

Abstract
The authors report the case of a 56-year-old male patient with neurofibromatosis type 1 (NF1) diagnosed during adolescence and with an insidious clinical evolution, characterised by an exuberant cutaneous involvement, referred to the orthopaedics outpatient clinic presenting with carpal tunnel syndrome secondary to a plexiform neurofibroma of the median nerve. A comprehensive clinical approach is discussed, considering the natural history of the disease and its potential complications, as well as the lack of consensus regarding standard therapeutic options for the compressive peripheral neuropathies in the NF1 disease.
Background
Peripheral neuropathy due to nerve compression is a rare complication of neurofibromatosis type 1 (NF1) described in up to 4% of all cases.1 Nevertheless, it can represent a severe complication of NF1 associated with frequent morbidity including the development of malignant peripheral nerve sheath tumours (MPNSTs).2
Surgical treatment of plexiform neurofibromas is often difficult due to their serpiginous growth, significant vascularisation and multiple nerve fibres involvement, which makes complete surgical removal in most cases, impossible. There are only few case reports in the literature describing this type of approach, which is usually associated with a considerable number of complications including frequent relapse, hypertrophic scaring and neurological deficits.3
Considering the often indolent and stable clinical course of plexiform neurofibromas among patients with NF1, a more conservative management should, therefore, be considered, including regular clinical follow-up of these tumours to allow early diagnosis and timely treatment of its complications.
In authors’ opinion, the case report described represents a useful clinical reminder on how to approach and safely manage a rare clinical manifestation related to a common genetic disorder that is NF1.
Case presentation
We report the case of a 56-year-old male patient referred to our orthopaedics outpatient clinic with a history of predominant nocturnal numbness during the previous month before his observation, located in the right hand, including the palmar surface of the thumb, second, third and lateral half of the fourth finger and over the thenar eminence.
He had a personal history of NF1 disease diagnosed at the age of 17, with insidious but exuberant cutaneous involvement, characterised by multiple neurofibromas, measuring between 3 mm and 3 cm of diameter, located mainly along the trunk and also with scattered lesions along the limbs, face and scalp. Considering the aesthetic deformity associated with some of the cutaneous neurofibromas, the patient, during the course of the disease, was submitted to surgical resection of three neurofibromas located in the scalp, back and leg, which allowed the histopathological diagnosis.
On physical examination multiple neurofibromas were observed, scattered along the forearm and wrist of the right-upper limb (figure 1). Clinical examination of the median nerve showed positive Tinel and Phalen tests, preserved abduction of the thumb and absence of hypoesthesia along the median nerve territory (Pinprick test). The remaining neurological examination was normal, with symmetrical osteotendinous reflexes and showing no motor or sensory dysfunction as well as muscular atrophy.
Figure 1.
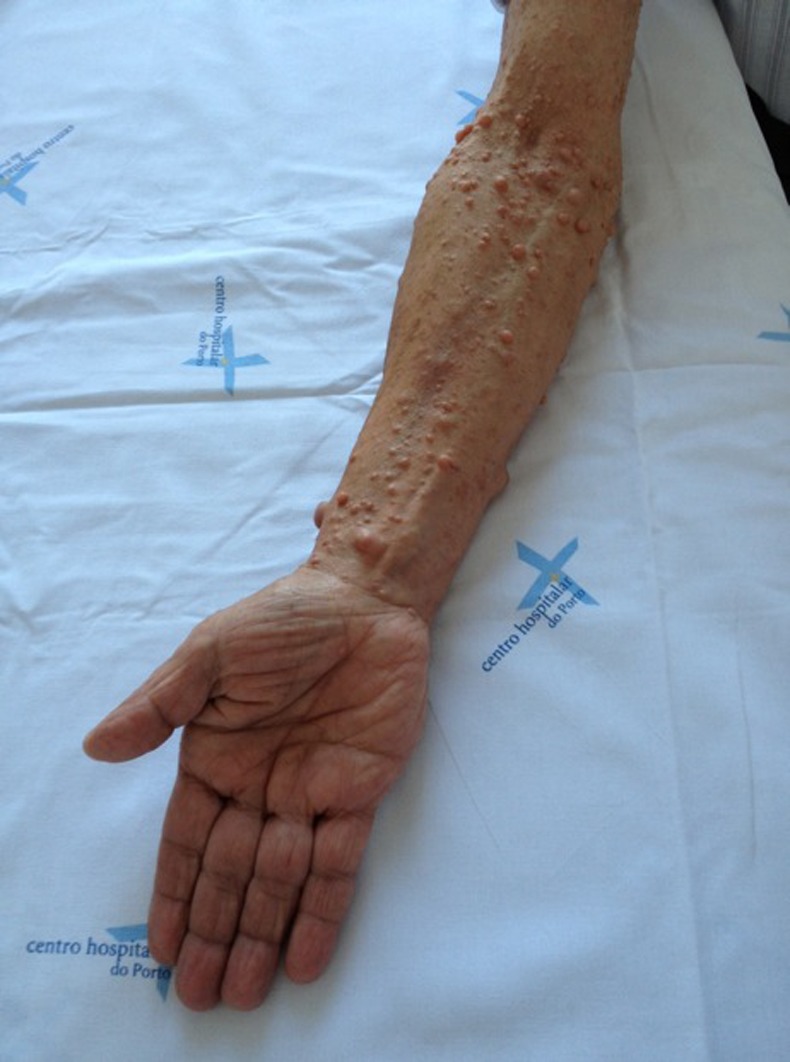
Multiple neurofibromas scattered along the forearm and wrist of the right upper limb.
Investigations
Electrodiagnostic tests such as electromyography (EMG) and nerve conduction studies (NCS) confirmed a discreet impairment of the sensory conduction velocity of the right median nerve between the wrist and the third digit (42.0 m/s (56.2±5.8(44))) with normal sensory latency (2.5 ms (2.84±0.34(3.5))) and amplitude (23.0 μV (38.5±15.6(19))) measured at the right wrist. The right median nerve motor response was normal, including motor nerve conduction velocity between the elbow and wrist (60.0 m/s (57.7±4.9(48))), latency (3.5 ms (3.49±0.34(4.2))) and amplitude of compound motor action potential (11.2 mV (7.0±3.0(3.5))) measured at the wrist.
Median NCS of the left-upper limb showed a normal sensory conduction velocity (55.0 m/s (56.2±5.8(44))), sensory distal latency (2.8 ms (2.84±0.34(3.5))) and amplitude (52.0 μV (38.5±15.6(19))).
No sensory abnormalities were found on the right and contralateral ulnar nerve, including their sensory conduction velocity (right: 56.8 m/s; left: 61.1 m/s (58.8±5.3(44))), sensory distal latency (right: 2.40 ms; left: 2.37 ms (2.54±0.29(3.1))) and amplitude (right: 19.0 μV; left: 34.0 μV (35.0±14.7(18))).
Considering the absence of neurological signs and symptoms suggesting NF1-related polyneuropathy, EMG and NCS of the lower limbs were not performed.
The MRI confirmed the presence of multiple subcutaneous neurofibromas in the forearm and wrist, with a 26×12×13 mm lobulated nodule following the median nerve trajectory detected on the palmar side of the transition between the forearm and wrist (figure 2). The lesion was isointense to the muscle on T1-weighted images and hyperintense on T2, with enhancement following contrast administration. Both its morphological and magnetic findings were compatible with the diagnosis of neurofibroma of the median nerve.
Figure 2.
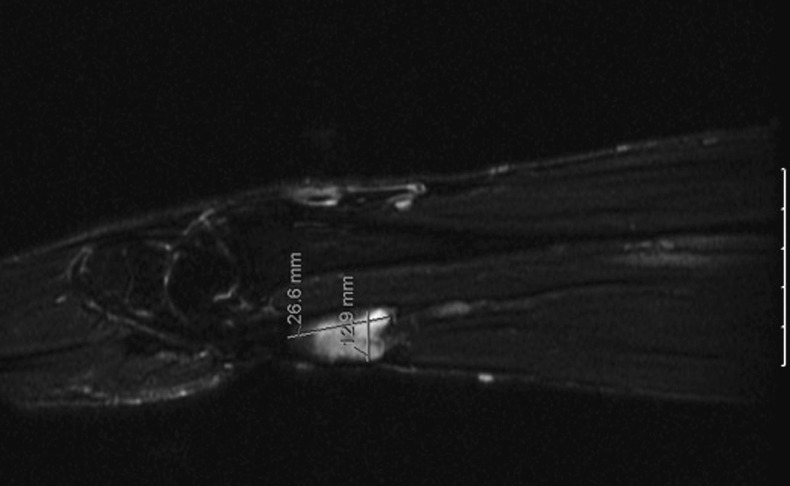
MRI showing a lobulated nodule (26×12×13 mm) following the median nerve trajectory on the palmar side of the transition between the forearm and wrist of the right upper limb.
Treatment
A non-surgical approach was decided with close clinical follow-up, including symptomatic treatment by using nocturnal wrist splint in the neutral position and clinical recommendations such as avoiding extreme flexion and extension of the wrist. The decision was based on the absence of clinical signs of malignancy (no significant neurological symptoms and pain associated with the neurofibroma; non-progressive dimensions) as well as MRI findings suggesting a benign nature of the lesion (<5 cm of diameter, well-defined margins, no evidence of adjacent adipose tissue invasion).
Outcome and follow-up
The patient became asymptomatic 3 months after the diagnosis with subsequent annual follow-up showing no complications or recurrent clinical manifestations.
Discussion
Peripheral neuropathy due to nerve compression, although rare, can represent a severe complication of NF1 with its morbidity including the potential development of MPNST.2
In the case presented, distal peripheral mononeuropathy is more likely the result of a combination of intrinsic factors related to the tumour such as involvement of the median nerve fibres responsible for its progressive dysfunction and also extrinsic factors, including median nerve compression due to its increased thickness associated with the presence of a neurofibroma located in the carpal tunnel proximal section. However, since the median nerve is involved at the wrist, it is unclear which clinical symptoms and diagnostic findings are due to tumour involvement, nerve compression or other mechanisms.
When managing these patients, regular and systematic clinical evaluation appears to be the most useful way to detect NF1 complications, even when compared with screening imaging.3 Therefore, close monitoring of plexiform neurofibromas should be included in their long-term follow-up, since they represent a potential risk for malignant transformation.
Several case studies have demonstrated that symptoms and clinical manifestations in patients with peripheral nerve involvement generally tend to be stable or slowly progressive.1
Although the estimated risk of MPNST among patients with NF1 remains considerably high (approximately 5–13%, according to published series), its peak of incidence usually occurs between the age of 15 and 40 years.4 5 Thus, the option for a non-surgical approach in the case reported seems to us well substantiated by current knowledge of the disease.
When approaching patients with NF1 another tool to be considered is the whole body MRI, which allows a complete evaluation of the entire body in a relatively short period of time without the use of ionising radiation and detects even relatively small or asymptomatic tumours located in all body regions, provides a more comprehensive knowledge of tumour burden in these patients and allows the analysis of tumour distribution across body parts.6
With the new developments of high resolution and electronic broadband transducers, ultrasound proved to be an optimal imaging technique to evaluate disorders of peripheral nerves, as it allows a quick and detailed imaging of the entire length of the major peripheral nerves, offering the possibility of performing a dynamic, non-invasive and low cost examination. Nevertheless, the limitations of this technique include the difficulty in assessing small nerves and deep masses, as well as its inability to allow a definite differentiation between two hystotypes (schwannomas and neurofibromas) of the extremities.7
During the patient clinical follow-up, an MRI was immediately performed when neurological abnormalities, although discrete, were detected, which enabled median nerve neurofibroma diagnosis and MPNST exclusion. Consequently, the patient underwent close follow-up during the first 6 months in order to detect possible clinical signs or symptoms suggesting malignancy, such as rapid and progressive growth or changes in the neurofibroma soft consistency, progressive neurological deterioration and significant, refractory pain. In the presence of such clinical findings an MRI should be repeated. Imaging findings, such as lesion diameter greater than 5 cm, ill-defined margins, lesion heterogeneity, surrounding oedema and invasion of contiguous adipose tissue, would then suggest MPNST.8 A multifocal core needle biopsy would be the procedure of choice by the authors to establish the histopathological diagnosis of MPNST.
Surgical management of plexiform neurofibromas in patients with NF1 should only be considered if malignant transformation of the neurofibroma occurs and in the presence of significant neurological deterioration or persistency of symptoms due to specific location or increased dimension of the neurofibroma.3 4 In this case, the options would include either classical release of the median nerve in the carpal tunnel with section of the transverse ligament, which would avoid possible neurological complications by preserving the nerve although total recovery of tumour-induced neuropathy would not be assured, or complete surgical excision of the neurofibroma if there is clinical suspicion and radiological evidence of malignant transformation. The latter approach has, nevertheless, a higher risk of surgical complications.3
This case report aims to emphasise the safety of a conservative therapeutic approach among patients with NF1 with plexiform neurofibromas, including strict clinical follow-up, even when associated with some degree of peripheral neuropathy, as its natural history usually reveals an indolent course, often stable and, therefore, more predictable. Considering these assumptions when managing patients with NF1, surgical treatment, often complicated by even more disabling sequelae, can be avoided.
Learning points.
Peripheral neuropathy due to nerve compression is a rare complication of neurofibromatosis type 1 (NF1), although associated with frequent morbidity.
Peripheral nerve involvement among patients with NF1 most frequently tends to be stable or slowly progressive with an indolent and often predictable course.
Close clinical follow-up should remain the first choice when approaching these patients, avoiding potentially disabling surgical complications.
Surgical management in patients with NF1 with peripheral nerve involvement should only be considered if clinical deterioration or if persistent/recurrent symptoms occur.
Acknowledgments
The authors would like to thank Dr António Viana Pinheiro (Department of Neurophysiology—Centro Hospitalar do Porto) for excellent technical and clinical assistance.
Footnotes
Contributors: DF was responsible for the case report design, data analysis and writing. PC was responsible for the initial diagnosis and follow-up as well as for the overall content of the article. MS and RA were responsible for the patient’s recent follow-up and also for case-related data collection. VO and LC were responsible for the initial management of the patient as well as for relevant literature research, imaging and photos selection. All coauthors have contributed to the planning, conduct and reporting of the case report presented in the article.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Créange A, Zeller J, Rostaing-Rigattieri S, et al. Neurological complications of neurofibromatosis type 1 in adulthood. Brain 1999;2013(Pt 3):473. [DOI] [PubMed] [Google Scholar]
- 2.Drouet A, Wolkenstein P, Lefaucheur JP, et al. Neurofibromatosis 1-associated neuropaties: a reappraisal. Brain 2004;2013:1993. [DOI] [PubMed] [Google Scholar]
- 3.Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;2013:51. [PubMed] [Google Scholar]
- 4.Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;2013:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res 2002;2013:1573–7 [PubMed] [Google Scholar]
- 6.Plotkin SR, Bredella MA, Cai W, et al. Quantitative Assessment of Whole-Body Tumor Burden in Adult Patients with Neurofibromatosis. PLoS ONE 2012;7:e35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bianchi S. Ultrasound of the peripheral nerves. Joint Bone Spine 2008;2013:643–9 [DOI] [PubMed] [Google Scholar]
- 8.Bhargava R, Parham DM, Lasater OE, et al. MR imaging differentiation of benign and malignant peripheral nerve sheath tumors: use of the target sign. Pediatr Radiol 1997;2013:124–9 [DOI] [PubMed] [Google Scholar]