert administrative assistance.
Funding Sources This work was supported by the NIH (KO8 grant), the Lerner, Harris, McArthur-Radovsky, and Watkins Funds, Gilead Research Scholars Fund, and the Pulmonary Hypertension Association (S. Y. C.).
Abbreviations
- [Ca2+]i
intracellular calcium concentration
- APD
action potential duration
- BMPR2
bone morphogenic protein receptor II
- COX
cytochrome c oxidase
- DCA
dichloroacetate
- ER
endoplasmic reticulum
- ET-1
endothelin-1
- FAO
fatty acid oxidation
- GLUT
glucose transporter
- GSK3B
glycogen synthase kinase-3b
- HIF-1
hypoxia inducible factor-1
- HK-II
hexokinase-II
- HPV
hypoxic vasoconstriction
- IDH
isocitrate dehydrogenase
- ISCU
iron sulfur cluster assembly protein
- LDHA
lactate dehydrogenase A
- MAPK
mitogen-activated protein kinase
- miRNA
microRNA
- MPT
mitochondrial permeability transition pore
- NFAT
nuclear factor of activated T-cells
- NOS
nitric oxide synthase
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary arterial smooth muscle cells
- PDH
pyruvate dehydrogenase
- PHD
prolyl hydroxylase
- PDK
pyruvate dehydrogenase kinase
- PET
positron emission tomography
- PH
pulmonary hypertension
- ROS
reactive oxygen species
- RV
right ventricle
- RVH
right ventricular hypertrophy
- TNF-α
tumor necrosis factor alpha
- UPR
unfolded protein response
- VDAC
voltage-dependent anion channel
- VHL
von Hippel-Lindau
- WHO
World Health Organization
Footnotes
Disclosures None
References
- 1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J, Harrington RA, Anderson JL, Bates ER, Bridges CR, Eisenberg MJ, Ferrari VA, Grines CL, Hlatky MA, Jacobs AK, Kaul S, Lichtenberg RC, Moliterno DJ, Mukherjee D, Pohost GM, Schofield RS, Shubrooks SJ, Stein JH, Tracy CM, Weitz HH, Wesley DJ. Accf/aha, accf/aha 2009 expert consensus document on pulmonary hypertension: A report of the american college of cardiology foundation task force on expert consensus documents and the american heart association: Developed in collaboration with the american college of chest physicians, american thoracic society, inc., and the pulmonary hypertension association. Circulation. 2009;119:2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. 2003;21:892–905. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 4.Lourenço AP, Fontoura D, Henriques-Coelho T, Leite-Moreira AF. Current pathophysiological concepts and management of pulmonary hypertension. Int J Cardiol. 2012;155:350–361. doi: 10.1016/j.ijcard.2011.05.066. [DOI] [PubMed] [Google Scholar]
- 5.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation. 2010;121:2045–2066. doi: 10.1161/CIRCULATIONAHA.108.847707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuder RM, Groves BM, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 7.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, McMahon GG, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death–dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. Faseb J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 8.Decker I, Ghosh S, Comhair SA, Farha S, Tang WH, Park M, Wang S, Lichtin AE, Erzurum SC. High levels of zinc-protoporphyrin identify iron metabolic abnormalities in pulmonary arterial hypertension. Clin Transl Sci. 2011;4:253–258. doi: 10.1111/j.1752-8062.2011.00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michelakis ED, Thebaud B, Weir EK, Archer SL. Hypoxic pulmonary vasoconstriction: Redox regulations of o2-sensitive k+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J Mol Cell Cardiol. 2004;37:1119–1136. doi: 10.1016/j.yjmcc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Aisenberg AC, Potter VR. Studies on the pasteur effect. J Biol Chem. 1957;224:1115–1127. [PubMed] [Google Scholar]
- 11.Pozeg ZI, Michelakis ED, McMurtry MS, Thebaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A, Archer SL. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation. 2003;107:2037–2044. doi: 10.1161/01.CIR.0000062688.76508.B3. [DOI] [PubMed] [Google Scholar]
- 12.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- 13.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 14.Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Augmented K+ currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol. 2001;281:L887–L894. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- 15.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Crit Care Med. 2011;185:260–266. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warburg O, Posener K, Negelein E. Ueber den stoffwechsel der carcinomzelle. Biochem Z. 1924;152:319–344. [Google Scholar]
- 18.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducuble factor 1 alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir K, Archer SL. An abnormal micochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 21.Salceda S, Caro J. Hypoxia-inducible factor 1 alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 22.Seagroves TN, Ryan HE, Lu H, Wouters BH, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the Pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30:142–150. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Kim JW, Tchernyshyov I, Semenza GL, Dang CCV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Kolb TM, Damico RL, Hassoun PM. Linking new and old concepts: Inflammation meets the warburg phenomenon in pulmonary arterial hypertension. J Mol Med. 2011;89:729–732. doi: 10.1007/s00109-011-0783-x. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase a, enolase 1, and lactate dehydrogenase a gene promotors contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 27.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda R, Zhang H, Kim JW, Shimoda LA, Dang CV, Semenza GL. Regulation of COX subunit composition by HIF-1: A mechanism for optimizing the efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 30.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agoston-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microrna signature of hypoxia. Mol Cell Biol. 2007;27 doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutteman M, Kadenbach B, Grossman LI. Mammalian subunit IV isoforms of COX. Gene. 2001;267:111–123. doi: 10.1016/s0378-1119(01)00385-7. [DOI] [PubMed] [Google Scholar]
- 32.Chan SY, Loscalzo J. Microrna-210: A unique and pleiotropic hypoxamir. Cell Cycle. 2010;9:1072–1083. doi: 10.4161/cc.9.6.11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del Toro M, Labayru MT, Landa J, Font A, Garcia-Villoria J, Merinero B, Ugarte M, Gutierrez-Solana LG, Campistol J, Garcia-Cazorla A, Vaquerizo J, Riudor E, Briones P, Elpeleg O, Ribes A, Lill R. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 36.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–679. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–C1853. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 39.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion cannel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 40.Zamzami N, Kroemer G. The mitochondrion in apoptosis: How pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 41.Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, Michelakis ED. Pyruvate dehydrogenase inhibition by the inflammatory cytokine tnfalpha contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med. 2011;89:771–783. doi: 10.1007/s00109-011-0762-2. [DOI] [PubMed] [Google Scholar]
- 42.Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol Med. 2011;17:641–649. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ. 2012;2:201–213. doi: 10.4103/2045-8932.97606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, Linehan WM, Neckers L. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;88:88ra55. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizzuto R, Duchen MR, Pozzan R. Flirting in little space: The ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004:re1. doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- 47.Szanda G, Koncz P, Várnai P, Spät A. Mitochondrial Ca2+ uptake with and without the formation of high-Ca2+ microdomains. Cell Calcium. 2006;40:527–537. doi: 10.1016/j.ceca.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 48.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 49.Belmont PJ, A. T, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–14021. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann Intern Med. 1991;114:464–469. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- 51.Yeager ME, Belchenko DD, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Endothelin-1, the unfolded protein response, and persistent inflammation role of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2012;46:14–22. doi: 10.1165/rcmb.2010-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med (Berl) 2010;88:1011–1020. doi: 10.1007/s00109-010-0679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleiman RB, Houser SR. Outward currents in normal and hypertrophied feline ventricular monocytes. Am J Physiol. 1989;256:H1450–H1461. doi: 10.1152/ajpheart.1989.256.5.H1450. [DOI] [PubMed] [Google Scholar]
- 54.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 55.Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus WJ, Simonides WS. Right-ventricular failure is associated with increased mitochondrial complex ii activity and production of reactive oxygen species. Cardiovasc Res. 2007;75:770–781. doi: 10.1016/j.cardiores.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J, Shirato K. Increased [18f]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–1855. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 57.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld C, Kasanmoentalib S, Visser FC, Ylstra B, Simonides WS. Microarray analysis reveals pivotal divergent mRNA expression profiles early in the development of either compensated ventricular hypertrophy or heart failure. Physiol Genomics. 2005;21:314–323. doi: 10.1152/physiolgenomics.00185.2004. [DOI] [PubMed] [Google Scholar]
- 59.Macchia A, Marchioli R, Marfisi R, Scarano M, Levantesi G, Tavazzi L, Tognoni G. A meta-analysis of trials of pulmonary hypertension: A clinical condition looking for drugs and research methedology. Am Heart J. 2007;153:1037–1047. doi: 10.1016/j.ahj.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 60.Bersin RM, Stacpoole PW. Dichloroacetate as a metabolic therapy for myocardial ischemia and failure. Am Heart J. 1997;134:841–855. doi: 10.1016/s0002-8703(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 61.Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia. 1980;19:109–113. doi: 10.1007/BF00421855. [DOI] [PubMed] [Google Scholar]
- 62.Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, et al. Dichloroacetate causes toxic nephropathy in melas: A randomized, controlled clinical trial. Neurology. 2006;66:324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 63.Lewis JF, DaCosta M, Wargowich T, Stacpoole P. Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol. 1998;21:888–892. doi: 10.1002/clc.4960211206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: A new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 65.Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin a-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- 66.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 67.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain-3-ketoacyl coenzyme a thiolase. Circ Res. 2000;86:580–588. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 68.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glycose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135–142. doi: 10.1161/01.cir.93.1.135. [DOI] [PubMed] [Google Scholar]
- 69.Euler US, Liljsjestrand G. Observations of the pulmonary arterial blood pressure on the cat. Acta Physiol Scand. 1946;12:301–320. [Google Scholar]
- 70.Grabczewska Z, Bialoszynski T, Szymanski P, Sukiennik A, Swiatkiewicz I, Kozinski M, Kochman W, Grzesk G, Kubica J. The effect of trimetazidine added to maximal anti-ischemic therapy in patients with advanced coronary artery disease. Cardiol J. 2008;15:344–350. [PubMed] [Google Scholar]
- 71.Wang P, Fraser H, Lloyd SG, McVeigh JJ, Belardinelli L, Chatham JC. A comparison between ranolazine and cvt-4325, a novel inhibitor of fatty acid oxidation, on cardiac metabolism and left ventricular function in rat isolated perfused heart during ischemia and reperfusion. J Pharmacol Exp Ther. 2004;321:213–220. doi: 10.1124/jpet.106.115519. [DOI] [PubMed] [Google Scholar]
- 72.Fragasso G, Palloshi A, Puccetti P, Sillpigni C, Rossodivita A, Pala M, Calori G, Alferi O, Margonato A. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. 2006;48:992–998. doi: 10.1016/j.jacc.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 73.Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J, Simard MJ, Bonnet S. Role for mir-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–548. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S, Tian H, Jiang C, Zhu D. The microrna-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and l-type calcium channel-α1c. Hypertension. 2012;59:1006–1013. doi: 10.1161/HYPERTENSIONAHA.111.185413. [DOI] [PubMed] [Google Scholar]
- 75.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Shelagh A, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 76.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Heiden MGV, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2:44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 78.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins iscu1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chan SY, Loscalzo J. The emerging paradigm of network medicine in the study of human disease. Circ Res. 2012;111:359–374. doi: 10.1161/CIRCRESAHA.111.258541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.White K, Loscalzo J, Chan SY. Holding our breath: The emerging and anticipated roles of microrna in pulmonary hypertension. Pulm Circ. 2012;2:278–290. doi: 10.4103/2045-8932.101395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernandez-Friera L, Garcia-Alvarez A, Guzman G, Bagheriannejad-Esfahani F, Malick W, Nair A, Fuster V, Garcia MJ, Sanz J. Apical right ventricular dysfunction in patients with pulmonary hypertension demonstrated with magnetic resonance. Heart. 2011;97:1250–1256. doi: 10.1136/hrt.2010.216101. [DOI] [PubMed] [Google Scholar]