

SUMMARY
To investigate the functions of signal transducers and activators of transcription 1 (STAT1)-induced anti-hepatitis C viral (HCV) effects, a stable Huh7.5 cell line (Huh7.5-STAT1ER) was established that constitutively expresses a fusion protein (STAT1ER) of STAT1 and the mouse oestrogen receptor (ER), which forms STAT1ER homodimers after 4-hydroxytamoxifen (4-HT) treatment. This inducible and cytokine/receptor-independent STAT1 activation system allowed us to investigate the anti-HCV effects of STAT1ER activation after inducing IFN-stimulated gene (ISG) expression. The anti-HCV effects of dimerized STAT1ER fusion protein were determined by real-time PCR in a time-dependent fashion post-HCV (JFH-1) infection. HCV (JFH-1) RNA decreased 48% at 72 h after 4-HT treatment. To distinguish the inhibitory effects of STAT1ER activation on HCV RNA replication or HCV internal ribosomal entry site (IRES)–mediated translation, a dicistronic pRL-HL construct was used in the studies. Both cellular (Cap-dependent) and HCV IRES–mediated (Cap-independent) translation were decreased by 63% and 57% at 72 h post-STAT1ER activation in the STAT1ER cell line. In our previous studies, interferon-induced transmembrane protein 3 [(IFITM3) (1-8U)] was found to inhibit HCV RNA replication. Subsequently, elevated expression of the 1-8U gene was confirmed by Western blotting in the Huh7.5-STAT1ER cell line. To further investigate the 1-8U function with both in vivo and in vitro studies, the 1-8U gene was found to suppress cellular and HCV IRES–mediated translation.
Keywords: HCV IRES, hepatitis C virus, IFITM3 (1-8U), mouse oestrogen receptor, STAT1 dimerization
INTRODUCTION
Hepatitis C virus causes chronic liver disease, and currently, 170 million people worldwide and nearly 4 million people in the United States are infected [1]. In the United States, HCV-related mortality will increase 2–3 fold over the next decade and become a leading cause of liver cancer [2]. Treatment of HCV has undergone significant improvements with a standard therapy of IFN and ribavirin. However, the sustained virological response rate in most patients to IFN-based treatment is <50% despite the fact that many patients are not candidates for IFN-based regimens because of comorbidities [3]. Host gene polymorphisms, innate/adaptive immune responses and viral quasispecies are considered to be important mechanisms involved in both HCV persistence and IFN nonresponse [4]. Thus, much effort is being focused on developing a better understanding of interactions between HCV virus and host antiviral defence pathways [5,6].
The HCV is a plus-sense RNA virus containing an IRES domain to mediate HCV translation by a unique mechanism [7-9]. The HCV IRES contains a long stretch of RNA with stem-loop structures and requires eukaryotic initiation factors (eIFs) as well as the IRES-specific cellular transacting factors (ITAFs) to initiate the HCV translation. IFN treatment is known to reduce the translation efficiency for all HCV genotypes [10,11]. However, the molecular basis and regulatory mechanism of HCV IRES–mediated translation are not well defined. Investigating potential interactions between the HCV IRES element and host genes offers an attractive target for understanding the mechanism of HCV IRES–mediated translation and drug discovery [12].
The key step for IFN-mediated intracellular anti-HCV state is the ligand-dependent formation of STAT dimers with subsequent changes in ISG expression. In our previous studies, the 1-8U gene was found to be stimulated by STAT3 activation in the HCV subgenomic replicon cell line FCA1 and was both necessary and sufficient for the inhibitory effects on HCV replication [13,14]. In this study, a STAT1ER fusion protein was used to establish a stable Huh7.5-STAT1ER cell line to imitate both the type I and type II IFN-dependent common activation pathways [15]. The STAT1ER fusion protein was activated by 4-HT treatment to induce the expression of ISGs. Together with our previous investigations of ISGs on HCV replication, the 1-8U gene was further demonstrated to inhibit HCV replication through its inhibitory effects on cellular and HCV IRES–mediated translation in this paper.
MATERIALS AND METHODS
Plasmids
A mammalian expression vector containing the STAT1ER cDNA and expressing a STAT1ER fusion protein was constructed by inserting the STAT1 coding region (84 Kd) adjacent to the C-terminal sequence of mouse ER (310 a.a.) in the pEF6/V5-His TOPO® TA vector (Invitrogen, Carlsbad, CA, USA). The 1-8U gene was cloned into the pEF6/V5-His TOPO® TA vector as pEF6TOPO-1-8U. The plasmid pJFH-1 contains a full-length HCV genomic cDNA [16]. The plasmid pRL-HL is a dicistronic construct that mediates Cap-dependent and HCV IRES–dependent translation [17]. Synthetic 4-HT was purchased from Sigma (Saint Louis, MO, USA) and dissolved in ethanol as a 5 mm stock solution.
Cell lines
Human hepatoma Huh7.5 cells [18] were grown in Dulbecco’s modified Eagle’s medium (Invitrogen). To establish the Huh7.5-STAT1ER cell line, Huh7.5 cells were transfected with the STAT1ER cDNA plasmid. Positive clones of Huh7.5-STAT1ER cells were selected with blasticidin (Invitrogen) for 14 days and amplified to examine STAT1ER dimer formation after 4-HT induction by Western blotting.
Detection of STAT1ER dimers and 1-8U protein by Western blotting
Huh7.5-STAT1ER cells were washed in phosphate-buffered saline (PBS) post-4-HT treatment. Cell lysates were obtained in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor cocktail (Sigma). The Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA, USA) was used to determine cellular protein concentrations. One hundred micrograms of total protein per well was loaded either on a 6% sodium dodecylsulfate (SDS) polyacrylamide gel (for STAT1ER dimers) or a 12% SDS-polyacrylamide gel (for 1-8U). Antibodies for actin (Santz Cruz Biotechnology Inc., Santa Cruz, CA, USA), STAT1 (Santa Cruz) and 1-8U (Abnova, Taipei City, Taiwan) were used to detect actin, STAT1ER homodimers or 1-8U proteins with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA).
HCV JFH-1 stocks and HCV infection
The pJFH-1 virus stock was generated accordingly [16] with a MEGAscript kit (Ambion, Austin, TX, USA) and Huh7.5 cells. For examining anti-HCV effects, Huh7.5-STAT1ER cells were incubated with 0.5 MOI JFH-1 HCV for 14 days to achieve fully infected Huh7.5-STAT1ER cells [19]. Huh7.5-STAT1ER cells were then treated with 4-HT for 72, 48 and 24 h prior to the end-point for sample collection. Control Huh7.5-STAT1ER cells were incubated for 72 h in the absence of 4-HT. Total cellular RNA was isolated at each end time point for detecting HCV RNA.
Measurement of HCV replication by quantitative real-time PCR
Total cellular RNA was isolated from infected Huh7.5-STAT1ER cells by Trizol (Invitrogen). Superscript II (200 U reverse transcriptase per reaction) and a RT-PCR kit (Invitrogen) were used to prime with oligo (dT) 12–18 (Invitrogen) for first-strand synthesis according to the manufacturer’s instructions. Taqman primers were obtained from Applied Biosystems (Foster City, CA, USA). Reactions were conducted in a 96-well spectrofluorometric thermal cycler (ABI PRISM 7900 Sequence Detector System; Applied Biosystems). Fluorescence was monitored during every PCR cycle at the annealing step. The primers for HCV JFH-1 are as follows: 5′-CGGAATTGCCGGGAAGAC-3′, 5′-CAAATGGCCGGGCATAGAG-3′, FAM probe: 5′-CTTTCTTGGATAAACCC-3′; GADPH primers were purchased from Applied Biosystems. PCR was performed with the following conditions: 50 °C, 2 min; 95 °C, 10 min; (95 °C, 15 s; 60 °C, 1 min) × 40 cycles. Results were analysed with the SDS 2.0 software (Applied Biosystems) in triplicate and expressed as the mean ± SEM.
Luciferase assays
Huh7.5-STAT1ER cells were transfected with the plasmid pRL-HL and Lipofectamine 2000 (Invitrogen). After 24 h of transfection, Huh7.5-STAT1ER cells were treated with 4-HT for 48, and 72 h for harvesting the cell lysates. Control cells were incubated for 72 h in the absence of 4-HT. Co-transfection of the plasmids pRL-HL (2 μg) and pEF6TOPO-1-8U (2 μg) was performed with the same conditions into Huh7.5 cells. All samples were analysed for luciferase activity using the Dual-Luciferase Reporter Assay System Kit (Promega, Madison, WI, USA) in triplicate. Luciferase activities of in vitro translation reactions using the HeLa cell extracts were analysed as described previously [20].
In vitro translation with HeLa cell extracts
Preparation of HeLa cell extract and in vitro translation conditions were reported previously [20]. The 1-8U-GST protein (a full-length 1-8U protein fused with GST) and GST protein were purchased from Abnova. For each translation reaction, 200 ng of pRL-HL mRNA [17] was translated in HeLa cell extract with different concentrations of 1-8U-GST or GST protein (0.1, 0.2 and 0.4 μg). Translation reactions (performed in triplicate) were harvested and assayed for luciferase activity after incubation at 30 °C for 7 h. Assays were performed in duplicate for each translation reaction to detect the Renilla and firefly luciferase activity. Translation efficiency for adding 1-8U-GST or GST protein was calculated as the proportion of positive control (100%).
Statistical analysis
Student’s t-test for independent variables was used to analyse the differences between translation reactions from control cells and after addition of 1-8U-GST. P values <0.05 were considered significant.
RESULTS
STAT1ER dimerization induced by 4-HT in Huh7.5-STAT1ER cells
The IFN-Jak/STAT pathway is a complicated system involved in the activation of many genes that carry out multiple functions. It is extremely difficult to activate a single gene without interference by the activation of other genes in the IFN-Jak/STAT pathway. Inducible dimerization of STATs fused with mouse ER provides a cytokine receptor-independent STAT activation system to study the functions of STATs during HCV replication [14,21]. In previous studies from us and others, concentration of 4-HT was titrated as 1 um to achieve the highest expression of STAT3ER dimerization, 1-8U gene, and the strongest inhibitory effects on HCV RNA replication [14]. In our previous studies, 4-HT was also demonstrated without anti-HCV effects [14]. In this study, similar sequences from the mouse ER C-terminal domain were fused to the C-terminus of the STAT1 gene. Homodimerization of STAT1ER was detected by Western blotting after treatment with 4-HT (1 μm). Our data indicated that the stable Huh7.5-STAT1ER cells could express the STAT1ER fusion protein and form homodimers after 4-HT treatment. In Fig. 1, endogenous STAT1 protein is 84 Kd (lane 1–3), the STAT1ER monomer is 119 Kd (lane 2 and 3) and the STAT1ER dimer is about 240 Kd (lane 3). The density of STAT1ER dimers was less than the density of STAT1ER monomers as shown in Fig. 1 (lane 2 and 3), which could be explained by the denaturing conditions used in the analysis, including SDS-polyacrylamide gel electrophoresis, RIPA lysis buffer and boiling during Western blotting to detect the STAT1ER dimers. This experiment enabled us to detect STAT1ER dimers as reported previously [14,21].
Fig. 1.
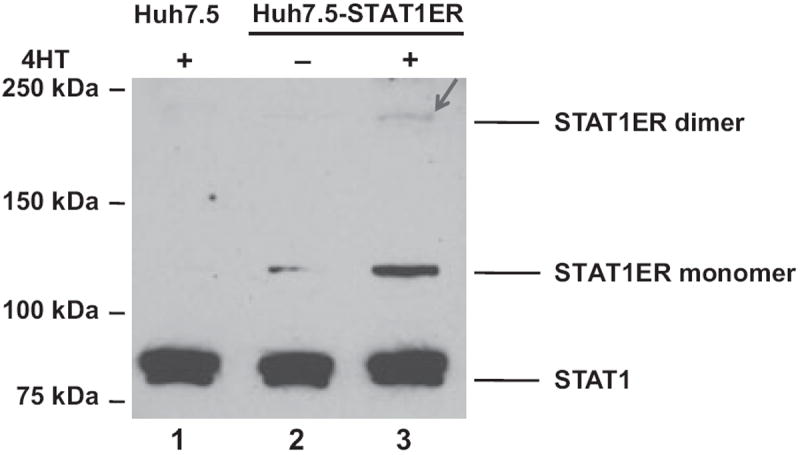
STAT1ER dimerization in Huh7.5-STAT1ER cells. Huh7.5-STAT1ER cells were treated with 4-HT (1 um) for 48 h. Total protein was extracted and analysed by Western blotting with anti-STAT1 antibody. The positions of the endogenous STAT1, STAT1ER fusion protein monomer and STAT1ER fusion protein dimer are indicated. Lane 1, Huh7.5 cells treated with 4-HT; lane 2, Huh7.5-STAT1ER cells without 4-HT treatment; lane 3, Huh7.5-STAT1ER cells with 4-HT treatment. Arrowhead indicates the STAT1ER dimers.
Anti-HCV activities induced by STAT1ER dimerization in Huh7.5-STAT1ER cells
Anti-HCV activities of IFNs are mediated predominantly by the Jak/STAT pathway through induction of downstream ISG expression. To examine the potential anti-HCV activity of the STAT1ER fusion protein, Huh7.5-STAT1ER cells were treated with 4-HT and examined for STAT1ER dimerization as well as anti-HCV activities. Because of activation of STAT1ER fusion protein in the absence of interfering cellular signalling systems, any anti-HCV function generated by 4-HT treatment should be solely initiated from STAT1ER dimerization. Figure 2 shows important roles for STAT1ER activation in IFN signalling pathways that inhibit HCV replication. HCV (JFH-1) RNA replication was decreased after 4-HT treatment in Huh7.5-STAT1ER cells in a time-dependent fashion. At 72 h, JFH-1 RNA level was reduced to 48% of control. However, it is not clear if the decrease in JFH-1 RNA replication after activation of STAT1ER is because of the induction of ISG expression on HCV RNA replication or HCV IRES–mediated translation. Here, we conclude that dimerization of STAT1ER fusion protein is a key step to carry out anti-HCV functions.
Fig. 2.
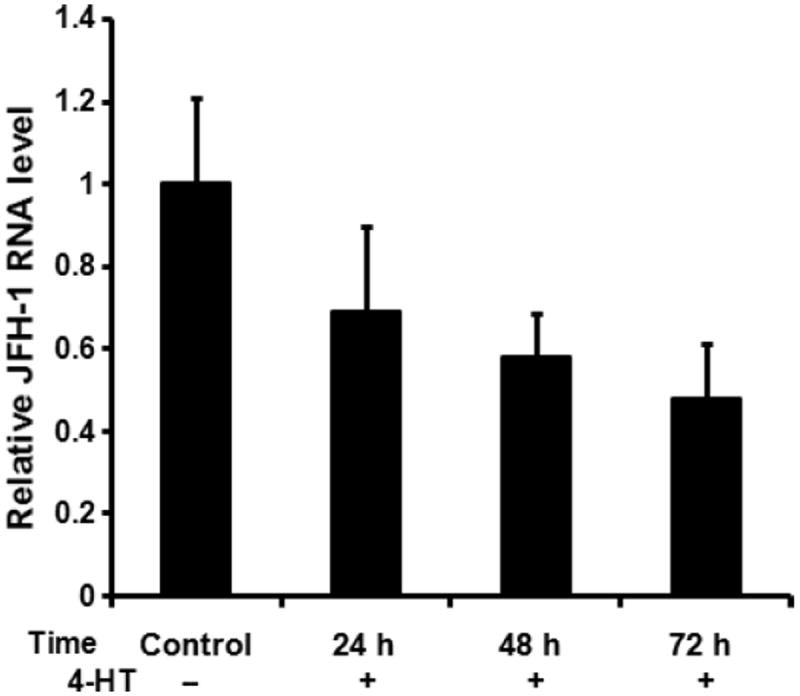
Anti-HCV effects induced by 4-HT in Huh7.5-STAT1ER cells. Huh7.5-STAT1ER cells were inoculated with HCV JFH-1 at a MOI of 0.5 for 14 days and then added 4-HT (1 μm) at 72, 48 and 24 h prior to end-point for sample collection. Control indicates the JFH-1-infected Huh7.5-STAT1ER cells without 4-HT treatment for 72 h. HCV JFH-1 RNA levels were measured by quantitative real-time PCR in triplicate. Relative JFH-1RNA level was calculated as proportion of control (1.0). The data are presented after normalization with an internal GAPDH control. The error bars indicate the variation present in three independent assays.
Inhibitory effects of STAT1ER dimerization on Cap-dependent and HCV IRES–dependent translation
The dicistronic pRL-HL plasmid was used to separate HCV RNA replication and IRES-mediated translation. The pRL-HL plasmid contains a dicistronic CMV transcriptional cassette in which an upstream Renilla luciferase gene and a downstream firefly luciferase gene represent both types of translation [17]. In Fig. 3, the dual luciferase assays show inhibitory effects on both types of translations after STAT1ER dimerization. At 72 h post-4-HT treatment, Cap-dependent translation was reduced to 63% and HCV IRES–dependent translation was decreased to 57%. These results indicate that dimerization of STAT1ER plays an important role in inhibiting Cap-dependent and HCV IRES–mediated translation.
Fig. 3.
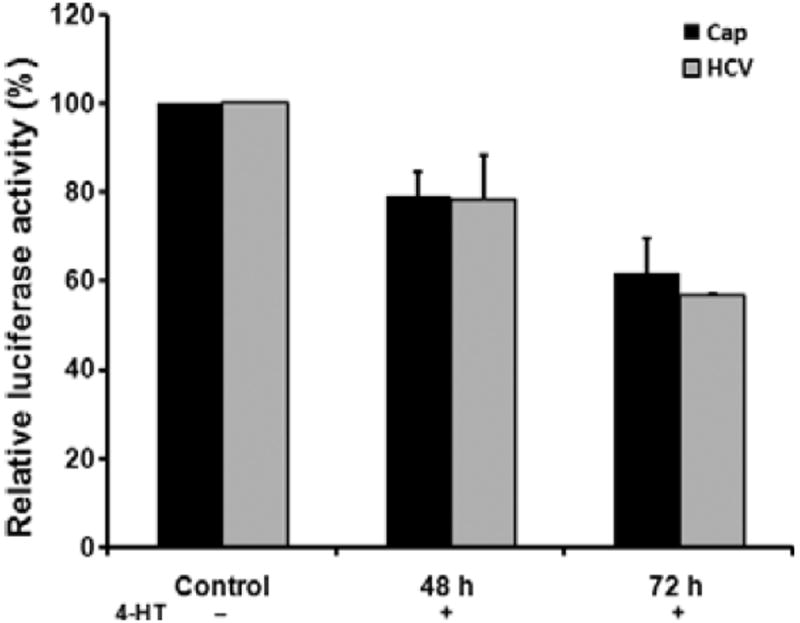
Inhibitory effects of STAT1ER dimerization on Cap-dependent and HCV IRES–dependent translation. Huh7.5-STAT1ER cell monolayers were transfected with the plasmid pRL-HL. After 24 h of incubation at 37 °C and 5% CO2, transfected Huh7.5-STAT1ER cells were treated with 4-HT for 48 and 72 h for analyses of luciferase activity. Control of Huh7.5-STAT1ER cells received the pRL-HL cDNA plasmid but did not receive 4-HT treatment. Translation efficiency for each sample was calculated as proportion of control (100%). The error bars indicate the variation of three independent assays.
Expression of the 1-8U gene and its inhibitory effects on HCV IRES–mediated translation
The 1-8U family proteins are IFN inducible polypeptides involved in the control of cell proliferation [22]. In our previous study, the 1-8U gene was stimulated by activation of a STAT3ER fusion protein to inhibit HCV RNA replication [14]. In our current study, we again show that the 1-8U gene is induced after activation of the STAT1ER fusion protein. In Fig. 4, 1-8U gene expression after STAT1ER dimerization was examined by Western blotting at 0, 24 and 48 h post-4-HT treatment. Expression of the 1-8U gene was increased in 4-HT-treated Huh7.5-STAT1ER cells in a manner similar to 1-8U gene induction by STAT3ER or IL-28A in treated GSB cells [23,24].
Fig. 4.
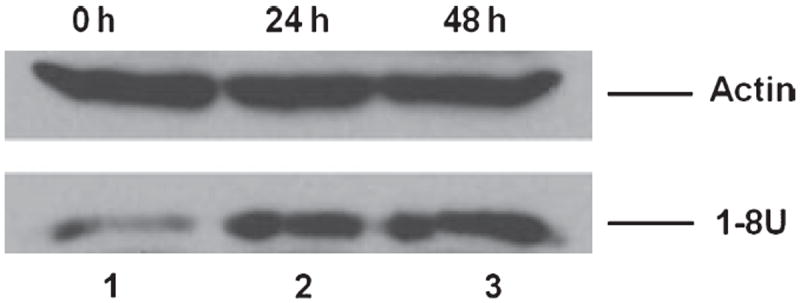
1-8U gene expression in Huh7.5-STAT1ER cells with 4-HT treatment. Huh7.5-STAT1ER cell lysates were harvested at 0, 24 and 48 h after adding 4-HT to induce STAT1ER dimerization. Total proteins were extracted and analysed by Western blotting with an anti-1-8U antibody. Lane 1, Huh7.5-STAT1ER cell lysate from cells treated with 4-HT for 0 h; lane 2 and 3, Huh7.5-STAT1ER cell lysates from cells treated with 4-HT for 24 and 48 h. Actin protein was used as an internal control.
To determine whether the 1-8U protein inhibits HCV replication, the plasmid pRL-HL was co-transfected with pEFT6TOPO-1-8U into Huh7.5 cells. As shown in Fig. 5a, both Cap-dependent and HCV IRES–mediated translations were reduced by co-transfection of the 1-8U expression plasmid and the pRL-HL cDNA plasmid. Translation efficiency of Cap-dependent translation and HCV IRES–mediated translation was only 50% and 35% of the positive control, respectively. To further examine inhibitory effects of the 1-8U protein, in vitro translation reactions were carried out with HeLa cell extract and different concentrations of 1-8U-GST and GST proteins as well as in vitro transcribed pRL-HL RNA transcripts. In Fig. 5b, both Cap-dependent and HCV IRES–mediated translations were inhibited by adding 1-8U-GST fusion protein (0.1, 0.2 and 0.4 μg). Statistical significance was observed in both Cap-dependent translation (P = 0.026) and HCV IRES–mediated translation (P = 0.029) after addition of 0.4 μg of the 1-8U-GST protein (final concentration 0.032 μg/μL). The GST protein does not exhibit any inhibitory effect on either mode of translation (Fig. 5c). This data indicate that the 1-8U protein inhibits Cap-dependent translation, an observation consistent with a previous study [22]. Inhibition of HCV IRES–mediated translation by the 1-8U protein provides a new avenue to reveal the mechanisms by which IFNs inhibit HCV replication and HCV IRES–mediated translation.
Fig. 5.
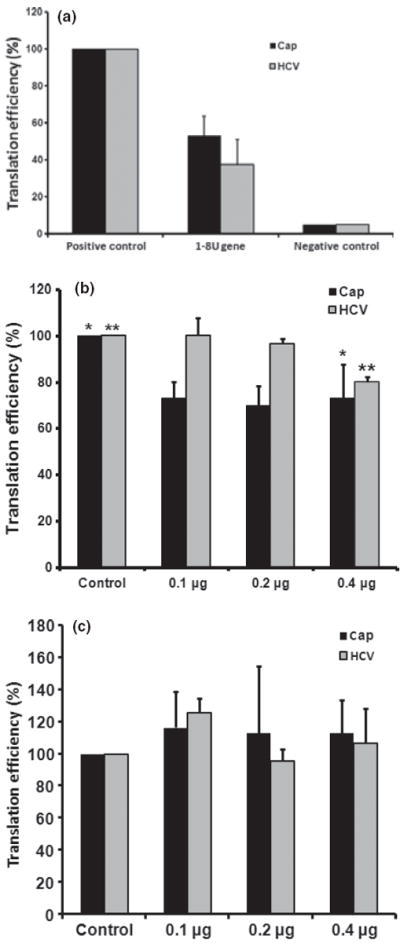
In vivo and in vitro inhibitory effects of 1-8U on Cap-dependent and HCV IRES–mediated translation. (a) Co-transfection of pEF6TOPO-1-8U and pRL-HL plasmids was carried out in Huh7.5 cells in triplicate at 37 °C for 24 h. Cell lysates were harvested and luciferase assays were performed in duplicate. The positive control consists of cells transfected with the plasmid pRL-HL as well as the plasmid pEF6TOPO. Negative controls are mock-transfected cells receiving no plasmid cDNA. (b) Inhibition of 1-8U protein on Cap-dependent and HCV IRES–mediated translation in HeLa cell extracts. HeLa cell extract was used to examine the translation efficiency of pRL-HL mRNA with different concentrations of 1-8U-GST protein (0.1, 0.2 and 0.4 μg). Control translation reactions contained HeLa cell extract without 1-8U-GST protein. Luciferase assays in duplicate were performed in triplicate after incubation at 30 °C for 7 h. *P = 0.026; **P = 0.029. (c) Cap-dependent and HCV IRES–mediated translation in the presence of 0.1, 0.2 and 0.4 μg of GST using the conditions described in fig. 5b. Translation efficiency of each experiment in this figure (a, b and c) was calculated as a proportion of controls (100%). The error bars indicate the variation of three independent assays.
DISCUSSION
The biological activities of IFNs are initiated when they bind to IFN receptors on the cell surface, which results in activation of the Jak/STAT pathway. The Jak/STAT pathway activates a large number of cellular ISGs that are normally quiescent or present at low level. Those ISGs play important roles at inhibiting HCV replication at multiple points in the HCV replication cycle. In this study, an inducible and cytokine/receptor-independent STAT1 activation system was established in Huh7.5 cells, which possesses inhibitory effects on HCV replication (Figs 1 and 2). This system provides a valuable tool to understand the molecular events triggering intracellular anti-HCV activities, such as 1-8U gene expression, that are involved in cellular and HCV IRES–mediated translation.
The HCV IRES consists of four highly conserved secondary structures (I–IV) that direct the binding of ribosomes in close proximity to the start codon of the HCV ORF. The interaction of HCV IRES sequence with host genes has been explored as a potential therapeutic target [20,25]. More and more cellular proteins have been identified that affect HCV translation after IFN treatment. They include the 2′–5′ oligoadenylate synthetase/Rnase L, PKR-eIF2α, P56-eIF3, all of which increase with IFN treatment, and ITAFs, such as La autoantigen, which have reduced expression post-IFN treatment [26-28]. Here, we describe a new pathway by which IFN treatment inhibits HCV IRES–mediated translation through the 1-8U protein (Figs 3 and 5a, b). The 1-8U protein contains two putative trans-membrane domains [29]. A unique leucine zipper domain (a.a.106–127) within the 1-8U protein suggests that it interacts with either DNA or RNA [29]. The biological function of the 1-8U gene is still unknown. A comprehensive gene expression profile study shows that the 1-8U gene is highly expressed in normal liver, cultured HepG2 cells and other tissues [30]. Constitutive expression of the 1-8U protein suppressed host cell proliferation and reduced DNA and protein synthesis [22]. Our data clearly demonstrate that the 1-8U protein is involved in inhibition of Cap-dependent and HCV IRES–mediated translation.
A number of in vitro studies have identified several ITAFs that enhance HCV IRES–mediated translation, but they are not essential for 48S complex formation [31]. Most notable among these ITAFs are the PTB [32], La protein [33], heat shock protein 70 [34], NSAP1 [35], insulin-like growth factor-II mRNA-binding protein 1 (IGF2BP1) [36] and hnRNP L and D [37-39]. An in vivo study demonstrated that decreased HCV IRES–mediated translation was correlated with reduced expression of La protein after IFN treatment in Huh7 cells [27]. Other ITAFs, such as NSAP1, hnRNP L and D, and heat shock protein 70 were found to enhance HCV IRES–mediated translation [34,35,39,40]. The relations of these ITAFs with IFN treatment are not yet well defined. Recently, another ISG, 6–16, was suggested to inhibit HCV IRES–mediated but not Cap-dependent translation in IL-28A-treated Huh7.5 cells [23]. Noteworthy, both 1-8U and 6-16 belong to the ISG family and have inhibitory effects on HCV IRES–mediated translation. This suggests that we need to characterize how both proteins are involved in suppressing HCV IRES–mediated translation in the future. Here, we demonstrate the 1-8U protein inhibits HCV IRES–mediated translation; however, the mechanism by which 1-8U decreases HCV IRES–mediated translation remains unknown and is under investigation.
Acknowledgments
DISCLOSURES
Xiaoyu Li is supported by the Junior Faculty Start-up Funding from the University of Florida-Jacksonville. David Nelson was supported by NIH UL1RR029890-0, R01 AI061158 and NIH-NCI K24CA139570-0. The authors appreciate Dr. Takaji Wakita who provided the HCV JFH-1 plasmids, Dr. Charles Rice who provided the Huh7.5 cells and Dr. Stanley Lemon provided us with the pRL-HL plasmid. Authors appreciate the proofreading and suggestions for this manuscript by Dr. Michael Haas. The authors who have taken part in the research described in this paper declare that they do not have a relationship with the pharmaceutical manufactures in this study.
Abbreviations
- CMV
Cytomegalovirus
- eIFs
Eukaryotic initiation factors
- HCV
Hepatitis C viral
- IRES
Internal ribosomal entry site
- ISG
IFN-stimulated gene
- PBS
Phosphate-buffered saline
- RBV
Ribavirin
- RIPA
Radioimmunoprecipitation assay
- SVR
Sustained virological response
References
- 1.Centers for Disease Control and Prevention. Recommendation for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep. 1998;47:1–39. [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma: an epidemiologic view. J Clin Gastroenterol. 2002;35:S72–S78. doi: 10.1097/00004836-200211002-00002. [DOI] [PubMed] [Google Scholar]
- 3.Di Bisceglie AM, Hoofnagle JH. Optimal therapy of hepatitis C. Hepatology. 2002;36:S121–S127. doi: 10.1053/jhep.2002.36228. [DOI] [PubMed] [Google Scholar]
- 4.Asselah T, Bieche I, Sabbagh A, et al. Gene expression and hepatitis C virus infection. Gut. 2009;58:846–858. doi: 10.1136/gut.2008.166348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang D, Guo H, Xu C, et al. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J Virol. 2008;82:1665–1678. doi: 10.1128/JVI.02113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato J, Kato N, Moriyama M, et al. Interferons specifically suppress the translation from the internal ribosome entry site of hepatitis C virus through a double-stranded RNA-activated protein kinase-independent pathway. J Infect Dis. 2002;186:155–163. doi: 10.1086/341467. [DOI] [PubMed] [Google Scholar]
- 7.Honda M, Ping LH, Rijnbrand RC, et al. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31–42. doi: 10.1006/viro.1996.0395. [DOI] [PubMed] [Google Scholar]
- 8.Lu HH, Wimmer E. Poliovirus chimeras replicating under the translational control of genetic elements of hepatitis C virus reveal unusual properties of the internal ribosomal entry site of hepatitis C virus. Proc Natl Acad Sci U S A. 1996;93:1412–1417. doi: 10.1073/pnas.93.4.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao WD, Wimmer E, Lahser FC. Poliovirus/Hepatitis C virus (internal ribosomal entry site-core) chimeric viruses: improved growth properties through modification of a proteolytic cleavage site and requirement for core RNA sequences but not for core-related polypeptides. J Virol. 1999;73:1546–1554. doi: 10.1128/jvi.73.2.1546-1554.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dash S, Prabhu R, Hazari S, et al. Interferons alpha, beta, gamma each inhibit hepatitis C virus replication at the level of internal ribosome entry site-mediated translation. Liver Int. 2005;25:580–594. doi: 10.1111/j.1478-3231.2005.01082.x. [DOI] [PubMed] [Google Scholar]
- 11.Hazari S, Patil A, Joshi V, et al. Alpha interferon inhibits translation mediated by the internal ribosome entry site of six different hepatitis C virus genotypes. J Gen Virol. 2005;86:3047–3053. doi: 10.1099/vir.0.81132-0. [DOI] [PubMed] [Google Scholar]
- 12.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat Chem Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu H, Liu C. Interleukin-1 inhibits hepatitis C virus subgenomic RNA replication by activation of extracellular regulated kinase pathway. J Virol. 2003;77:5493–8. doi: 10.1128/JVI.77.9.5493-5498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H, Shang X, Terada N, Liu C. STAT3 induces anti-hepatitis C viral activity in liver cells. Biochem Biophys Res Commun. 2004;324:518–528. doi: 10.1016/j.bbrc.2004.09.081. [DOI] [PubMed] [Google Scholar]
- 15.Uddin S, Lekmine F, Sharma N, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275:27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 16.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honda M, Kaneko S, Matsushita E, Kobayashi K, Abell GA, Lemon SM. Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology. 2000;118:152–162. doi: 10.1016/s0016-5085(00)70424-0. [DOI] [PubMed] [Google Scholar]
- 18.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu H, Dong H, Eksioglu E, et al. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology. 2007;133:1649–1659. doi: 10.1053/j.gastro.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Mueller S, Wimmer E. Inhibition of hepatitis C virus IRES-mediated translation by oligonucleotides. Virus Res. 2009;146:29–35. doi: 10.1016/j.virusres.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda T, Nakamura T, Nakao K, et al. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO. 1999;18:4261–4269. doi: 10.1093/emboj/18.15.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brem R, Oraszlan-Szovik K, Foser S, Bohrmann B, Certa U. Inhibition of proliferation by 1-8U in interferon-alpha-responsive and non-responsive cell lines. Cell Mol Life Sci. 2003;60:1235–1248. doi: 10.1007/s00018-003-3016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu H, Butera M, Nelson DR, Liu C. Novel type I interferon IL-28A suppresses hepatitis C viral RNA replication. Virol J. 2005;2:80. doi: 10.1186/1743-422X-2-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Zhao H, Collins CD, et al. Gene expression associated with interferon alfa antiviral activity in an HCV replicon cell line. Hepatology. 2003;37:1180–1188. doi: 10.1053/jhep.2003.50184. [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi K, Umehara T, Nishikawa F, Fukuda K, Hasegawa T, Nishikawa S. Increased inhibitory ability of conjugated RNA aptamers against the HCV IRES. Biochem Biophys Res Commun. 2009;386:118–123. doi: 10.1016/j.bbrc.2009.05.135. [DOI] [PubMed] [Google Scholar]
- 26.Guo JT, Sohn JA, Zhu Q, Seeger C. Mechanism of the interferon alpha response against hepatitis C virus replicons. Virology. 2004;325:71–81. doi: 10.1016/j.virol.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 27.Shimazaki T, Honda M, Kaneko S, Kobayashi K. Inhibition of internal ribosomal entry site-directed translation of HCV by recombinant IFN-alpha correlates with a reduced La protein. Hepatology. 2002;35:199–208. doi: 10.1053/jhep.2002.30202. [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Pflugheber J, Sumpter R, Jr, et al. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martensen PM, Justesen J. Small ISGs coming forward. J Interferon Cytokine Res. 2004;24:1–19. doi: 10.1089/107999004772719864. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita T, Hashimoto S, Kaneko S, et al. Comprehensive gene expression profile of a normal human liver. Biochem Biophys Res Commun. 2000;269:110–116. doi: 10.1006/bbrc.2000.2272. [DOI] [PubMed] [Google Scholar]
- 31.Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ non-translated regions of hepatitis C virus and classical swine fever virus RNAs. J Virol. 1998;72:4775–4782. doi: 10.1128/jvi.72.6.4775-4782.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali N, Siddiqui A. Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J Virol. 1995;69:6367–6375. doi: 10.1128/jvi.69.10.6367-6375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali N, Siddiqui A. The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc Natl Acad Sci U S A. 1997;94:2249–2254. doi: 10.1073/pnas.94.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez O, Fontanes V, Raychaudhuri S, et al. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology. 2009;50:1756–1764. doi: 10.1002/hep.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JH, Paek KY, Ha SH, et al. A cellular RNA-binding protein enhances internal ribosomal entry site-dependent translation through an interaction downstream of the hepatitis C virus polyprotein initiation codon. Mol Cell Biol. 2004;24:7878–7890. doi: 10.1128/MCB.24.18.7878-7890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinlich S, Huttelmaier S, Schierhorn A, Behrens SE, Ostareck-Le-derer A, Ostareck DH. IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3′UTR. RNA. 2009;15:1528–1542. doi: 10.1261/rna.1578409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domitrovich AM, Diebel KW, Ali N, Sarker S, Siddiqui A. Role of La autoantigen and polypyrimidine tract-binding protein in HCV replication. Virology. 2005;335:72–86. doi: 10.1016/j.virol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Hahm B, Kim YK, Kim JH, Kim TY, Jang SK. Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitis C virus. J Virol. 1998;72:8782–8788. doi: 10.1128/jvi.72.11.8782-8788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang B, Lim JH, Hahm B, Jang SK, Lee SW. hnRNP L is required for the translation mediated by HCV IRES. Biochem Biophys Res Commun. 2009;378:584–588. doi: 10.1016/j.bbrc.2008.11.091. [DOI] [PubMed] [Google Scholar]
- 40.Paek KY, Kim CS, Park SM, Kim JH, Jang SK. RNA-binding protein hnRNP D modulates internal ribosome entry site-dependent translation of hepatitis C virus RNA. J Virol. 2008;82:12082–12093. doi: 10.1128/JVI.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]