

Abstract
The mangrove endophytic fungus Aspergillus terreus (No. GX7-3B) was cultivated in potato dextrose liquid medium, and one rare thiophene compound (1), together with anhydrojavanicin (2), 8-O-methylbostrycoidin (3), 8-O-methyljavanicin (4), botryosphaerone D (5), 6-ethyl-5-hydroxy-3,7-dimethoxynaphthoquinone (6), 3β,5α-dihydroxy-(22E,24R)-ergosta-7,22-dien-6-one (7), 3β,5α,14α-trihydroxy-(22E,24R)-ergosta-7,22-dien-6-one (8), NGA0187 (9) and beauvericin (10), were isolated. Their structures were elucidated by analysis of spectroscopic data. This is the first report of a natural origin for compound 6. Moreover, compounds 3, 4, 5, 7, 8 and 10 were obtained from marine microorganism for the first time. In the bioactive assays in vitro, compounds 2, 3, 9 and 10 displayed remarkable inhibiting actions against α-acetylcholinesterase (AChE) with IC50 values 2.01, 6.71, 1.89, and 3.09 μM, respectively. Furthermore, in the cytotoxicity assays, compounds 7 and 10 exhibited strong or moderate cytotoxic activities against MCF-7, A549, Hela and KB cell lines with IC50 values 4.98 and 2.02 (MCF-7), 1.95 and 0.82 (A549), 0.68 and 1.14 (Hela), and 1.50 and 1.10 μM (KB), respectively; compound 8 had weak inhibitory activities against these tumor cell lines; compounds 1, 2, 3, 4, 5, 6 and 9 exhibited no inhibitory activities against them.
Keywords: mangrove endophytic fungi, thiophene, secondary metabolites, cytotoxicity, AChE
1. Introduction
Marine fungi have been proven to be an important source of structurally novel and bioactive secondary metabolites [1,2,3,4,5], and it is well-known that under various culture conditions, many marine fungi can produce various secondary metabolites, which possess unique structures and bioactivities [6,7]. Aspergillus terreus (No. GX7-3B) is a mangrove endophytic fungal strain from the South China Sea. We have previously reported that four sesquiterpenes together with cyclo [IIe–IIe] dipeptide, ergosterol and ergosterol peroxide were isolated from this strain using glucose yeast-extract peptone (GYP) as the cultivation medium. The sesquiterpenes included botryosphaerin F, a new bioactive compound [8]. However, on further investigation, when the fungal strain was fermented on potato dextrose broth (PDB) medium, many different metabolites from those cultivated on GYP medium were obtained, including compound (1), as well as anhydrojavanicin (2), 8-O-methylbostrycoidin (3), 8-O-methyljavanicin (4), botryosphaerone D (5), 6-ethyl-5-hydroxy-3,7-dimethoxynaphthoquinone (6), 3β,5α-dihydroxy-(22E,24R)-ergosta-7,22-dien-6-one (7), 3β,5α,14α-trihydroxy-(22E,24R)-ergosta-7,22-dien-6-one (8), NGA0187 (9) and beauvericin (10) (see Figure 1) [9,10,11,12,13,14,15,16,17,18,19]. Compound 1 possessed the naphtho[2,3-b]thiophene-4,9-dione system, such a structure hasn’t been encountered in the described yet from nature. This is the first time compound 6 has been isolated from natural sources. In addition, compounds 3, 4, 5, 7, 8 and 10 were isolated from marine microorganism for the first time. Their inhibitory activities against AChE and their cytotoxic activities against MCF-7, A549, Hela and KB cell lines were examined in vitro. Herein, we report the isolation, structure elucidation and biological activities of these compounds.
Figure 1.

Structures of 1–10.
2. Results and Discussion
The crude extract was subjected to a combination of column chromatography on silica gel, sephadex LH-20, C18 reversed phase silica gel, HPLC and reverse phase HPLC.
Compound 1 was isolated as a reddish-orange solid. The HR-EI-MS analysis provided the molecular formula C16H14O6S (obsd m/z = 334.0501 [M]+, calcd 334.0506), indicating the presence of one sulfur atom, and requiring 10 degrees of unsaturation. The UV spectrum absorption band at 259 nm suggested a benzene ring chromophore. 1H NMR spectrum had showed two hydroxy groups (δH = 3.49, s, and δH = 13.53, s), one of which was H-bonded, one methyl group (δH = 1.71, d, J = 6.4 Hz), two O-bearing methyl groups (δH = 4.03 and 4.04, s), two aromatic protons (δH = 6.84, s and δH = 8.09, s), one O-bearing methine (δH = 5.65, q, J = 6.4 Hz). The 13C NMR spectrum showed two carbonyl signals (δC = 177.5 and 187.3), ten aromatic carbon signals (δC = 104.2, 112.0, 117.2, 128.2, 129.4, 140.2, 149.6, 155.6, 156.5 and 162.7), accounting for 7 of 10 degrees of unsaturation required by the molecular formula. These data revealed that compound 1 was a three-ring compound, and was probably a naphthoquinone compound (see Table 1). This was supported by comparison with the reported NMR data for naphthoquinones [20]. The sulfur atom should be in the third ring according to the analysis of the chemical shifts of remaining carbons, especially the downfield methine at δC 65.6, which could only be connected to a hydroxy group and not to a sulfydryl thiol. The 2D NMR spectra (HMBC and COSY) enabled the determination of the overall structure of compound 1 (see Figure 2). The coupled signals from H-10 to H-11 in the 1H–1H COSY spectrum, and HMBC correlations of H-10 with C-11, of H-11 with C-2 and C-10, and of H-3 with C-2, C-3a and C-4 constructed the contiguous sequence from C-11 to C-4. The hydroxy group (δH = 3.49, OH) was located at C-10 (δC = 65.6). So the sulfur atom must be positioned between C-2 and C-9a. The HMBC correlations of H-6 with C-4a, C-7 and C-8, the correlations of H-12 with C-5, of H-13 with C-7, and of 8-OH with C-7, C-8 and C-8a established the positions of two methoxyl groups and downfield hydroxy group. In general, the chemical shift of a H-bonded carbonyl carbon is at lower field than one that isn’t H-bonded, and the assignment of two carbonyl carbons (δC = 177.4, C-4, δC = 187.3, C-9) could also be adequately presented by comparison with naphtoquinones data from literature. The amount of compound 1 was very small, so its absolute stereochemistry has not been determined. All data indicated that compound 1 is a rare thiophene compound, named 8-hydroxy-2-[1-hydroxyethyl]-5,7-dimethoxynaphtho[2,3-b] thiophene-4,9-dione.
Table 1.
NMR spectroscopic data of compound 1 a (in chloroform-d, δ in ppm, J in Hz).
| No. | δC | δH | 1H–1H Cosy | HMBC (H to C) |
|---|---|---|---|---|
| 2 | 162.7 | |||
| 3 | 128.2 | 8.09 (1H, s) | C-2, C-3a, C-4 | |
| 3a | 129.4 | |||
| 4 | 177.5 | |||
| 4a | 112.0 | |||
| 5 | 156.5 | |||
| 6 | 104.2 | 6.84 (1H, s) | C-4a, C-7, C-8 | |
| 7 | 155.6 | |||
| 8 | 149.6 | |||
| 8a | 117.2 | |||
| 9 | 187.3 | |||
| 9a | 140.2 | |||
| 10 | 65.6 | 5.65 (1H, q, J = 6.4) | H-11 | C-11 |
| 11 | 23.3 | 1.71 (3H, d, J = 6.4) | C-2, C-10 | |
| 12 | 57.4 | 4.03 (3H, s) | C-5 | |
| 13 | 56.6 | 4.04 (3H, s) | C-7 | |
| 8-OH | 13.53 (s) | C-7, C-8, C-8a | ||
| 10-OH | 3.49 (s) |
a Measured at 400 MHz (for 1H) and 101 MHz (for 13C).
Figure 2.

Key 1H–1H COSY and HMBC correlations for compound 1.
The known compounds were identified as compounds 2–10 by spectral analysis and comparison with reported literature data. Isolated of compound 6 from natural sources hasn’t previously been reported, although it has been synthesized [14]. In addition, compounds 3, 4, 5, 7, 8 and 10 were obtained from marine microorganism for the first time.
All compounds 1–10 were evaluated as AChE inhibitors, following the method described by Ellman [21], using Huperzine A as reference. Compounds 2, 3, 9 and 10 displayed inhibitory activity of this enzyme with the IC50 values 2.01, 6.71, 1.89, and 3.09 μM respectively (see Table 2).
Table 2.
The inhibitory activities against AChE and cytototicities towards tumor cell lines of compounds 1–10 in vitro.
| Compounds | Inhibition of AChE | Cytotoxicity | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | |||||
| Hela | A549 | MCF-7 | KB | |||
| 1 | - | - | - | - | - | |
| 2 | 2.01 | - | - | - | - | |
| 3 | 6.71 | - | - | - | - | |
| 4 | - | - | - | - | - | |
| 5 | - | - | - | - | - | |
| 6 | - | - | - | - | - | |
| 7 | - | 4.98 | 1.95 | 0.68 | 1.50 | |
| 8 | - | 25.4 | 27.1 | 24.4 | 19.4 | |
| 9 | 1.89 | - | - | - | - | |
| 10 | 3.09 | 2.02 | 0.82 | 1.14 | 1.10 | |
| Huperzine A a | 0.003 | |||||
| Epirubicin a | 1.07 | 0.79 | 0.42 | 0.05 | ||
a as a positive control; “-“ as “no action”.
Compounds 1–10 were further evaluated for inhibitory activities against human breast cancer cells (MCF-7), lung cancer cells (A549), cervix carcinoma cells (Hela) and human nasopharyngeal carcinoma cells (KB). These cell lines were incubated for 72 h with increasing concentrations of compounds 1–10 respectively, and Epirubicin was used as a positive control. IC50 of these compounds were determined by the MTT assay [22] (see Table 2). The results showed compounds 7 and 10 exhibited strong or moderate cytotoxic activies against all of MCF-7, A549, Hela, and KB cell lines; compound 8 had weak inhibitory activities against these tumor cell lines; compounds 1, 2, 3, 4, 5, 6 and 9 exhibited no inhibitory activities against MCF-7, A549, Hela and KB cells lines.
3. Experimental Section
3.1. General
Column chromatography (CC) was carried out on silica gel (200–300 mesh, Qingdao marine Chemical) and sephadex LH-20 (GE Healthcare, Pharmacia, Sweden). The HPLC system consisted of a Waters 2010 series. A mini ODS column (250 × 10 mm, 10 μm particle size) was used. Melting point (m.p.) was detected on Fisher-Johns hot-stage apparatus and was uncorrected. Optical rotation was measured on a Schmidt Haensch Polartronic HH W5 polarimeter and was uncorrected. NMR data were recorded in chloroform-d, using TMS as internal reference on a Varian Inova 400 MHz NMR spectrometer (1H, 400 MHz; 13C, 101MHz). EIMS were on a Thermo DSQ EI-mass spectrometer. LC/MS data were acquired using an Applied Biosystems/MDS Sciex and ESI source. HR-EIMS were measured on a Thermo MAT95XP. UV spectra were measured on a Shimadzu UV-2501 PC spectrophotometer. IR spectra were measured on a Bruker Vector 22 spectrophotometer.
3.2. Strain Isolation, Taxonomic Classification and Endophyte Fermentation
The strain of mangrove endophytic fungus Aspergillus terreus (No. GX7-3B) was isolated from a branch of Bruguiera gymnoihiza (Linn.) Savigny, growing in the coastal salt marsh of the South China Sea in Guangxi province. The strain was stored at School of the Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, China. It was identified according to a molecular biological protocol by DNA amplification and sequencing of the ITS region as described previously with an ITS sequence GenBank ID: KC 461499. The fungal strain was cultivated on potato dextrose liquid medium (20 g of dextrose and 3 g of crude sea salt in 1 L of potato infusion). Starter cultures were maintained on cornmeal seawater agar. Plugs of agar supporting mycelia growth were cut and transferred aseptically into a 500 mL Erlenmeyer flask containing 200 mL of liquid medium, and incubated at 28 °C on a rotary shaker for 5–7 days. The mycelium was aseptically transferred into 1000 mL Erlenmeyer flasks containing 300 mL PDB medium and incubated at 28 ± 1 °C for 30 days under stationary conditions.
3.3. Extraction and Separation of Metabolites
The cultures (130 L) were filtered through cheesecloth. The filtrate was concentrated to about 5 L below 55 °C, and extracted three times with an equal volume of ethyl acetate. The mycelium was air-dried first, and then extracted three times with methanol (5 L × 3). After concentration in vacuo, the combined extract (161.9 g) was chromatographed on silica gel CC using gradient elution with petroleum ether (PE) and ethyl acetate (EA) mixture (90:10–0:100, v/v) to give five fractions (1–5). Fraction 2 (15.3 g) was purified by gradient elution with PE-EA mixture (90:10–30:70, v/v) to give compound 4 (11.6 mg), compound 5 (20.4 mg) and compound 6 (15.7 mg). Fraction 3 (10.8 g) was sequentially purified by gradient elution with PE-EA mixture (30:70–50:50, v/v) to give 3-1 (12.6 mg), 3-2 (15.4 mg) and 3-3 (20.1 mg). These three subfractions were further purified by Sephadex LH-20 gel CC with CHCl3–MeOH (50:50, v/v) as a mobile phase and by preparative HPLC with an ODS column (10 mm × 250 mm), eluting with MeOH–H2O (85:15, v/v) to give compound 2 (2.7 mg), compound 8 (8.2 mg), compound 9 (13.5 mg) and compound 10 (16.5 mg). Fraction 4 (20.7 g) was similarly purified by gradient elution with PE-EA mixture (50:50–70:30, v/v) to give 4-1 (21.6 mg), 4-2 (30.4 mg) and 4-3 (13.7 mg). These three subfractions were further purified by Sephadex LH-20 gel CC with CHCl3–MeOH (50:50, v/v) as a mobile phase and by preparative HPLC with an ODS column (10 mm × 250 mm), eluting with MeOH–H2O (75:25, v/v) to give compound 1 (2.1 mg), compound 3 (2.4 mg) and compound 7 (5.1 mg).
Compound 1: Reddish-orange solid. M.p: 131–132 °C. [α]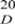 = +271.80 (c = 0.883 mg/mL, MeOH). 1H NMR (400 MHz, CDCl3): δ 1.71 (d, J = 6.4 Hz, 3H, H-11), δ 4.03 (s, 3H, H-12), δ 4.04 (s, 3H, H-13), δ 5.65 (q, J = 6.4 Hz, 1H, H-10), δ 6.84 (s, 1H, H-6), δ 8.09 (s, 1H, H-3), δ 3.49 (s, OH-10), δ 13.53 (s, OH-8). 13C NMR (101 MHz, CDCl3): δ 23.3 (C-11), 56.6 (C-13), 57.4 (C-12), 65.6 (C-10), 104.2 (C-6), 112.0 (C-4a), 117.2 (C-8a), 128.2 (C-3), 129.4 (C-3a), 140.2 (C-9a), 149.6 (C-8), 155.6 (C-7), 156.5 (C-5), 162.7 (C-2), 177.5 (C-4), 187.3 (C-9). EI-MS at m/z = 334, ESI-MS at m/z = 335 [M + H]+; HR-EI-MS m/z: 334.0501 [M]+, (calcd. for C16H14O6S, 334.0506). UV (MeOH): λmax (log ε) = 259 (0.32) nm. IR (KBr): νmax = 3428, 2921, 2852, 1652, 1623, 1544, 1515, 1508, 1460, 1434, 1371, 1316, 1216, 1028, 896, 758, 581, 469 cm−1.
= +271.80 (c = 0.883 mg/mL, MeOH). 1H NMR (400 MHz, CDCl3): δ 1.71 (d, J = 6.4 Hz, 3H, H-11), δ 4.03 (s, 3H, H-12), δ 4.04 (s, 3H, H-13), δ 5.65 (q, J = 6.4 Hz, 1H, H-10), δ 6.84 (s, 1H, H-6), δ 8.09 (s, 1H, H-3), δ 3.49 (s, OH-10), δ 13.53 (s, OH-8). 13C NMR (101 MHz, CDCl3): δ 23.3 (C-11), 56.6 (C-13), 57.4 (C-12), 65.6 (C-10), 104.2 (C-6), 112.0 (C-4a), 117.2 (C-8a), 128.2 (C-3), 129.4 (C-3a), 140.2 (C-9a), 149.6 (C-8), 155.6 (C-7), 156.5 (C-5), 162.7 (C-2), 177.5 (C-4), 187.3 (C-9). EI-MS at m/z = 334, ESI-MS at m/z = 335 [M + H]+; HR-EI-MS m/z: 334.0501 [M]+, (calcd. for C16H14O6S, 334.0506). UV (MeOH): λmax (log ε) = 259 (0.32) nm. IR (KBr): νmax = 3428, 2921, 2852, 1652, 1623, 1544, 1515, 1508, 1460, 1434, 1371, 1316, 1216, 1028, 896, 758, 581, 469 cm−1.
3.4. Method of α-Acetylcholinesterase Inhibitory Activities
α-Acetylcholinesterase inhibitory activity was assayed using 50 mM phosphate buffer (pH 7.0). Compounds 1–10 were dissolved in DMSO to different concentrations. 10 μL of sample solution and 10 μL of α-acetylcholinesterase solution (2 units mL−1, in the phosphate buffer at pH 7.0) were added to 960 μL of phosphate buffer and incubated at 37 °C for 30 min, and then 20 μL of substrate were added to initiate the enzyme reaction. The enzyme reaction was carried out at 37 °C for 30 min. Product was monitored spectrophotometrically by measuring the absorbance (λ = 400 nm). Huperzine A was used as reference standard inhibitor for comparison. Dose-response curves were obtained by performing assays in the presence of increasing concentrations of inhibitors. IC50 value was determined by interpolation of the dose-response curves. For all tests, the inhibition assays were performed in triplicate.
3.5. Cytotoxicity Assays
The cytotoxic activities were determined using MCF-7, A549, Hela, KB human tumor cells by the MTT colorimetric method. Cells were harvested during logarithmic growth phase and seeded in 96-well plates at a density of 1 × 104 cells/mL, and cultured at 37 °C in a humidified incubator (5% CO2) for 24 h, followed by exposure to various concentrations of compounds tested for 48 h. Subsequently 20 μL of MTT solution (5 mg/mL) were added to each well and mixed, the cells were then incubated for an additional 4 h. Culture supernatant were removed, 150 μL of DMSO were added to each well to fully dissolve the MTT—formazan crystals. Cell growth inhibition was determined by measuring the absorbance (Abs) at λ = 570 nm using a microplate reader and calculated according to the following equation:
| Growth inhibition = (1 − OD of treated cells/OD of control cells) × 100% | (1) |
The half maximal inhibitory concentrations (IC50) were obtained from linear regression analysis of the concentration-response curves plotted for each tested compound (Bliss’s software). Results were expressed as the mean value of triplicate data points.
4. Conclusions
This work showed that Aspergillus terreus (No. GX7-3B) could produce a rich variety of secondary metabolites, and small changes in the cultivation conditions could completely shift the metabolic profile of this strain. Its metabolites obtained on PDB medium were very different from those of on GYP medium. When the strain was cultivated on PDB medium, one novel sulfur-containing compound, as well as nine known compounds were isolated, which had exhibited antitumor or AChE inhibition activities.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (21172271, 20772162), and the National Science Foundation of Guangdong Province, China (S2011020001231).
Supplementary Files
Supplementary Information (PDF, 285 KB)
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Huang C.H., Pan J.H., Chen B., Yu M., Huang H.B., Zhu X., Lu Y.J., She Z.G., Lin Y.C. Three bianthraquinone derivatives from the mangrove endophytic fungus Alternaria sp. ZJ9-6B from the South China Sea. Mar. Drugs. 2011;9:832–843. doi: 10.3390/md9050832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J., Zhang D.M., Zhu X., He Z.J., Liu S., Li M.F., Pang J.Y., Lin Y.C. Studies on synthesis and structure-activity relationship (SAR) of derivatives of a new natural product from marine fungi as inhibitors of influenza virus neuraminidase. Mar. Drugs. 2011;9:1887–1901. doi: 10.3390/md9101887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen G.Y., Lin Y.C., Wen L., Vrijmoed L.L.P., Jones E.B.G. Two new metabolites of a marine endophytic fungus (No. 1893) from an estuarine mangrove on the South China Sea Coast. Tetrahedron. 2003;59:4907–4909. doi: 10.1016/S0040-4020(03)00703-8. [DOI] [Google Scholar]
- 4.Huang H.B., Feng X.J., Liu L., Chen B., Lu Y.J., Ma L., She Z.G., Lin Y.C. Three dimeric naphtho-y-pyrones from the mangrove endophytic fungus Aspergillus tubingensis isolated from pongamia pinnata. Planta Med. 2010;76:1–4. doi: 10.1055/s-0029-1240671. [DOI] [PubMed] [Google Scholar]
- 5.Huang H., She Z., Lin Y., Vrijmoed L.L.P., Lin W. Cyclic peptides from an endophytic fungus obtained from a mangrove leaf (Kandelia candel) J. Nat. Prod. 2007;70:1696–1699. doi: 10.1021/np0605891. [DOI] [PubMed] [Google Scholar]
- 6.Xia X.K., Li Q., Li J., Shao C.L., Zhang J.Y., Zhang Y.G., Liu X., Lin Y.C., Liu C.H., She Z.G. Two new derivatives of griseofulvin from the mangrove endophytic fungus nigrospora sp. (No. 1403) from Kandelia Candel (L.) Druce. Planta Med. 2011;77:1735–1738. doi: 10.1055/s-0030-1271040. [DOI] [PubMed] [Google Scholar]
- 7.Xia X.K., Huang H.R., She Z.G., Shao C.L., Liu F., Cai X.L., Vrijmoed L.L.P., Lin Y.C. 1H and 13C NMR assignments for five anthraquinones from the mangrove endophytic fungus Halorosellinia sp. (No. 1403) Magn. Reson. Chem. 2007;45:1006–1009. doi: 10.1002/mrc.2078. [DOI] [PubMed] [Google Scholar]
- 8.Deng C.M., Huang C.H., Wu Q.L., Pang J.Y., Lin Y.C. A new sesquiterpene from the mangrove endophytic fungus Aspergillus terreus (No. GX7-3B) Nat. Prod. Res. 2013 doi: 10.1080/14786419.2013.778847. [DOI] [PubMed] [Google Scholar]
- 9.Yasuo K., Atsumi S., Hiromitsu N., Hamasaki T. Structures of naphthoquinones produced by the fungus, Fusarium sp., and their biological activity toward Pollen Germination. Agric. Biol. Chem. 1988;52:1253–1259. doi: 10.1271/bbb1961.52.1253. [DOI] [Google Scholar]
- 10.Tatum J.H., Baker R.A., Berry R.E. Metabolites of Fusarium solani. Phytochemistry. 1988;28:283–284. doi: 10.1016/0031-9422(89)85062-9. [DOI] [Google Scholar]
- 11.Cameron D.W., Deutscher K.R., Feutrill G.I. Nucleophilic Alkenes. IX. Addition of 1,l-dimethoxyethene to azanaphthoquinones: synthesis of bostrycoidin and 8-O-methylbostrycoidin. Aust. J. Chem. 1982;35:1439–1450. doi: 10.1071/CH9821439. [DOI] [Google Scholar]
- 12.Kimura Y., Hamasaki T., Nakajima H. Isolation, identification and biological activities of 8-O-methyljavanicin produced by Fusarium solani. Agric. Biol. Chem. 1981;45:2653–2654. doi: 10.1271/bbb1961.45.2653. [DOI] [Google Scholar]
- 13.Xu Y.H., Lu C.H., Zheng Z.H., Shen Y.M. New polyketides isolated from Botryosphaeria australis Strain ZJ12-1A. Helv. Chim. Acta. 2011;94:897–902. doi: 10.1002/hlca.201000345. [DOI] [Google Scholar]
- 14.Caron B., Brassard P. Regiospecific α-substitution of crotonic esters: Synthesis of naturally occurring derivatives of 6-ethyljuglone. Tetrahedron. 1991;47:4287–4298. doi: 10.1016/S0040-4020(01)87099-X. [DOI] [Google Scholar]
- 15.Ishizuka T., Yaoita Y., Kikuchi M. Sterol constituents from the fruit bodies of Grifola frondosa (Fr.) SF Gray. Chem. Pharm. Bull. 1997;45:1756–1760. doi: 10.1248/cpb.45.1756. [DOI] [PubMed] [Google Scholar]
- 16.Valisolalao J., Luu B., Ourisson G. Chemical and biochemical study of chinese drugs. Part VIII. Cytotoxic steroids from Polyporus versicolor. Tetrahedron. 1983;39:2779–2785. doi: 10.1016/S0040-4020(01)82446-7. [DOI] [Google Scholar]
- 17.Nozawa Y., Sakai N., Matsumoto K., Mizoue K. A novel neuritogenic compound, NGA0187. J. Antibiot. 2002;55:629–634. doi: 10.7164/antibiotics.55.629. [DOI] [PubMed] [Google Scholar]
- 18.Hamill R.L., Higgens C.E., Boaz H.E., Gorman M. The structure of beauvericin, a new depsipeptide antibiotic toxic to Artemia salina. Tetrahedron Lett. 1969;49:4255–4258. [Google Scholar]
- 19.Li C.Y., Chen S., Zuo C.W., Kuang R.B., Yi G.J. Identification of beauvericin, a novel mycotoxin from Fusarium oxysporum f. sp. Cubense. Acta Hortic. Sin. 2011;38:2092–2098. [Google Scholar]
- 20.Yu B.Z., Song C.Z., Du Z.Z., Cai X.H., Huang S.X., Luo X.D. Naphthaquinones from Fusarium sp. 1RGa-1b, an endophytic fungus associated with Trewia nudiflora (Euphorbiaceae) Nat. Prod. Res. Dev. 2009;21:574–576. [Google Scholar]
- 21.Ellman G.L., Courtney K.D., Andres V.J., Feather-Stone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J.Y., Tao L.Y., Liang Y.J., Yan Y.Y., Dai C.L., Xia X.K., She Z.G., Lin Y.C., Fu L.W. Secalonic acid D induced leukemia cell apoptosis and cell cycle arrest of G(1) with involvement of GSK-3beta/beta-catenin/c-Myc pathway. Cell Cycle. 2009;8:2444–2450. doi: 10.4161/cc.8.15.9170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information (PDF, 285 KB)